
Overcoming the Limitations of CRISPR-Cas9 Systems in Saccharomyces cerevisiae: Off-Target Effects, Epigenome, and Mitochondrial Editing

Abstract
1. Introduction
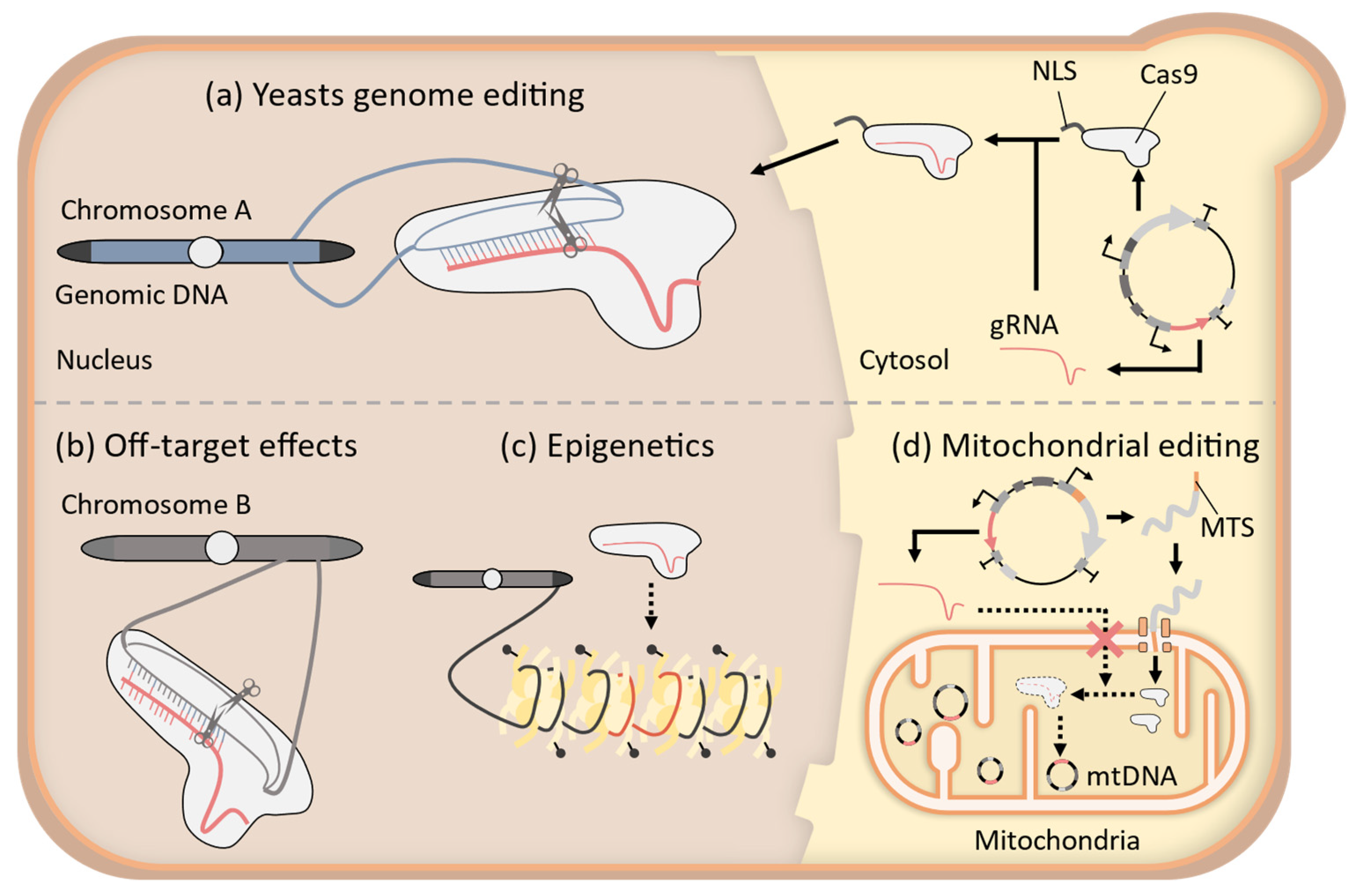
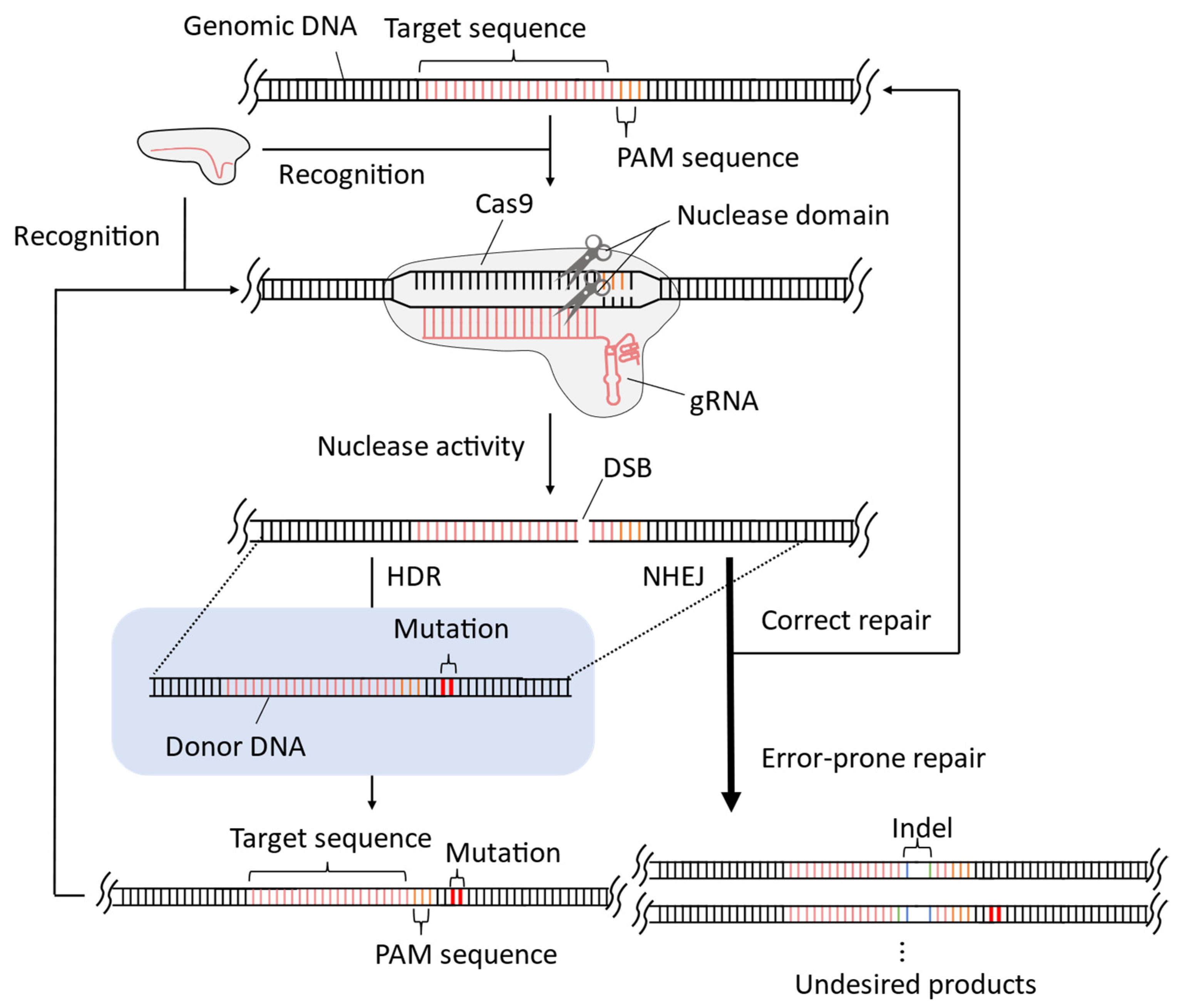
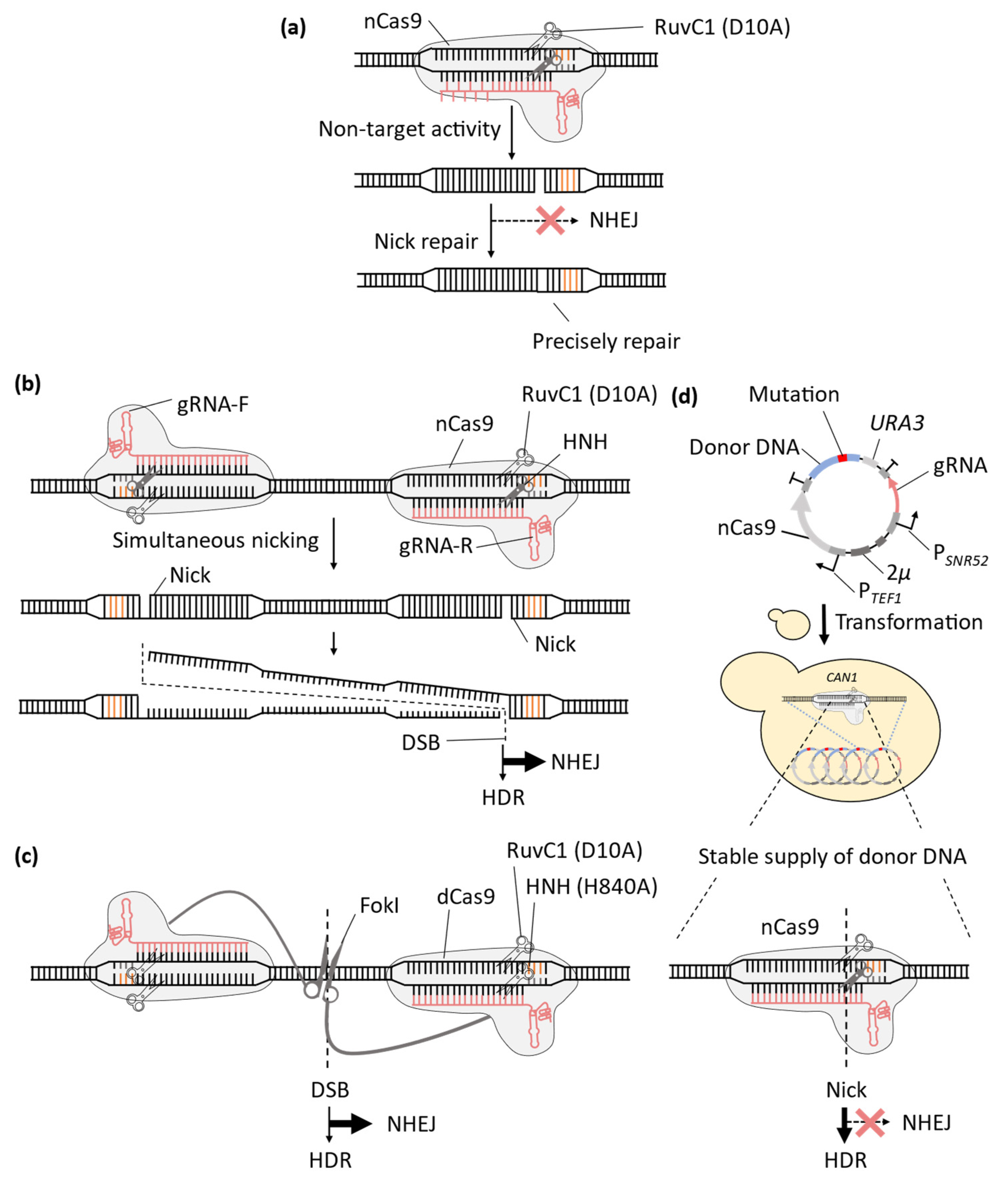
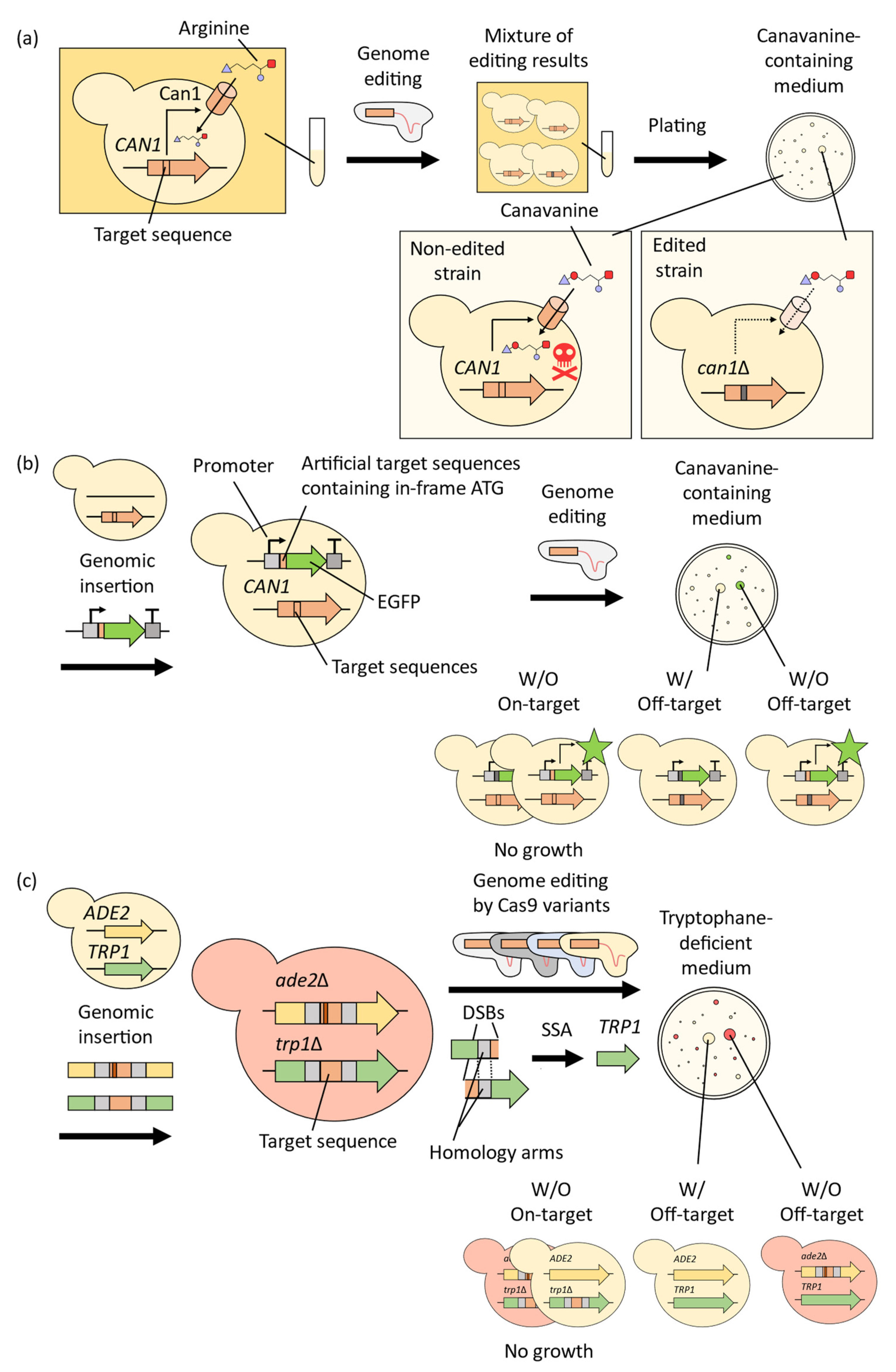
2. Improvement of CRISPR-Cas9 System for Precise Genome-Wide Editing in Yeast
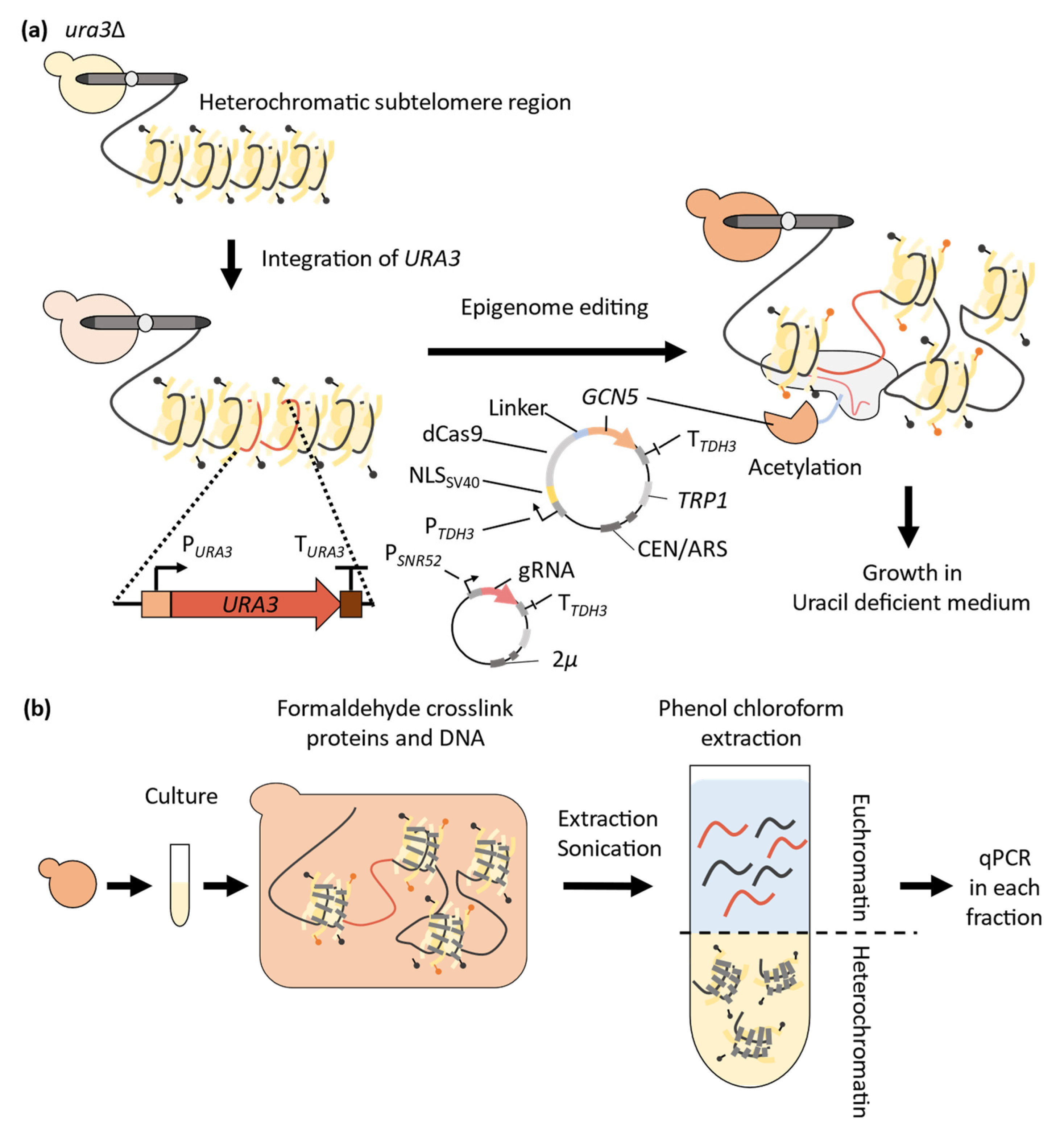
3. Application of CRISPR-Cas9 System to Epigenome Editing in Yeast
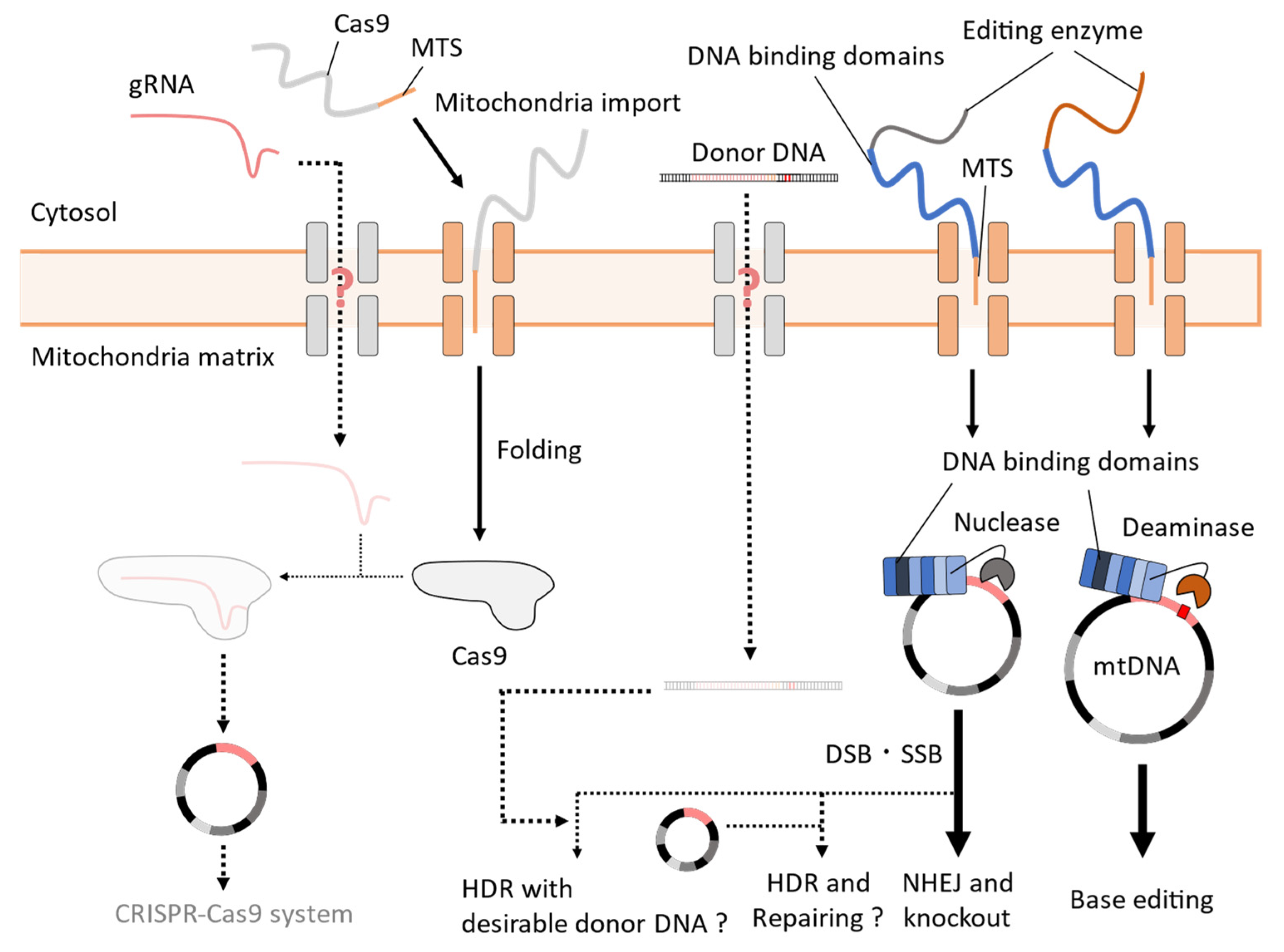
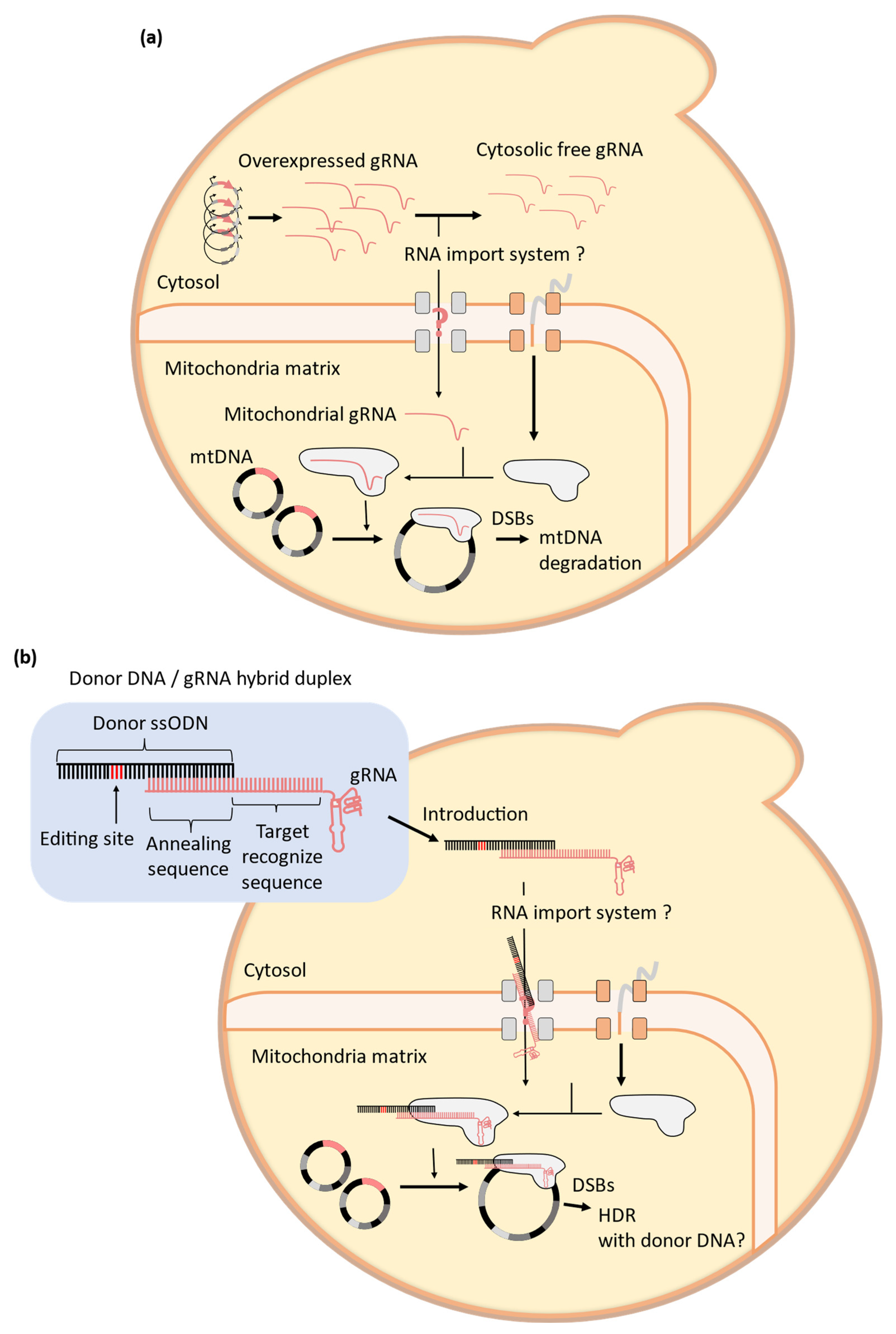
4. Challenges to the Establishment of Mitochondrial CRISPR-Cas9 Systems in Yeast

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Editing Enzymes | DNA Targeting Elements | Types of Mutations | Editing Efficiency | Off-Target Editing Frequency of mtDNA | Refs |
|---|---|---|---|---|---|
| DdCBEs | TALE repeats | 5′-TC-3′ to 5′-TT-3′ | 5–50% | 0.024–0.049% | [111] |
| TALEDs | TALE repeats | A to G | 49% | 0.008–0.013% | [17] |
| HiFi-DdCBEs | TALE repeats | 5′-TC-3′ to 5′-TT-3′ | 53–61% | 0.001–0.0175% | [103] |
| MTS-Cas9 | gRNA | Reduction of mtDNA | 30–60% | Not reported | [22,98,100] |
| mito-Cas9 system | gRNA | Small sequence insertion | 0.03–0.23% | Low | [20] |
5. Summary and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Baudin, A.; Ozier-kalogeropoulos, O.; Denouel, A.; Lacroute, F.; Cullin, C. A Simple and Efficient Method for Direct Gene Deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993, 21, 3329. [Google Scholar] [CrossRef]
- DiCarlo, J.E.; Norville, J.E.; Mali, P.; Rios, X.; Aach, J.; Church, G.M. Genome Engineering in Saccharomyces cerevisiae Using CRISPR-Cas Systems. Nucleic Acids Res. 2013, 41, 4336–4343. [Google Scholar] [CrossRef]
- Jakočiunas, T.; Jensen, M.K.; Keasling, J.D. CRISPR/Cas9 Advances Engineering of Microbial Cell Factories. Metab. Eng. 2016, 34, 44–59. [Google Scholar] [CrossRef]
- Shaw, W.M.; Yamauchi, H.; Mead, J.; Gowers, G.O.F.; Bell, D.J.; Öling, D.; Larsson, N.; Wigglesworth, M.; Ladds, G.; Ellis, T. Engineering a Model Cell for Rational Tuning of GPCR Signaling. Cell 2019, 177, 782–796.e27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hansen, L.G.; Gudich, O.; Viehrig, K.; Lassen, L.M.M.; Schrübbers, L.; Adhikari, K.B.; Rubaszka, P.; Carrasquer-Alvarez, E.; Chen, L.; et al. A Microbial Supply Chain for Production of the Anti-Cancer Drug Vinblastine. Nature 2022, 609, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Schultz, C.; Cao, M.; HamediRad, M.; Zhao, H. Multi-Functional Genome-Wide CRISPR System for High Throughput Genotype–Phenotype Mapping. Nat. Commun. 2019, 10, 5794. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA Targeting Specificity of RNA-Guided Cas9 Nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-Seq Enables Genome-Wide Profiling of off-Target Cleavage by CRISPR-Cas Nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.S.; Hinz, J.M.; Wyrick, J.J. Tips, Tricks, and Potential Pitfalls of CRISPR Genome Editing in Saccharomyces Cerevisiae. Front. Bioeng. Biotechnol. 2022, 10, 924914. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target Specificity of the CRISPR-Cas9 System. Quant. Biol. 2014, 2, 59–70. [Google Scholar] [CrossRef]
- Nicoglou, A.; Merlin, F. Epigenetics: A Way to Bridge the Gap between Biological Fields. Stud. Hist. Philos. Biol. Biomed. Sci. 2017, 66, 73–82. [Google Scholar] [CrossRef]
- Cavalli, G.; Heard, E. Advances in Epigenetics Link Genetics to the Environment and Disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.L.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA Mutations and Human Disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 113–128. [Google Scholar] [CrossRef]
- Cring, M.R.; Sheffield, V.C. Gene Therapy and Gene Correction: Targets, Progress, and Challenges for Treating Human Diseases. Gene Ther. 2022, 29, 3–12. [Google Scholar] [CrossRef]
- Cho, S.I.; Lee, S.; Mok, Y.G.; Lim, K.; Lee, J.; Lee, J.M.; Chung, E.; Kim, J.S. Targeted A-to-G Base Editing in Human Mitochondrial DNA with Programmable Deaminases. Cell 2022, 185, 1764–1776.e12. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; Kotrys, A.V.; Raguram, A.; Huang, T.P.; Mootha, V.K.; Liu, D.R. CRISPR-Free Base Editors with Enhanced Activity and Expanded Targeting Scope in Mitochondrial and Nuclear DNA. Nat. Biotechnol. 2022, 40, 1378–1387. [Google Scholar] [CrossRef]
- Schmiderer, L.; Yudovich, D.; Oburoglu, L.; Hjort, M.; Larsson, J. Site-Specific CRISPR-Based Mitochondrial DNA Manipulation Is Limited by gRNA Import. Sci. Rep. 2022, 12, 18687. [Google Scholar] [CrossRef] [PubMed]
- Bi, R.; Li, Y.; Xu, M.; Zheng, Q.; Zhang, D.-F.; Li, X.; Ma, G.; Xiang, B.; Zhu, X.; Zhao, H.; et al. Direct Evidence of CRISPR-Cas9-Mediated Mitochondrial Genome Editing. Innov. J. 2022, 3, 100329. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Luo, J.; Huang, D.; Li, H. Current Progress of Mitochondrial Genome Editing by CRISPR. Front. Physiol. 2022, 13, 884. [Google Scholar] [CrossRef] [PubMed]
- Loutre, R.; Heckel, A.M.; Smirnova, A.; Entelis, N.; Tarassov, I. Can Mitochondrial DNA Be CRISPRized: Pro and Contra. IUBMB Life 2018, 70, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.C.; Yadav, N.S.; Orozco, E.M.; Sakai, H. Cas9/gRNA-Mediated Genome Editing of Yeast Mitochondria and Chlamydomonas Chloroplasts. PeerJ 2020, 8, e8362. [Google Scholar] [CrossRef]
- Zheng, T.; Hou, Y.; Zhang, P.; Zhang, Z.; Xu, Y.; Zhang, L.; Niu, L.; Yang, Y.; Liang, D.; Yi, F.; et al. Profiling Single-Guide RNA Specificity Reveals a Mismatch Sensitive Core Sequence. Sci. Rep. 2017, 7, 40638. [Google Scholar] [CrossRef]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 Transcriptional Activators for Target Specificity Screening and Paired Nickases for Cooperative Genome Engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef]
- Bravo, J.P.K.; Liu, M.S.; Hibshman, G.N.; Dangerfield, T.L.; Jung, K.; McCool, R.S.; Johnson, K.A.; Taylor, D.W. Structural Basis for Mismatch Surveillance by CRISPR–Cas9. Nature 2022, 603, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural Basis of PAM-Dependent Target DNA Recognition by the Cas9 Endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef]
- Legut, M.; Daniloski, Z.; Xue, X.; McKenzie, D.; Guo, X.; Wessels, H.H.; Sanjana, N.E. High-Throughput Screens of PAM-Flexible Cas9 Variants for Gene Knockout and Transcriptional Modulation. Cell Rep. 2020, 30, 2859–2868. [Google Scholar] [CrossRef] [PubMed]
- Gooden, A.A.; Evans, C.N.; Sheets, T.P.; Clapp, M.E.; Chari, R. DbGuide: A Database of Functionally Validated Guide RNAs for Genome Editing in Human and Mouse Cells. Nucleic Acids Res. 2021, 49, D871–D876. [Google Scholar] [CrossRef] [PubMed]
- Waldrip, Z.J.; Jenjaroenpun, P.; DeYoung, O.; Nookaew, I.; Taverna, S.D.; Raney, K.D.; Tackett, A.J. Genome-Wide Cas9 Binding Specificity in Saccharomyces cerevisiae. PeerJ 2020, 8, e9442. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of Double-Strand Breaks Induced by CRISPR–Cas9 Leads to Large Deletions and Complex Rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Satomura, A.; Nishioka, R.; Mori, H.; Sato, K.; Kuroda, K.; Ueda, M. Precise Genome-Wide Base Editing by the CRISPR Nickase System in Yeast. Sci. Rep. 2017, 7, 41538. [Google Scholar] [CrossRef]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient Introduction of Specific Homozygous and Heterozygous Mutations Using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Vriend, L.E.M.; Krawczyk, P.M. Nick-Initiated Homologous Recombination: Protecting the Genome, One Strand at a Time. DNA Repair 2017, 50, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-Guided FokI Nucleases for Highly Specific Genome Editing. Nat. Biotechnol. 2014, 32, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of Catalytically Inactive Cas9 to FokI Nuclease Improves the Specificity of Genome Modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef]
- Saleh-Gohari, N.; Bryant, H.E.; Schultz, N.; Parker, K.M.; Cassel, T.N.; Helleday, T. Spontaneous Homologous Recombination Is Induced by Collapsed Replication Forks That Are Caused by Endogenous DNA Single-Strand Breaks. Mol. Cell. Biol. 2005, 25, 7158–7169. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.C.; Takeuchi, R.; Pellenz, S.; Davis, L.; Maizels, N.; Monnat, R.J.; Stoddard, B.L. Generation of a Nicking Enzyme That Stimulates Site-Specific Gene Conversion from the I-Anil LAGLIDADG Homing Endonuclease. Proc. Natl. Acad. Sci. USA 2009, 106, 5099–5104. [Google Scholar] [CrossRef]
- Strathern, J.N.; Weinstock, K.G.; Higgins, D.R.; McGill, C.B. A Novel Recombinator in Yeast Based on Gene II Protein from Bacteriophage F1. Genetics 1991, 127, 61–73. [Google Scholar] [CrossRef]
- Casini, A.; Olivieri, M.; Petris, G.; Montagna, C.; Reginato, G.; Maule, G.; Lorenzin, F.; Prandi, D.; Romanel, A.; Demichelis, F.; et al. A Highly Specific SpCas9 Variant Is Identified by in vivo Screening in Yeast. Nat. Biotechnol. 2018, 36, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Lafrance, B.; Kaplan, M.; Doudna, J.A. Conformational Control of DNA Target Cleavage by CRISPR–Cas9. Nature 2015, 527, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally Engineered Cas9 Nucleases with Improved Specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-Fidelity CRISPR-Cas9 Nucleases with No Detectable Genome-Wide off-Target Effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced Proofreading Governs CRISPR-Cas9 Targeting Accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef]
- Lee, J.K.; Jeong, E.; Lee, J.; Jung, M.; Shin, E.; Kim, Y.H.; Lee, K.; Jung, I.; Kim, D.; Kim, S.; et al. Directed Evolution of CRISPR-Cas9 to Increase Its Specificity. Nat. Commun. 2018, 9, 3048. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, H.K.; Lee, S.; Seo, J.H.; Choi, J.W.; Park, J.; Min, S.; Yoon, S.; Cho, S.R.; Kim, H.H. Prediction of the Sequence-Specific Cleavage Activity of Cas9 Variants. Nat. Biotechnol. 2020, 38, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, J.; Janssen, J.M.; Le Bouteiller, M.; Frock, R.L.; Gonçalves, M.A.F.V. Precise and Broad Scope Genome Editing Based on High-Specificity Cas9 Nickases. Nucleic Acids Res. 2021, 49, 1173–1198. [Google Scholar] [CrossRef]
- Ahmad, M.; Bussey, H. Yeast Arginine Permease: Nucleotide Sequence of the CAN1 Gene. Curr. Genet. 1986, 10, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bai, Y.; Cheng, X.; Kalds, P.G.T.; Sun, B.; Wu, Y.; Lv, H.; Xu, K.; Zhang, Z. Efficient SSA-Mediated Precise Genome Editing Using CRISPR/Cas9. FEBS J. 2018, 285, 3362–3375. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, S.; Bruschi, C.V. The Red/White Colony Color Assay in the Yeast Saccharomyces cerevisiae: Epistatic Growth Advantage of White Ade8-18, Ade2 Cells over Red Ade2 Cells. Curr. Genet. 1996, 30, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-Guided Activation of Endogenous Human Genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Li, X.; Fu, X.-D. Chromatin-Associated RNAs as Facilitators of Functional Genomic Interactions. Nat. Rev. Genet. 2019, 20, 503–519. [Google Scholar] [CrossRef]
- Kim, J.H.; Rege, M.; Valeri, J.; Dunagin, M.C.; Metzger, A.; Titus, K.R.; Gilgenast, T.G.; Gong, W.; Beagan, J.A.; Raj, A.; et al. LADL: Light-Activated Dynamic Looping for Endogenous Gene Expression Control. Nat. Methods 2019, 16, 633–639. [Google Scholar] [CrossRef]
- Chen, T.; Dent, S.Y.R. Chromatin Modifiers and Remodellers: Regulators of Cellular Differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef]
- Mirabella, A.C.; Foster, B.M.; Bartke, T. Chromatin Deregulation in Disease. Chromosoma 2016, 125, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y. Transcriptional Silencing in Saccharomyces cerevisiae and Schizosaccharomyces pombe. Nucleic Acids Res. 2002, 30, 1465–1482. [Google Scholar] [CrossRef]
- Bouyx, C.; Schiavone, M.; François, J.M. FLO11, a Developmental Gene Conferring Impressive Adaptive Plasticity to the Yeast Saccharomyces cerevisiae. Pathogens 2021, 10, 1509. [Google Scholar] [CrossRef]
- Canzonetta, C.; Leo, M.; Guarino, S.R.; Montanari, A.; Francisci, S.; Filetici, P. SAGA Complex and Gcn5 Are Necessary for Respiration in Budding Yeast. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 3160–3168. [Google Scholar] [CrossRef]
- Reardon, R.M.; Walsh, A.K.; Larsen, C.I.; Schmidberger, L.A.H.; Morrow, L.A.; Thompson, A.E.; Wellik, I.M.; Thompson, J.S. An Epigenetically Inherited UV Hyper-Resistance Phenotype in Saccharomyces cerevisiae. Epigenetics Chromatin 2022, 15, 31. [Google Scholar] [CrossRef]
- Jensen, K.T.; Fløe, L.; Petersen, T.S.; Huang, J.; Xu, F.; Bolund, L.; Luo, Y.; Lin, L. Chromatin Accessibility and Guide Sequence Secondary Structure Affect CRISPR-Cas9 Gene Editing Efficiency. FEBS Lett. 2017, 591, 1892–1901. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-Wide Binding of the CRISPR Endonuclease Cas9 in Mammalian Cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef]
- Yarrington, R.M.; Verma, S.; Schwartz, S.; Trautman, J.K.; Carroll, D. Nucleosomes Inhibit Target Cleavage by CRISPR-Cas9 in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, 9351–9358. [Google Scholar] [CrossRef]
- Turman; Gaea; Pollard, D. “NucJuke—A gRNA Re-Evaluation Tool for CRISPR-Cas9 Experiments in Yeast”, Ver. 1.0.0, April 2019. Available online: https://nucjuke.biol.wwu.edu (accessed on 11 April 2023).
- Hilton, I.B.; Vockley, C.M.; Pratiksha, I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A.; Carolina, N.; States, U.; Biology, C.; Carolina, N. Epigenome Editing by a CRISPR/Cas9-Based Acetyltransferase Activates Genes from Promoters and Enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Yamanashi, Y.; Fujimura, A.; Sato, Y.; Kujirai, T.; Kurumizaka, H.; Kimura, H.; Yamatsugu, K.; Kawashima, S.A.; Kanai, M. Live-Cell Epigenome Manipulation by Synthetic Histone Acetylation Catalyst System. Proc. Natl. Acad Sci. USA 2021, 118, e2019554118. [Google Scholar] [CrossRef]
- Dekker, F.J.; Van Den Bosch, T.; Martin, N.I. Small Molecule Inhibitors of Histone Acetyltransferases and Deacetylases Are Potential Drugs for Inflammatory Diseases. Drug Discov. Today 2014, 19, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Olejar, K.J.; On, S.L.W.; Winefield, C.; Wescombe, P.A.; Brennan, C.S.; Hider, R.N.; Chelikani, V. Epigenetic Changes in Saccharomyces cerevisiae Alters the Aromatic Profile in Alcoholic Fermentation. Appl. Environ. Microbiol. 2022, 88, e0152822. [Google Scholar] [CrossRef] [PubMed]
- Torres-Garcia, S.; Yaseen, I.; Shukla, M.; Audergon, P.N.C.B.; White, S.A.; Pidoux, A.L.; Allshire, R.C. Epigenetic Gene Silencing by Heterochromatin Primes Fungal Resistance. Nature 2020, 585, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Garber, M.; Biology, I. Functional Annotation of Native Enhancers with a Cas9-Histone Demethylase Fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA Demethylation in vIvo Using dCas9-Peptide Repeat and sCFv-TET1 Catalytic Domain Fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef]
- Kwon, D.Y.; Zhao, Y.-T.; Lamonica, J.M.; Zhou, Z. Locus-Specific Histone Deacetylation Using a Synthetic CRISPR-Cas9-Based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef]
- Park, M.; Patel, N.; Keung, A.J.; Khalil, A.S. Engineering Epigenetic Regulation Using Synthetic Read-Write Modules. Cell 2019, 176, 227–238.e20. [Google Scholar] [CrossRef] [PubMed]
- Proffitt, J.H.; Davie, J.R.; Swinton, D.; Hattman, S. 5-Methylcytosine Is Not Detectable in Saccharomyces cerevisiae DNA. Mol. Cell. Biol. 1984, 4, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gao, X.-D.; Wang, Y.; Yuan, B.-F.; Feng, Y.-Q. Widespread Existence of Cytosine Methylation in Yeast DNA Measured by Gas Chromatography/Mass Spectrometry. Anal. Chem. 2012, 84, 7249–7255. [Google Scholar] [CrossRef]
- Capuano, F.; Mülleder, M.; Kok, R.; Blom, H.J.; Ralser, M. Cytosine DNA Methylation Is Found in Drosophila melanogaster but Absent in Saccharomyces cerevisiae, Schizosaccharomyces pombe, and Other Yeast Species. Anal. Chem. 2014, 86, 3697–3702. [Google Scholar] [CrossRef]
- Sugiyama, K.-I.; Furusawa, H.; Grúz, P.; Honma, M. Functional Role of DNA Methylation at the FLO1 Promoter in Budding Yeast. FEMS Microbiol. Lett. 2017, 364, fnx221. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.; Cui, L.; Xiong, Z.; Qi, Q.; Hou, J. CRISPR-Mediated Protein-Tagging Signal Amplification Systems for Efficient Transcriptional Activation and Repression in Saccharomyces cerevisiae. Nucleic Acids Res. 2022, 50, 5988–6000. [Google Scholar] [CrossRef] [PubMed]
- Farzadfard, F.; Perli, S.D.; Lu, T.K. Tunable and Multifunctional Eukaryotic Transcription Factors Based on CRISPR/Cas. ACS Synth. Biol. 2013, 2, 604. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Sato, G.; Amai, T.; Ueda, M.; Kuroda, K. Development of Artificial System to Induce Chromatin Loosening in Saccharomyces cerevisiae. Biomolecules 2022, 12, 1138. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.S.; Gottschling, D.E. TLC1:Template RNA Component of Saccharomyces cerevisiae Telomerase. Science 1994, 266, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE) to Isolate Active Regulatory DNA. Nat. Protoc. 2012, 7, 256–267. [Google Scholar] [CrossRef]
- Segorbe, D.; Wilkinson, D.; Mizeranschi Alexandruand Hughes, T.; Aaløkken, R.; Váchová, L.; Palková, Z.; Gilfillan, G.D. An Optimized FAIRE Procedure for Low Cell Numbers in Yeast. Yeast 2018, 35, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Paduch, M.; Kim, S.A.; Kramer, R.M.; Barrios, A.F.; Lu, V.; Luke, J.; Usatyuk, S.; Kossiakoff, A.A.; Tan, S. Structural Basis for Activation of SAGA Histone Acetyltransferase Gcn5 by Partner Subunit Ada2. Proc. Natl. Acad. Sci. USA 2018, 115, 10010–10015. [Google Scholar] [CrossRef]
- Saraste, M. Oxidative Phosphorylation at the Fin de Siècle. Science 1999, 283, 1488–1493. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-Y.; Seol, D.-W. The Role of Mitochondria in Apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.P.; Rahman, H.S. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194. [Google Scholar] [CrossRef]
- Klucnika, A.; Ma, H. Mapping and Editing Animal Mitochondrial Genomes: Can We Overcome the Challenges? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190187. [Google Scholar] [CrossRef]
- Rahman, S. Mitochondrial Disease and Epilepsy. Dev. Med. Child Neurol. 2012, 54, 397–406. [Google Scholar] [CrossRef]
- Blass, J.P. The Mitochondrial Spiral: An Adequate Cause of Dementia in the Alzheimer’s Syndrome. Ann. N. Y. Acad. Sci. 2000, 924, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101. [Google Scholar] [CrossRef] [PubMed]
- Jo, A.; Ham, S.; Lee, G.H.; Lee, Y.I.; Kim, S.; Lee, Y.S.; Shin, J.H.; Lee, Y. Efficient Mitochondrial Genome Editing by CRISPR/Cas9. Biomed Res. Int. 2015, 2015, 305716. [Google Scholar] [CrossRef]
- Bian, W.P.; Chen, Y.L.; Luo, J.J.; Wang, C.; Xie, S.L.; Pei, D.S. Knock-In Strategy for Editing Human and Zebrafish Mitochondrial DNA Using Mito-CRISPR/Cas9 System. ACS Synth. Biol. 2019, 8, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.R.A.; Yalvac, M.E.; Khoo, B.; Eckardt, S.; McLaughlin, K.J. Adapting CRISPR/Cas9 System for Targeting Mitochondrial Genome. Front. Genet. 2021, 12, 402. [Google Scholar] [CrossRef]
- Antón, Z.; Mullally, G.; Ford, H.C.; van der Kamp, M.W.; Szczelkun, M.D.; Lane, J.D. Mitochondrial Import, Health and MtDNA Copy Number Variability Seen When Using Type II and Type V CRISPR Effectors. J. Cell Sci. 2021, 133, jcs248468. [Google Scholar]
- Wang, G.; Shimada, E.; Zhang, J.; Hong, J.S.; Smith, G.M.; Teitell, M.A.; Koehler, C.M. Correcting Human Mitochondrial Mutations with Targeted RNA Import. Proc. Natl. Acad. Sci. USA 2012, 109, 4840–4845. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, H.; Baek, G.; Kim, J.-S. Precision Mitochondrial DNA Editing with High-Fidelity DddA-Derived Base Editors. Nat. Biotechnol. 2022, 41, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Amai, T.; Tsuji, T.; Ueda, M.; Kuroda, K. Development of a Mito-CRISPR System for Generating Mitochondrial DNA-Deleted Strain in Saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 2021, 85, 895–901. [Google Scholar] [CrossRef]
- Silva-Pinheiro, P.; Minczuk, M. The Potential of Mitochondrial Genome Engineering. Nat. Rev. Genet. 2022, 23, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.; Dürr, M.; Westermann, B. Saccharomyces cerevisiae as a Model Organism to Study Mitochondrial Biology: General Considerations and Basic Procedures. Methods Mol. Biol. 2007, 372, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Tarassov, I.A.; Martin, R.P. Mechanisms of TRNA Import into Yeast Mitochondria: An Overview. Biochimie 1996, 78, 502–510. [Google Scholar] [CrossRef]
- Frechin, M.; Senger, B.; Braye, M.; Kern, D.; Martin, R.P.; Becker, H.D. Yeast Mitochondrial Gln-TRNAGln Is Generated by a GatFAB-Mediated Transamidation Pathway Involving Arc1p-Controlled Subcellular Sorting of Cytosolic GluRS. Genes Dev. 2009, 23, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Tonin, Y.; Heckel, A.M.; Dovydenko, I.; Meschaninova, M.; Comte, C.; Venyaminova, A.; Pyshnyi, D.; Tarassov, I.; Entelis, N. Characterization of Chemically Modified Oligonucleotides Targeting a Pathogenic Mutation in Human Mitochondrial DNA. Biochimie 2014, 100, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Gowher, A.; Smirnov, A.; Tarassov, I.; Entelis, N. Induced TRNA Import into Human Mitochondria: Implication of a Host Aminoacyl-TRNA-Synthetase. PLoS ONE 2013, 8, e66228. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A Bacterial Cytidine Deaminase Toxin Enables CRISPR-Free Mitochondrial Base Editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, E.S.; Chabbert, C.D.; Klaus, B.; Steinmetz, L.M. A Genome-Wide Map of Mitochondrial DNA Recombination in Yeast. Genetics 2014, 198, 755–771. [Google Scholar] [CrossRef] [PubMed]
- Law, S.S.Y.; Liou, G.; Nagai, Y.; Giménez-Dejoz, J.; Tateishi, A.; Tsuchiya, K.; Kodama, Y.; Fujigaya, T.; Numata, K. Polymer-Coated Carbon Nanotube Hybrids with Functional Peptides for Gene Delivery into Plant Mitochondria. Nat. Commun. 2022, 13, 2417. [Google Scholar] [CrossRef]
- Chernega, T.; Choi, J.; Salmena, L.; Andreazza, A.C. Mitochondrion-Targeted RNA Therapies as a Potential Treatment Strategy for Mitochondrial Diseases. Mol. Ther.-Nucleic Acids 2022, 30, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Aiba, W.; Amai, T.; Ueda, M.; Kuroda, K. Improving Precise Genome Editing Using Donor DNA/gRNA Hybrid Duplex Generated by Complementary Bases. Biomolecules 2022, 12, 1621. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Koblan, L.W.; Arbab, M.; Shen, M.W.; Hussmann, J.A.; Anzalone, A.V.; Doman, J.L.; Newby, G.A.; Yang, D.; Mok, B.; Replogle, J.M.; et al. Efficient C•G-to-G•C Base Editors Developed Using CRISPRi Screens, Target-Library Analysis, and Machine Learning. Nat. Biotechnol. 2021, 39, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, G.; Kuroda, K. Overcoming the Limitations of CRISPR-Cas9 Systems in Saccharomyces cerevisiae: Off-Target Effects, Epigenome, and Mitochondrial Editing. Microorganisms 2023, 11, 1040. https://doi.org/10.3390/microorganisms11041040
Sato G, Kuroda K. Overcoming the Limitations of CRISPR-Cas9 Systems in Saccharomyces cerevisiae: Off-Target Effects, Epigenome, and Mitochondrial Editing. Microorganisms. 2023; 11(4):1040. https://doi.org/10.3390/microorganisms11041040
Chicago/Turabian StyleSato, Genki, and Kouichi Kuroda. 2023. "Overcoming the Limitations of CRISPR-Cas9 Systems in Saccharomyces cerevisiae: Off-Target Effects, Epigenome, and Mitochondrial Editing" Microorganisms 11, no. 4: 1040. https://doi.org/10.3390/microorganisms11041040
APA StyleSato, G., & Kuroda, K. (2023). Overcoming the Limitations of CRISPR-Cas9 Systems in Saccharomyces cerevisiae: Off-Target Effects, Epigenome, and Mitochondrial Editing. Microorganisms, 11(4), 1040. https://doi.org/10.3390/microorganisms11041040