
Pseudomonas aeruginosa and the Complement System: A Review of the Evasion Strategies

 and
and 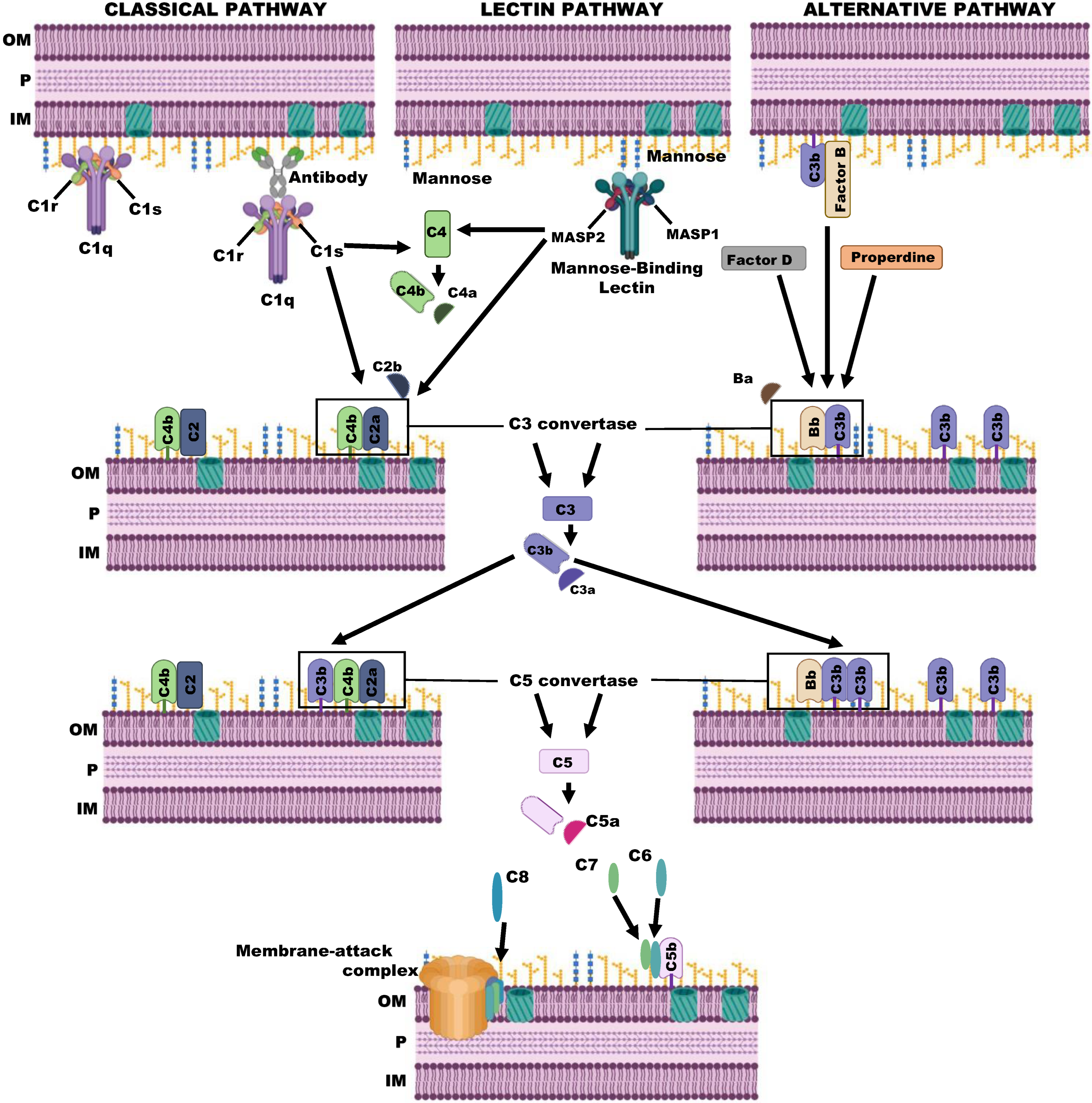{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Complement System
3. The Complement System in P. aeruginosa Infections
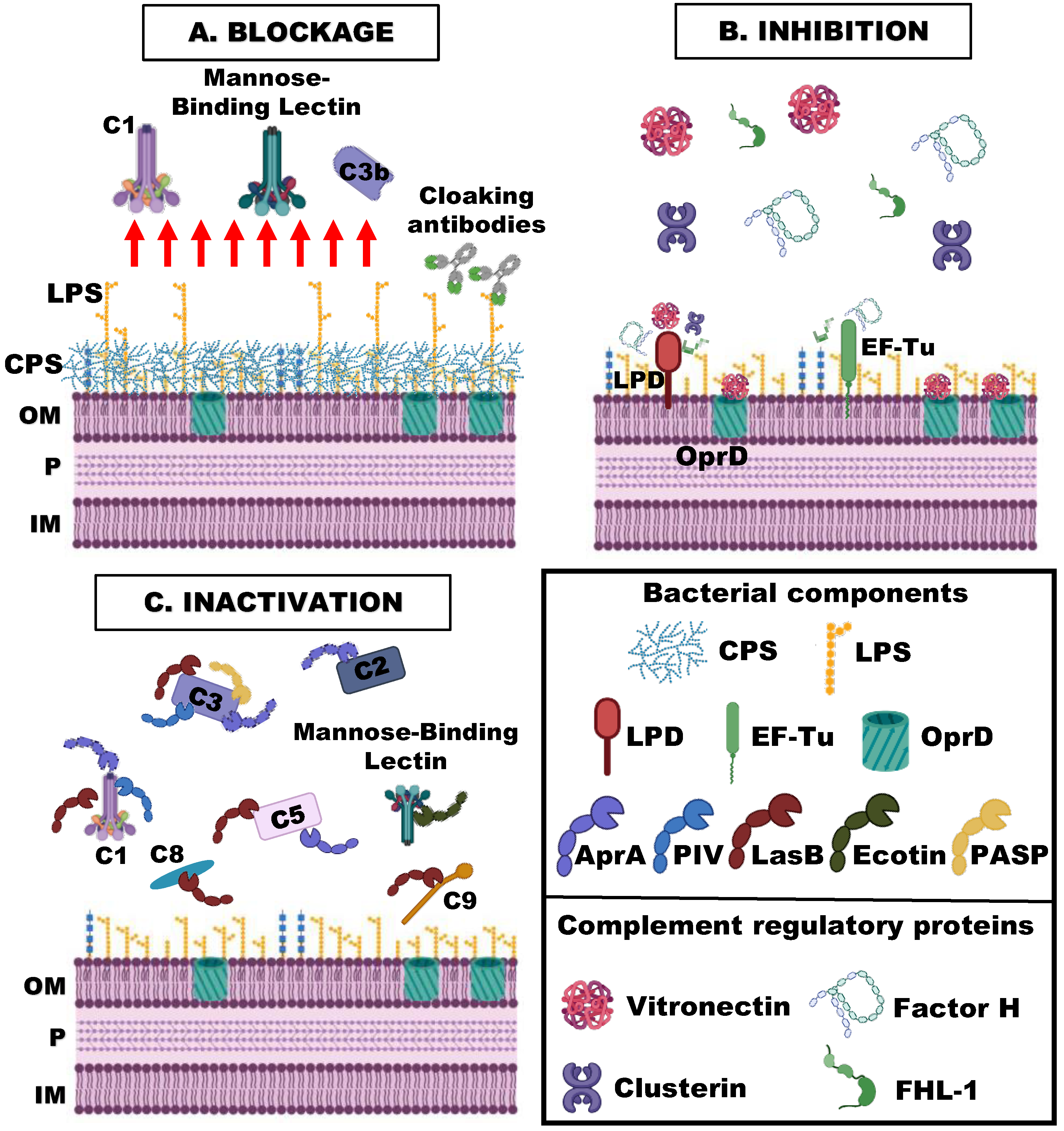
4. Complement Evasion Strategies of P. aeruginosa
4.1. Blockage of the Binding of the Complement Activating Components
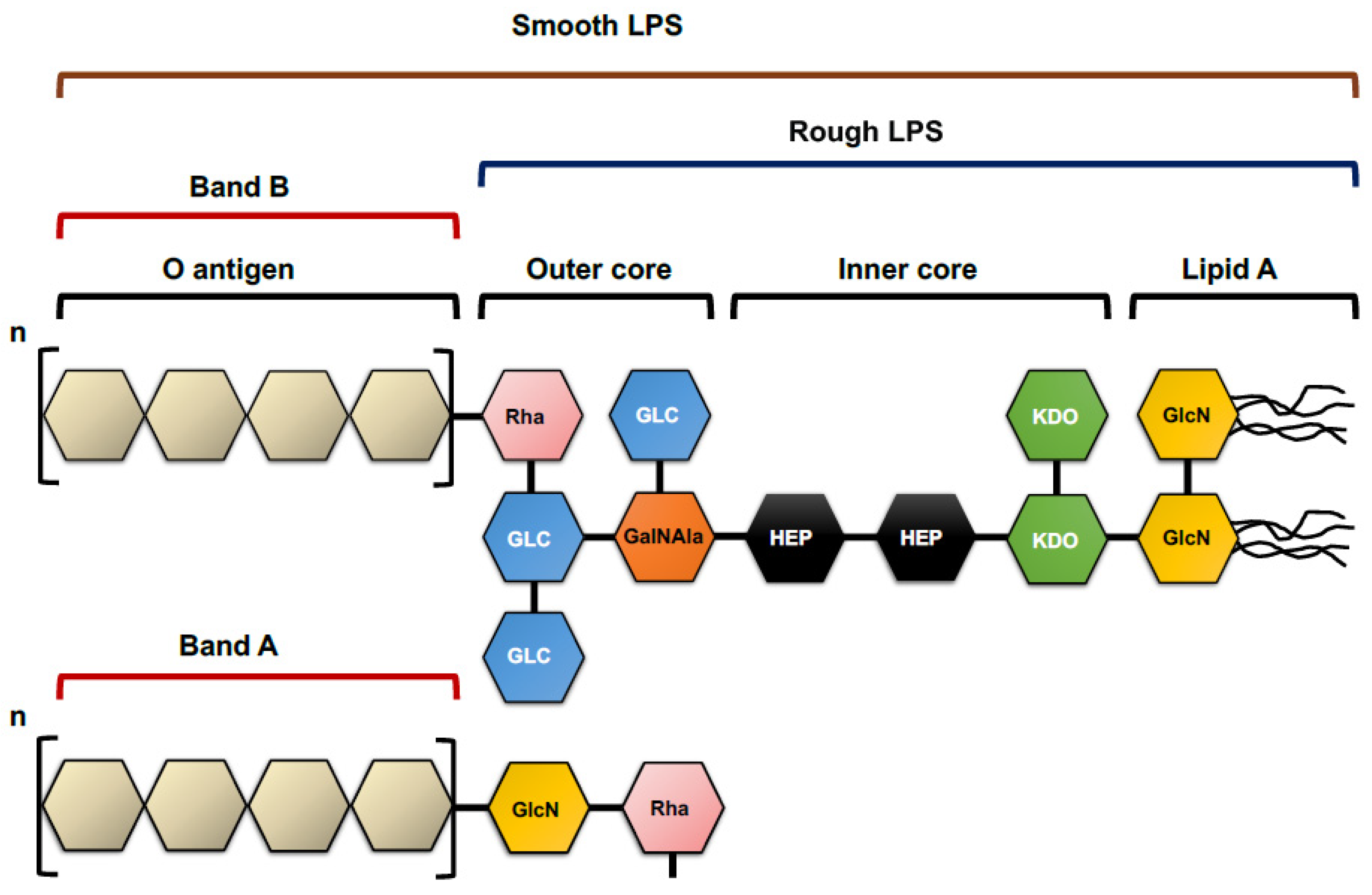
4.1.1. Lipopolysaccharide O Antigen
4.1.2. Capsule Polysaccharides
4.1.3. Biofilm
4.1.4. Cloaking Antibodies
4.2. Binding of Inhibitory Complement Regulatory Proteins
4.2.1. Elongation Factor Tu (EF-Tu)
4.2.2. Dihydrolipoamide Dehydrogenase (Lpd)
4.2.3. OprD
4.3. Inactivation of Complement Components
4.3.1. Exoproteases
4.3.2. Ecotin
4.3.3. Lytic Polysaccharide Monooxygenase CbpD
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Diekema, D.J.; Hsueh, P.R.; Mendes, R.E.; Pfaller, M.A.; Rolston, K.V.; Sader, H.S.; Jones, R.N. The microbiology of bloodstream infection: 20-year trends from the SENTRY antimicrobial surveillance program. Antimicrob. Agents Chemother. 2019, 63, e00355-19. [Google Scholar] [CrossRef]
- Holmes, C.L.; Anderson, M.T.; Mobley, H.L.T.; Bachman, M.A. Pathogenesis of Gram-negative bacteremia. Clin. Microbiol. Rev. 2021, 34, e00234-20. [Google Scholar] [CrossRef] [PubMed]
- Peña, C.; Suarez, C.; Gozalo, M.; Murillas, J.; Almirante, B.; Pomar, V.; Aguilar, M.; Granados, A.; Calbo, E.; Rodríguez-Baño, J.; et al. Prospective multicenter study of the impact of carbapenem resistance on mortality in Pseudomonas aeruginosa bloodstream infections. Antimicrob. Agents Chemother. 2012, 56, 1265–1272. [Google Scholar] [CrossRef]
- Peña, C.; Suarez, C.; Ocampo-Sosa, A.; Murillas, J.; Almirante, B.; Pomar, V.; Aguilar, M.; Granados, A.; Calbo, E.; Rodríguez-Baño, J.; et al. Effect of adequate single-drug vs combination antimicrobial therapy on mortality in Pseudomonas aeruginosa bloodstream infections: A post hoc analysis of a prospective cohort. Clin. Infect. Dis. 2013, 57, 208–216. [Google Scholar] [CrossRef]
- Chamot, E.; Boffi El Amari, E.; Rohner, P.; Van Delden, C. Effectiveness of combination antimicrobial therapy for Pseudomonas aeruginosa bacteremia. Antimicrob. Agents Chemother. 2003, 47, 2756–2764. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.I.; Kim, S.H.; Park, W.B.; Lee, K.D.; Kim, H.B.; Kim, E.C.; Oh, M.D.; Choe, K.W. Clinical features and outcome of patients with community-acquired Pseudomonas aeruginosa bacteraemia. Clin. Microbiol. Infect. 2005, 11, 415–418. [Google Scholar] [CrossRef]
- Peña, C.; Cabot, G.; Gomez-Zorrilla, S.; Zamorano, L.; Ocampo-Sosa, A.; Murillas, J.; Almirante, B.; Pomar, V.; Aguilar, M.; Granados, A.; et al. Influence of virulence genotype and resistance profile in the mortality of Pseudomonas aeruginosa bloodstream infections. Clin. Infect. Dis. 2015, 60, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Thaden, J.; Park, L.; Maskarinec, S.; Ruffin, F.; Fowler, V.J.; van Duin, D. Results from a 13-year prospective cohort study show increased mortality associated with bloodstream infections caused by Pseudomonas aeruginosa compared to other bacteria. Antimicrob. Agents Chemother. 2017, 6, e02671-16. [Google Scholar] [CrossRef]
- Poole, K. Pseudomonas aeruginosa: Resistance to the max. Front. Microbiol. 2011, 2, 65. [Google Scholar] [CrossRef]
- Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas aeruginosa: An audacious pathogen with an adaptble arsenal of virulence factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Hattemer, A.; Hauser, A.; Diaz, M.; Scheetz, M.; Shah, N.; Allen, J.P.; Porhomayon, J.; El-Solh, A.A. Bacterial and clinical characteristics of health care- and community-acquired bloodstream infections due to Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 3969–3975. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I—Molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part II: Role in immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Bajic, G.; Degn, S.E.; Thiel, S.; Andersen, G.R. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. 2015, 34, 2735–2757. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.N.; Rehm, S.R.; Pierce, A.K. The effect of complement depletion on lung clearance of bacteria. J. Clin. Investig. 1978, 62, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, T.S.; Zaidi, T.; Pier, G.B. Role of neutrophils, MyD88-mediated neutrophil recruitment, and complement in antibody-mediated defense against Pseudomonas aeruginosa keratitis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2085–2093. [Google Scholar] [CrossRef]
- Cerquetti, M.C.; Sordelli, D.O.; Bellanti, J.A.; Hooke, A.M. Lung defenses against Pseudomonas aeruginosa in C5-deficient mice with different genetic backgrounds. Infect. Immun. 1986, 52, 853–857. [Google Scholar] [CrossRef]
- Larsen, G.L.; Mitchell, B.C.; Harper, T.B.; Henson, P.M. The pulmonary response of C5 sufficient and deficient mice to Pseudomonas aeruginosa. Am. Rev. Respir. Dis. 1982, 126, 306–311. [Google Scholar]
- Mueller-Ortiz, S.L.; Drouin, S.M.; Wetsel, R.A. The alternative activation pathway and complement component C3 are critical for a protective immune response against Pseudomonas aeruginosa in a murine model of pneumonia. Infect. Immun. 2004, 72, 2899–2906. [Google Scholar] [CrossRef]
- Møller-Kristensen, M.; Eddie Ip, W.K.; Shi, L.; Gowda, L.D.; Hamblin, M.R.; Thiel, S.; Jensenius, J.C.; Ezekowitz, R.A.B.; Takahashi, K. Deficiency of mannose-binding lectin greatly increases susceptibility to postburn infection with Pseudomonas aeruginosa. J. Immunol. 2006, 176, 1769–1775. [Google Scholar] [CrossRef]
- Kenawy, H.I.; Ali, Y.M.; Rajakumar, K.; Lynch, N.J.; Kadioglu, A.; Stover, C.M.; Schwaeble, W.J. Absence of the lectin activation pathway of complement does not increase susceptibility to Pseudomonas aeruginosa infections. Immunobiology 2012, 217, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Pont, S.; Fraikin, N.; Caspar, Y.; Van Melderen, L.; Attrée, I.; Cretin, F. Bacterial behavior in human blood reveals complement evaders with some persister-like features. PLoS Pathog. 2020, 16, e1008893. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, E.; Nagano, K.; Nikaido, H. Alternative folding pathways of the major porin OprF of Pseudomonas aeruginosa. FEBS J. 2012, 279, 910–918. [Google Scholar] [CrossRef]
- Mishra, M.; Ressler, A.; Schlesinger, L.S.; Wozniak, D.J. Identification of OprF as a complement component C3 binding acceptor molecule on the surface of Pseudomonas aeruginosa. Infect. Immun. 2015, 83, 3006–3014. [Google Scholar] [CrossRef]
- Qadi, M.; Izquierdo-Rabassa, S.; Mateu Borrás, M.; Doménech-Sánchez, A.; Juan, C.; Goldberg, J.B.; Hancock, R.E.W.; Albertí, S. Sensing Mg2+ contributes to the resistance of Pseudomonas aeruginosa to complement-mediated opsonophagocytosis. Environ. Microbiol. 2017, 19, 4278–4286. [Google Scholar] [CrossRef] [PubMed]
- Joiner, K.A.; Grossman, N.; Schmetz, M.; Leive, L. C3 binds preferentially to long-chain lipopolysaccharide during alternative pathway activation by Salmonella montevideo. J. Immunol. 1986, 136, 710–715. [Google Scholar] [CrossRef]
- Albertí, S.; Alvarez, D.; Merino, S.; Casado, M.T.; Vivanco, F.; Tomás, J.M.; Benedí, V.J. Analysis of complement C3 deposition and degradation on Klebsiella pneumoniae. Infect. Immun. 1996, 64, 4726–4732. [Google Scholar] [CrossRef]
- Jensen, E.T.; Kharazmi, A.; Garred, P.; Kronborg, G.; Fomsgaard, A.; Mollnes, T.E.; Høiby, N. Complement activation by Pseudomonas aeruginosa biofilms. Microb. Pathog. 1993, 15, 377–388. [Google Scholar] [CrossRef]
- Schiller, N.L.; Hatch, R.A.; Joiner, K.A. Complement activation and C3 binding by serum-sensitive and serum-resistant strains of Pseudomonas aeruginosa. Infect. Immun. 1989, 57, 1707–1713. [Google Scholar] [CrossRef]
- Huszczynski, S.M.; Lam, J.S.; Khursigara, C.M. The role of Pseudomonas aeruginosa lipopolysaccharide in bacterial pathogenesis and physiology. Pathogens 2020, 9, 6. [Google Scholar] [CrossRef]
- Ernst, R.K.; Yi, E.C.; Guo, L.; Lim, K.B.; Burns, J.L.; Hackett, M.; Miller, S.I. Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science 1999, 286, 1561–1565. [Google Scholar] [CrossRef]
- Pier, G.B.; Coleman, F.; Grout, M.; Franklin, M.; Ohman, D.E. Role of alginate O acetylation in resistance of mucoid Pseudomonas aeruginosa to opsonic phagocytosis. Infect. Immun. 2001, 69, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, N.; Fernandez, R. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front. Cell. Infect. Microbiol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lim, K.B.; Poduje, C.M.; Daniel, M.; Gunn, J.S.; Hackett, M.; Miller, S.I. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 1998, 95, 189–198. [Google Scholar] [CrossRef]
- Hajjar, A.M.; Ernst, R.K.; Tsai, J.H.; Wilson, C.B.; Miller, S.I. Human toll-like receptor 4 recognizes host-specific LPS modifications. Nat. Immunol. 2002, 3, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Bystrova, O.V.; Shashkov, A.S.; Kocharova, N.A.; Knirel, Y.A.; Zähringer, U.; Pier, G.B. Elucidation of the structure of the lipopolysaccharide core and the linkage between the core and the O-antigen in Pseudomonas aeruginosa immunotype 5 using strong alkaline degradation of the lipopolysaccharide. Biochem. Biokhimiia 2003, 68, 918–925. [Google Scholar] [CrossRef]
- Bystrova, O.V.; Knirel, Y.A.; Lindner, B.; Kocharova, N.A.; Kondakova, A.N.; Zähringer, U.; Pier, G.B. Structures of the core oligosaccharide and O-units in the R- and SR-type lipopolysaccharides of reference strains of Pseudomonas aeruginosa O-serogroups. FEMS Immunol. Med. Microbiol. 2006, 46, 85–99. [Google Scholar] [CrossRef]
- Beckmann, F.; Moll, H.; Jäger, K.E.; Zähringer, U. Preliminary communication 7-O-Carbamoyl-L-Glycero-D-Manno-Heptose: A new core constituent in the lipopolysaccharide of Pseudomonas aeruginosa. Carbohydr. Res. 1995, 267, C3-7. [Google Scholar] [CrossRef]
- Pier, G.B. Pseudomonas aeruginosa lipopolysaccharide: A major virulence factor, initiator of inflammation and target for effective immunity. Int. J. Med. Microbiol. IJMM 2007, 297, 277–295. [Google Scholar] [CrossRef]
- Lam, J.S.; Taylor, V.L.; Islam, S.T.; Hao, Y.; Kocíncová, D. Genetic and functional diversity of Pseudomonas aeruginosa lipopolysaccharide. Front. Microbiol. 2011, 2, 118. [Google Scholar] [CrossRef]
- Kintz, E.; Goldberg, J.B. Regulation of lipopolysaccharide O antigen expression in Pseudomonas aeruginosa. Future Microbiol. 2008, 3, 191–203. [Google Scholar] [CrossRef]
- Kintz, E.; Scarff, J.M.; DiGiandomenico, A.; Goldberg, J.B. Lipopolysaccharide O-antigen chain length regulation in Pseudomonas aeruginosa serogroup O11 strain PA103. J. Bacteriol. 2008, 190, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Priebe, G.P.; Dean, C.R.; Zaidi, T.; Meluleni, G.J.; Coleman, F.T.; Coutinho, Y.S.; Noto, M.J.; Urban, T.A.; Pier, G.B.; Goldberg, J.B. The GalU Gene of Pseudomonas aeruginosa is required for corneal infection and efficient systemic spread following pneumonia but not for infection confined to the lung. Infect. Immun. 2004, 72, 4224–4232. [Google Scholar] [CrossRef]
- Schiller, N.L.; Joiner, K.A. Interaction of complement with serum-sensitive and serum-resistant strains of Pseudomonas aeruginosa. Infect. Immun. 1986, 54, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Pier, G.B.; Grout, M.; Zaidi, T.S.; Olsen, J.C.; Johnson, L.G.; Yankaskas, J.R.; Goldberg, J.B. Role of mutant CFTR in hypersusceptibility of cystic fibrosis patients to lung infections. Science 1996, 271, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.F.; Sá-Correia, I.; Valvano, M.A. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 2016, 40, 480–493. [Google Scholar] [CrossRef]
- Penketh, A.; Pitt, T.; Roberts, D.; Hodson, M.E.; Batten, J.C. The relationship of phenotype changes in Pseudomonas aeruginosa to the clinical condition of patients with cystic fibrosis. Am. Rev. Respir. Dis. 1983, 127, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Franklin, M.; Nivens, D.; Weadge, J.; Howell, P. Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, alginate, Pel, and Psl. Front. Microbiol. 2011, 2, 167. [Google Scholar] [CrossRef]
- Hogardt, M.; Heesemann, J. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int. J. Med. Microbiol. IJMM 2010, 300, 557–562. [Google Scholar] [CrossRef]
- Boucher, J.C.; Yu, H.; Mudd, M.H.; Deretic, V. Mucoid Pseudomonas aeruginosa in cystic fibrosis: Characterization of Muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect. Immun. 1997, 65, 3838–3846. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Byrd, M.S.; Sergeant, S.; Azad, A.K.; Parsek, M.R.; McPhail, L.; Schlesinger, L.S.; Wozniak, D.J. Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell. Microbiol. 2012, 14, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Pestrak, M.J.; Baker, P.; Dellos-Nolan, S.; Hill, P.J.; Passos da Silva, D.; Silver, H.; Lacdao, I.; Raju, D.; Parsek, M.R.; Wozniak, D.J.; et al. Treatment with the Pseudomonas aeruginosa glycoside hydrolase PslG combats wound infection by improving antibiotic efficacy and host innate immune activity. Antimicrob. Agents Chemother. 2019, 63, e00234-19. [Google Scholar] [CrossRef]
- Jones, C.J.; Wozniak, D.J. Psl produced by mucoid Pseudomonas aeruginosa contributes to the establishment of biofilms and immune evasion. mBio 2017, 8, e00864-17. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef]
- Whitchurch, C.B.; Tolker-Nielsen, T.; Ragas, P.C.; Mattick, J.S. Extracellular DNA required for bacterial biofilm formation. Science 2002, 295, 1487. [Google Scholar] [CrossRef]
- Matsukawa, M.; Greenberg, E.P. Putative exopolysaccharide synthesis genes influence Pseudomonas aeruginosa biofilm development. J. Bacteriol. 2004, 186, 4449–4456. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M. Biofilm formation: A clinically relevant microbiological process. Clin. Infect. Dis. 2001, 33, 1387–1392. [Google Scholar] [CrossRef]
- Sauer, K.; Stoodley, P.; Goeres, D.M.; Hall-Stoodley, L.; Burmølle, M.; Stewart, P.S.; Bjarnsholt, T. The biofilm life cycle: Expanding the conceptual model of biofilm formation. Nat Rev Microbiol. 2022, 20, 608–620. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, G.; Kaplan, H.B.; Kolter, R. Biofilm formation as microbial development. Annu. Rev. Microbiol. 2000, 54, 49–79. [Google Scholar] [CrossRef]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef] [PubMed]
- Glen, K.A.; Lamont, I.L. β-lactam Resistance in Pseudomonas aeruginosa: Current Status, Future Prospects. Pathogens. 2021, 10, 1638. [Google Scholar] [CrossRef]
- Lichtenberg, M.; Jakobsen, T.H.; Kühl, M.; Kolpen, M.; Jensen, P.; Bjarnsholt, T. The structure-function relationship of Pseudomonas aeruginosa in infections and its influence on the microenvironment. FEMS Microbiol Rev. 2022, 46, fuac018. [Google Scholar] [CrossRef]
- Hair, P.S.; Sass, L.A.; Vazifedan, T.; Shah, T.A.; Krishna, N.K.; Cunnion, K.M. Complement effectors, C5a and C3a, in cystic fibrosis lung fluid correlate with disease severity. PLoS ONE 2017, 12, e0173257. [Google Scholar] [CrossRef]
- Wells, T.J.; Whitters, D.; Sevastsyanovich, Y.R.; Heath, J.N.; Pravin, J.; Goodall, M.; Browning, D.F.; O’Shea, M.K.; Cranston, A.; De Soyza, A.; et al. Increased severity of respiratory infections associated with elevated anti-LPS IgG2 which inhibits serum bactericidal killing. J. Exp. Med. 2014, 211, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.; Ledger, E.L.; Coggon, C.F.; Henderson, I.R.; Reid, D.W.; Bell, S.C.; Smith, D.J.; Wells, T.J. Anti-LPS IgA and IgG can inhibit serum killing of Pseudomonas aeruginosa in patients with cystic fibrosis. Infect. Immun. 2021, 89, e0041221. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.L.; Jarocki, V.M.; Charles, I.G.; Djordjevic, S.P. The diverse functional roles of Elongation Factor Tu (EF-Tu) in microbial pathogenesis. Front. Microbiol. 2019, 10, 2351. [Google Scholar] [CrossRef]
- Kunert, A.; Losse, J.; Gruszin, C.; Hühn, M.; Kaendler, K.; Mikkat, S.; Volke, D.; Hoffmann, R.; Jokiranta, T.S.; Seeberger, H.; et al. Immune evasion of the human pathogen Pseudomonas aeruginosa: Elongation Factor Tuf is a Factor H and plasminogen binding protein. J. Immunol. 2007, 179, 2979–2988. [Google Scholar] [CrossRef] [PubMed]
- Hallström, T.; Mörgelin, M.; Barthel, D.; Raguse, M.; Kunert, A.; Hoffmann, R.; Skerka, C.; Zipfel, P.F. Dihydrolipoamide dehydrogenase of Pseudomonas aeruginosa is a surface-exposed immune evasion protein that binds three members of the Factor H family and plasminogen. J. Immunol. 2012, 189, 4939–4950. [Google Scholar] [CrossRef]
- Hallström, T.; Uhde, M.; Singh, B.; Skerka, C.; Riesbeck, K.; Zipfel, P.F. Pseudomonas aeruginosa uses dihydrolipoamide dehydrogenase (Lpd) to bind to the human terminal pathway regulators vitronectin and clusterin to inhibit terminal pathway complement attack. PLoS ONE 2015, 10, e0137630. [Google Scholar] [CrossRef]
- Paulsson, M.; Che, K.F.; Ahl, J.; Tham, J.; Sandblad, L.; Smith, M.E.; Qvarfordt, I.; Su, Y.-C.; Lindén, A.; Riesbeck, K. Bacterial outer membrane vesicles induce vitronectin release into the bronchoalveolar space conferring protection from complement-mediated killing. Front. Microbiol. 2018, 9, 1559. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Luo, Y.F.; Williams, B.J.; Blackwell, T.S.; Xie, C.M. Structure and function of OprD protein in Pseudomonas aeruginosa: From antibiotic resistance to novel therapies. Int. J. Med. Microbiol. 2012, 302, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.; Juan, C.; Cercenado, E.; Navarro, F.; Bouza, E.; Coll, P.; Pérez, J.L.; Oliver, A. Molecular epidemiology and mechanisms of carbapenem resistance in Pseudomonas aeruginosa isolates from spanish hospitals. Antimicrob. Agents Chemother. 2007, 51, 4329–4335. [Google Scholar] [CrossRef]
- Paulsson, M.; Singh, B.; Al-Jubair, T.; Su, Y.-C.; Høiby, N.; Riesbeck, K. Identification of outer membrane porin D as a vitronectin-binding factor in cystic fibrosis clinical isolates of Pseudomonas aeruginosa. J. Cyst. Fibros. 2015, 14, 600–607. [Google Scholar] [CrossRef]
- Tingpej, P.; Smith, L.; Rose, B.; Zhu, H.; Conibear, T.; Al Nassafi, K.; Manos, J.; Elkins, M.; Bye, P.; Willcox, M.; et al. Phenotypic characterization of clonal and nonclonal Pseudomonas aeruginosa strains isolated from lungs of adults with cystic fibrosis. J. Clin. Microbiol. 2007, 45, 1697–1704. [Google Scholar] [CrossRef]
- Alamu, J.; Kakithakara Vajravelu, L.; Venkatesan, B.; Thulukanam, J. Correlation of phenotypic and genotypic virulence markers, antimicrobial susceptibility pattern, and outcome of Pseudomonas aeruginosa sepsis infection. Microb Pathog. 2022, 170, 105716. [Google Scholar] [CrossRef]
- Thayer, M.M.; Flaherty, K.M.; McKay, D.B. Three-dimensional structure of the elastase of Pseudomonas aeruginosa at 1.5-A resolution. J. Biol. Chem. 1991, 266, 2864–2871. [Google Scholar] [CrossRef]
- Nouwens, A.S.; Beatson, S.A.; Whitchurch, C.B.; Walsh, B.J.; Schweizer, H.P.; Mattick, J.S.; Cordwell, S.J. Proteome analysis of extracellular proteins regulated by the las and rhl quorum sensing systems in Pseudomonas aeruginosa PAO1. Microbiology 2003, 149, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.R.; Miller, K.D. Elastase of Pseudomonas aeruginosa: Inactivation of complement components and complement-derived chemotactic and phagocytic factors. Infect. Immun. 1974, 10, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.Q.; Ghebrehiwet, B. Effect of Pseudomonas aeruginosa elastase and alkaline protease on serum complement and isolated components C1q and C3. Clin. Immunol. Immunopathol. 1992, 62, 133–138. [Google Scholar] [CrossRef]
- Mateu-Borrás, M.; Zamorano, L.; González-Alsina, A.; Sánchez-Diener, I.; Doménech-Sánchez, A.; Oliver, A.; Albertí, S. Molecular analysis of the contribution of alkaline protease A and elastase B to the virulence of Pseudomonas aeruginosa bloodstream infections. Front. Cell. Infect. Microbiol. 2021, 11, 816356. [Google Scholar] [CrossRef] [PubMed]
- Mateu-Borrás, M.; González-Alsina, A.; Doménech-Sánchez, A.; Querol-García, J.; Fernández, F.J.; Vega, M.C.; Albertí, S. Pseudomonas aeruginosa adaptation in cystic fibrosis patients increases C5a levels and promotes neutrophil recruitment. Virulence 2022, 13, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Baumann, U.; Wu, S.; Flaherty, K.M.; McKay, D.B. Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: A two-domain protein with a calcium binding parallel beta roll motif. EMBO J. 1993, 12, 3357–3364. [Google Scholar] [CrossRef]
- Gambello, M.J.; Kaye, S.; Iglewski, B.H. LasR of Pseudomonas aeruginosa is a transcriptional activator of the alkaline protease gene (Apr) and an enhancer of exotoxin A expression. Infect. Immun. 1993, 61, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Laarman, A.J.; Bardoel, B.W.; Ruyken, M.; Fernie, J.; Milder, F.J.; van Strijp, J.A.G.; Rooijakkers, S.H.M. Pseudomonas aeruginosa alkaline protease blocks complement activation via the classical and lectin pathways. J. Immunol. 2012, 188, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.; Hao, Y.; Walling, B.E.; Jeffries, J.L.; Ohman, D.E.; Lau, G.W. Pseudomonas aeruginosa elastase provides an escape from phagocytosis by degrading the pulmonary surfactant protein-A. PLoS ONE 2011, 6, e27091. [Google Scholar] [CrossRef]
- Bastaert, F.; Kheir, S.; Saint-Criq, V.; Villeret, B.; Dang, P.M.-C.; El-Benna, J.; Sirard, J.-C.; Voulhoux, R.; Sallenave, J.-M. Pseudomonas aeruginosa LasB subverts alveolar macrophage activity by interfering with bacterial killing through downregulation of innate immune defense, reactive oxygen species generation, and complement activation. Front. Immunol. 2018, 9, 1675. [Google Scholar] [CrossRef]
- Wretlind, B.; Pavlovskis, O.R. Pseudomonas aeruginosa elastase and its role in Pseudomonas infections. Rev. Infect. Dis. 1983, 5 (Suppl. 5), S998–S1004. [Google Scholar] [CrossRef]
- Caballero, A.; Thibodeaux, B.; Marquart, M.; Traidej, M.; O’Callaghan, R. Pseudomonas keratitis: Protease IV gene conservation, distribution, and production relative to virulence and other Pseudomonas proteases. Investig. Ophthalmol. Vis. Sci. 2004, 45, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Engel, L.S.; Hill, J.M.; Caballero, A.R.; Green, L.C.; O’Callaghan, R.J. Protease IV, a unique extracellular protease and virulence factor from Pseudomonas aeruginosa. J. Biol. Chem. 1998, 273, 16792–16797. [Google Scholar] [CrossRef]
- Tang, A.; Marquart, M.E.; Fratkin, J.D.; McCormick, C.C.; Caballero, A.R.; Gatlin, H.P.; O’Callaghan, R.J. Properties of PASP: A pseudomonas protease capable of mediating corneal erosions. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3794–3801. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tang, A.; Caballero, A.R.; Marquart, M.E.; O’Callaghan, R.J. Pseudomonas aeruginosa small protease (PASP), a keratitis virulence factor. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2821–2828. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nagy, Z.A.; Szakács, D.; Boros, E.; Héja, D.; Vígh, E.; Sándor, N.; Józsi, M.; Oroszlán, G.; Dobó, J.; Gál, P.; et al. Ecotin, a microbial inhibitor of serine proteases, blocks multiple complement dependent and independent microbicidal activities of human serum. PLoS Pathog. 2019, 15, e1008232. [Google Scholar] [CrossRef] [PubMed]
- Askarian, F.; Uchiyama, S.; Masson, H.; Sørensen, H.V.; Golten, O.; Bunæs, A.C.; Mekasha, S.; Røhr, Å.K.; Kommedal, E.; Ludviksen, J.A.; et al. The lytic polysaccharide monooxygenase CbpD promotes Pseudomonas aeruginosa virulence in systemic infection. Nat. Commun. 2021, 23, 1230. [Google Scholar] [CrossRef]
- Józsi, M. Factor H family proteins in complement evasion of microorganisms. Front. Immunol. 2017, 18, 571. [Google Scholar] [CrossRef]
- Olszak, T.; Shneider, M.M.; Latka, A.; Maciejewska, B.; Browning, C.; Sycheva, L.V.; Cornelissen, A.; Danis-Wlodarczyk, K.; Senchenkova, S.N.; Shashkov, A.S.; et al. The O-specific polysaccharide lyase from the phage LKA1 tailspike reduces Pseudomonas virulence. Sci. Rep. 2017, 7, 16302. [Google Scholar] [CrossRef]
- Abd El-Aziz, A.M.; Elgaml, A.; Ali, Y.M. Bacteriophage therapy increases complement-mediated lysis of bacteria and enhances bacterial clearance after acute lung infection with multidrug-resistant Pseudomonas aeruginosa. J. Infect. Dis. 2019, 219, 1439–1447. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Alsina, A.; Mateu-Borrás, M.; Doménech-Sánchez, A.; Albertí, S. Pseudomonas aeruginosa and the Complement System: A Review of the Evasion Strategies. Microorganisms 2023, 11, 664. https://doi.org/10.3390/microorganisms11030664
González-Alsina A, Mateu-Borrás M, Doménech-Sánchez A, Albertí S. Pseudomonas aeruginosa and the Complement System: A Review of the Evasion Strategies. Microorganisms. 2023; 11(3):664. https://doi.org/10.3390/microorganisms11030664
Chicago/Turabian StyleGonzález-Alsina, Alex, Margalida Mateu-Borrás, Antonio Doménech-Sánchez, and Sebastián Albertí. 2023. "Pseudomonas aeruginosa and the Complement System: A Review of the Evasion Strategies" Microorganisms 11, no. 3: 664. https://doi.org/10.3390/microorganisms11030664
APA StyleGonzález-Alsina, A., Mateu-Borrás, M., Doménech-Sánchez, A., & Albertí, S. (2023). Pseudomonas aeruginosa and the Complement System: A Review of the Evasion Strategies. Microorganisms, 11(3), 664. https://doi.org/10.3390/microorganisms11030664