
Bacterial-Artificial-Chromosome-Based Genome Editing Methods and the Applications in Herpesvirus Research


Abstract
1. Introduction
2. Genome Editing Techniques of Herpesviruses Based on BAC
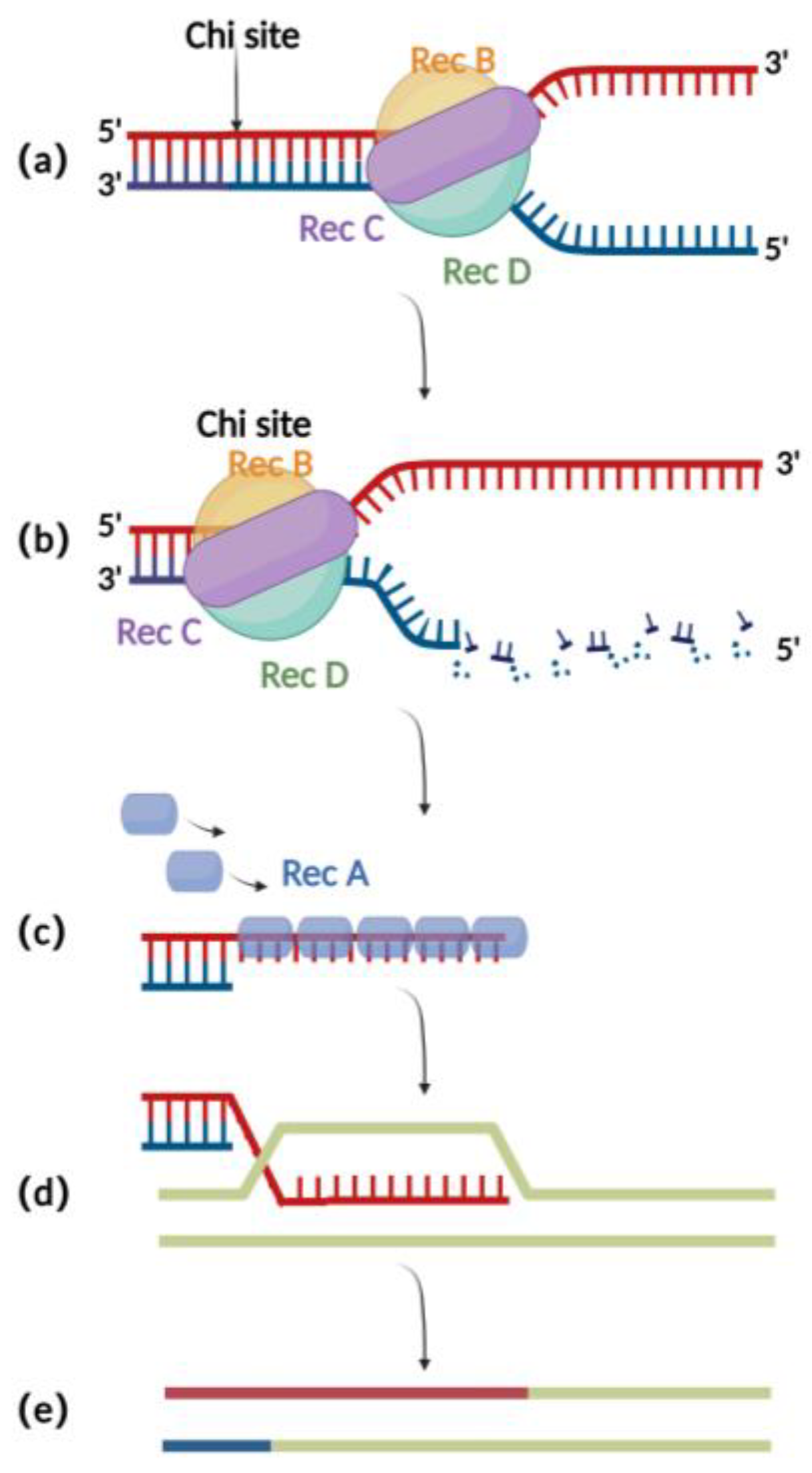
2.1. RecA Recombination Technique
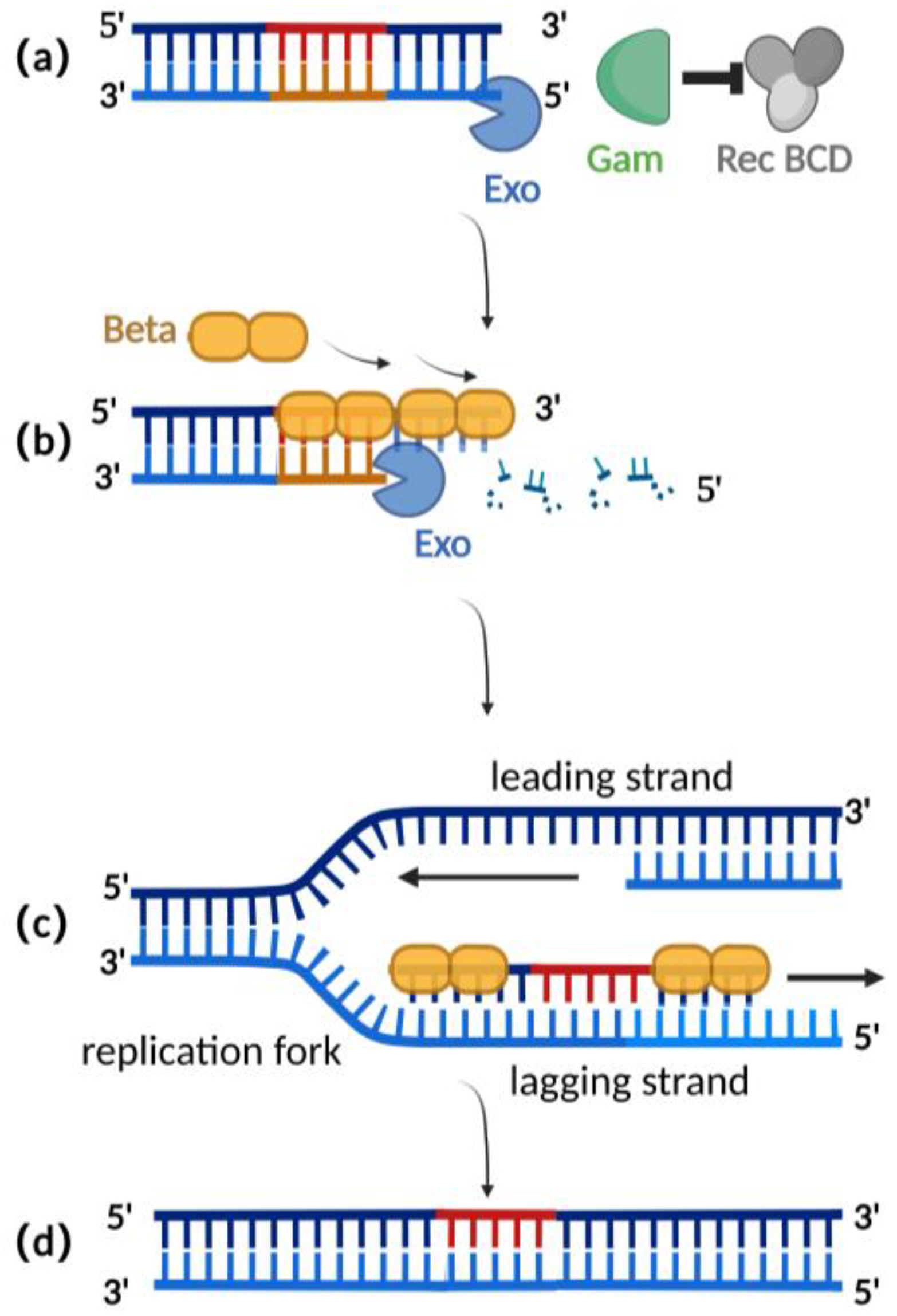
2.2. λ-Red Recombination Technique
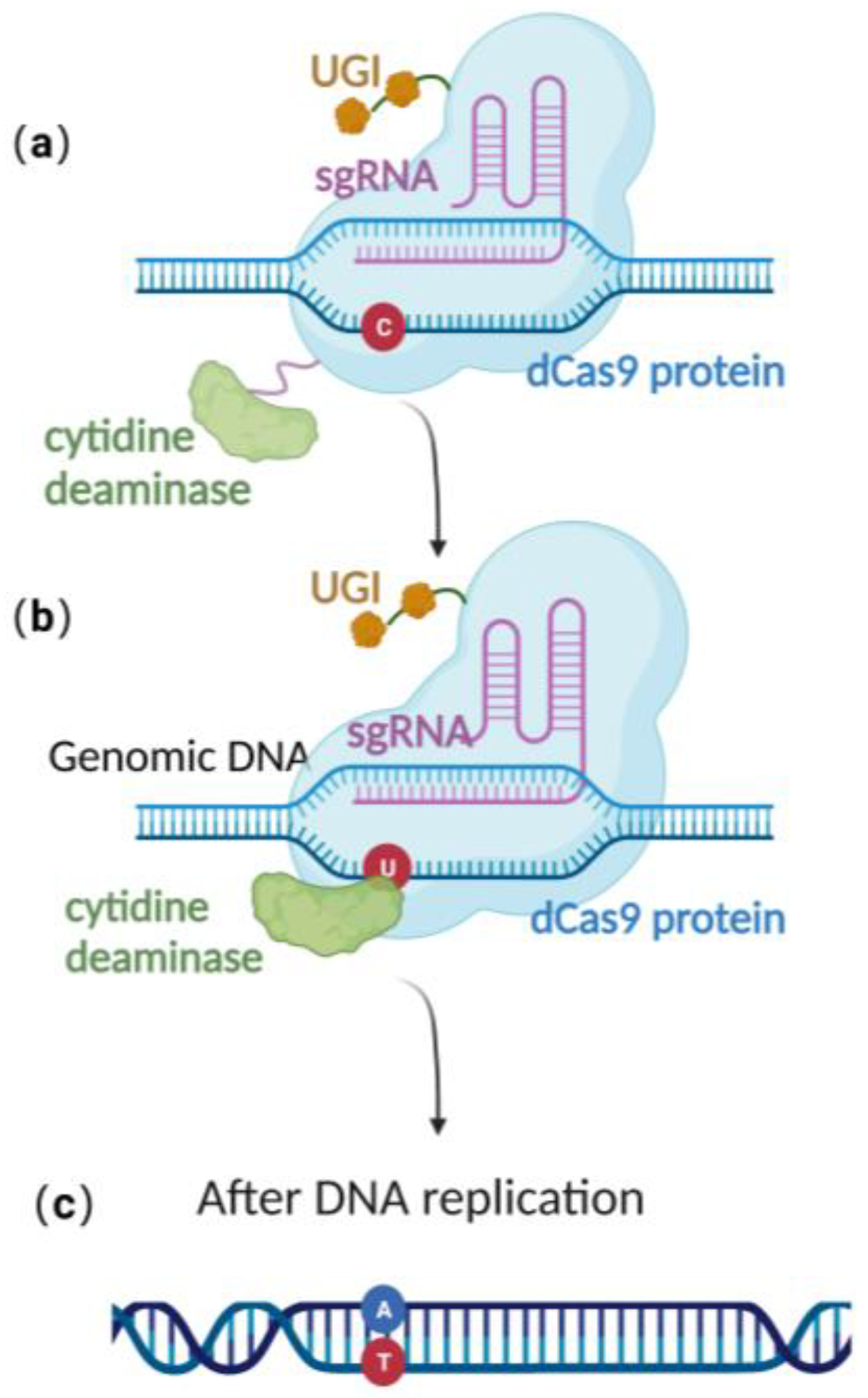
2.3. Base Editing Technique
3. Screening Methods of Herpesvirus Mutants
3.1. Single Selection Cassette
3.2. Selection Cassette in Combination with Site-Specific Recombinase Recognition Motif
3.3. Selection Cassette in Combination with I-sceI Endonuclease Recognition Site (En Passant Mutagenesis)
3.4. Positive and Negative (Dual) Selection Cassettes
4. Application of BAC-Based Gene Editing in Herpesvirus Research
4.1. Gene Function of Herpesvirus
4.2. Gene Therapy Vectors and Vaccine Vectors
4.3. Visualization of Herpes Virus
5. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Tischer, B.K.; Kaufer, B.B. Viral bacterial artificial chromosomes: Generation, mutagenesis, and removal of mini-F sequences. J. Biomed. Biotechnol. 2012, 2012, 472537. [Google Scholar] [CrossRef] [PubMed]
- Masud, H.M.A.A.; Watanabe, T.; Yoshida, M.; Sato, Y.; Goshima, F.; Kimura, H.; Murata, T. Epstein-Barr Virus BKRF4 Gene Product Is Required for Efficient Progeny Production. J. Virol. 2017, 91, e00975-17. [Google Scholar] [CrossRef]
- Delecluse, H.J.; Hammerschmidt, W. The genetic approach to the Epstein-Barr virus: From basic virology to gene therapy. Mol. Pathol. 2000, 53, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Wang, F.; Mannick, J.; Kieff, E. Epstein-Barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc. Natl. Acad. Sci. USA 1989, 86, 9558–9562. [Google Scholar] [CrossRef]
- Mannick, J.B.; Cohen, J.I.; Birkenbach, M.; Marchini, A.; Kieff, E. The Epstein-Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B-lymphocyte transformation. J. Virol. 1991, 65, 6826–6837. [Google Scholar] [CrossRef]
- Smiley, J.R. Construction in vitro and rescue of a thymidine kinase-deficient deletion mutation of herpes simplex virus. Nature 1980, 285, 333–335. [Google Scholar] [CrossRef]
- Post, L.E.; Roizman, B. A generalized technique for deletion of specific genes in large genomes: Alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell 1981, 25, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Shizuya, H.; Birren, B.; Kim, U.J.; Mancino, V.; Slepak, T.; Tachiiri, Y.; Simon, M. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl. Acad. Sci. USA 1992, 89, 8794–8797. [Google Scholar] [CrossRef]
- Tsai, M.-H.; Raykova, A.; Klinke, O.; Bernhardt, K.; Gärtner, K.; Leung, C.S.; Geletneky, K.; Sertel, S.; Münz, C.; Feederle, R.; et al. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep. 2013, 5, 458–470. [Google Scholar] [CrossRef]
- Messerle, M.; Crnkovic, I.; Hammerschmidt, W.; Ziegler, H.; Koszinowski, U.H. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 1997, 94, 14759–14763. [Google Scholar] [CrossRef]
- Borst, E.M.; Hahn, G.; Koszinowski, U.H.; Messerle, M. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: A new approach for construction of HCMV mutants. J. Virol. 1999, 73, 8320–8329. [Google Scholar] [CrossRef]
- Hosoda, F.; Nishimura, S.; Uchida, H.; Ohki, M. An F factor based cloning system for large DNA fragments. Nucleic. Acids. Res. 1990, 18, 3863–3869. [Google Scholar] [CrossRef]
- Delecluse, H.J.; Hilsendegen, T.; Pich, D.; Zeidler, R.; Hammerschmidt, W. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 1998, 95, 8245–8250. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Yajima, M.; Ahsan, N.; Tanaka, M.; Takada, K. Production of high-titer Epstein-Barr virus recombinants derived from Akata cells by using a bacterial artificial chromosome system. J. Virol. 2004, 78, 7004–7015. [Google Scholar] [CrossRef]
- Delecluse, H.J.; Kost, M.; Feederle, R.; Wilson, L.; Hammerschmidt, W. Spontaneous activation of the lytic cycle in cells infected with a recombinant Kaposi’s sarcoma-associated virus. J. Virol. 2001, 75, 2921–2928. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.; Wigley, D.B. Structural features of Chi recognition in AddAB with implications for RecBCD. Cell Cycle 2014, 13, 2812–2820. [Google Scholar] [CrossRef][Green Version]
- Krajewski, W.W.; Fu, X.; Wilkinson, M.; Cronin, N.B.; Dillingham, M.S.; Wigley, D.B. Structural basis for translocation by AddAB helicase-nuclease and its arrest at chi sites. Nature 2014, 508, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.M. Motoring along with the bacterial RecA protein. Nat. Rev. Mol. Cell. Biol. 2007, 8, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Court, D.L.; Sawitzke, J.A.; Thomason, L.C. Genetic engineering using homologous recombination. Annu. Rev. Genet. 2002, 36, 361–388. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.; Kozlov, A.G.; Lohman, T.M. Single-molecule views of protein movement on single-stranded DNA. Annu. Rev. Biophys. 2012, 41, 295–319. [Google Scholar] [CrossRef]
- Wyman, C.; Ristic, D.; Kanaar, R. Homologous recombination-mediated double-strand break repair. DNA Repair. (Amst.) 2004, 3, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Wang, Y.; Li, N.; Jiang, F.-F.; Wu, C.-X.; Liu, F.; Chen, H.-C.; Liu, Z.-F. Highly efficient base editing in bacteria using a Cas9-cytidine deaminase fusion. Commun. Biol. 2018, 1, 32. [Google Scholar] [CrossRef] [PubMed]
- Spaete, R.R.; Mocarski, E.S. Insertion and deletion mutagenesis of the human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 1987, 84, 7213–7217. [Google Scholar] [CrossRef]
- Mocarski, E.S.; Post, L.E.; Roizman, B. Molecular engineering of the herpes simplex virus genome: Insertion of a second L-S junction into the genome causes additional genome inversions. Cell 1980, 22, 243–255. [Google Scholar] [CrossRef]
- Murata, T.; Isomura, H.; Yamashita, Y.; Toyama, S.; Sato, Y.; Nakayama, S.; Kudoh, A.; Iwahori, S.; Kanda, T.; Tsurumi, T. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 2009, 389, 75–81. [Google Scholar] [CrossRef]
- Karu, A.E.; Sakaki, Y.; Echols, H.; Linn, S. The gamma protein specified by bacteriophage gamma. Structure and inhibitory activity for the recBC enzyme of Escherichia coli. J. Biol. Chem. 1975, 250, 7377–7387. [Google Scholar] [CrossRef]
- Murphy, K.C. Lambda Gam protein inhibits the helicase and chi-stimulated recombination activities of Escherichia coli RecBCD enzyme. J. Bacteriol. 1991, 173, 5808–5821. [Google Scholar] [CrossRef]
- Little, J.W. An exonuclease induced by bacteriophage lambda. II. Nature of the enzymatic reaction. J. Biol. Chem. 1967, 242, 679–686. [Google Scholar] [CrossRef]
- Karakousis, G.; Ye, N.; Li, Z.; Chiu, S.K.; Reddy, G.; Radding, C.M. The beta protein of phage lambda binds preferentially to an intermediate in DNA renaturation. J. Mol. Biol. 1998, 276, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Rybalchenko, N.; Golub, E.I.; Bi, B.; Radding, C.M. Strand invasion promoted by recombination protein beta of coliphage lambda. Proc. Natl. Acad. Sci. USA 2004, 101, 17056–17060. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Ellis, H.M.; Lee, E.C.; Jenkins, N.A.; Copeland, N.G.; Court, D.L. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 5978–5983. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Sharan, S.K.; Thomason, L.C.; Kuznetsov, S.G.; Court, D.L. Recombineering: A homologous recombination-based method of genetic engineering. Nat. Protoc. 2009, 4, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Cho, N.; Jung, D.; Bang, D. Genome-scale genetic engineering in Escherichia coli. Biotechnol. Adv. 2013, 31, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Coupling the CRISPR/Cas9 System with Lambda Red Recombineering Enables Simplified Chromosomal Gene Replacement in Escherichia coli. Appl. Environ. Microbiol. 2015, 81, 5103–5114. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.J.; Cummings, A.M.; Nguyen, J.N.; Hamm, D.C.; Donohue, L.K.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 2013, 194, 1029–1035. [Google Scholar] [CrossRef]
- Hou, Z.; Zhang, Y.; Propson, N.E.; Howden, S.E.; Chu, L.-F.; Sontheimer, E.J.; Thomson, J.A. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA 2013, 110, 15644–15649. [Google Scholar] [CrossRef]
- Li, J.-F.; Norville, J.E.; Aach, J.; McCormack, M.; Zhang, D.; Bush, J.; Church, G.M.; Sheen, J. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 2013, 31, 688–691. [Google Scholar] [CrossRef]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Altenbuchner, J. Editing of the Bacillus subtilis Genome by the CRISPR-Cas9 System. Appl. Environ. Microbiol. 2016, 82, 5421–5427. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zheng, G.; Jiang, W.; Hu, H.; Lu, Y. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta. Biochim. Biophys. Sin. (Shanghai) 2015, 47, 231–243. [Google Scholar] [CrossRef]
- Shuman, S.; Glickman, M.S. Bacterial DNA repair by non-homologous end joining. Nat. Rev. Microbiol. 2007, 5, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Bowater, R.; Doherty, A.J. Making ends meet: Repairing breaks in bacterial DNA by non-homologous end-joining. PLoS Genet. 2006, 2, e8. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Bikard, D.; Hatoum-Aslan, A.; Mucida, D.; Marraffini, L.A. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 2012, 12, 177–186. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef]
- Zerbini, F.; Zanella, I.; Fraccascia, D.; König, E.; Irene, C.; Frattini, L.F.; Tomasi, M.; Fantappiè, L.; Ganfini, L.; Caproni, E.; et al. Large scale validation of an efficient CRISPR/Cas-based multi gene editing protocol in Escherichia coli. Microb. Cell Fact. 2017, 16, 68. [Google Scholar] [CrossRef]
- Bassalo, M.C.; Garst, A.D.; Halweg-Edwards, A.L.; Grau, W.C.; Domaille, D.W.; Mutalik, V.K.; Arkin, A.P.; Gill, R.T. Rapid and Efficient One-Step Metabolic Pathway Integration in E. coli. ACS Synth. Biol. 2016, 5, 561–568. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Zhao, S.; Pi, Y.; Chen, W.; Chen, C.; Liu, Q.; Li, M.; Han, D.; Ji, Q. Highly efficient base editing in using an engineered CRISPR RNA-guided cytidine deaminase. Chem. Sci. 2018, 9, 3248–3253. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Jiang, F.-F.; Su, L.; Wang, X.; Chen, Y.-X.; Chen, H.-C.; Liu, Z.-F. Highly Efficient Base Editing in Viral Genome Based on Bacterial Artificial Chromosome Using a Cas9-Cytidine Deaminase Fused Protein. Virol. Sin. 2020, 35, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zong, Y.; Gao, Q.; Zhu, Z.; Wang, Y.; Qin, P.; Liang, C.; Wang, D.; Qiu, J.-L.; Zhang, F.; et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 2019, 364, 292–295. [Google Scholar] [CrossRef]
- Zuo, E.; Sun, Y.; Wei, W.; Yuan, T.; Ying, W.; Sun, H.; Yuan, L.; Steinmetz, L.M.; Li, Y.; Yang, H. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 2019, 364, 289–292. [Google Scholar] [CrossRef]
- Zuo, E.; Sun, Y.; Yuan, T.; He, B.; Zhou, C.; Ying, W.; Liu, J.; Wei, W.; Zeng, R.; Li, Y.; et al. A rationally engineered cytosine base editor retains high on-target activity while reducing both DNA and RNA off-target effects. Nat. Methods 2020, 17, 600–604. [Google Scholar] [CrossRef]
- Wrighton, K.H. Cytosine base editors go off-target. Nat. Rev. Genet. 2019, 20, 254–255. [Google Scholar] [CrossRef]
- Shukal, S.; Lim, X.H.; Zhang, C.; Chen, X. Metabolic engineering of Escherichia coli BL21 strain using simplified CRISPR-Cas9 and asymmetric homology arms recombineering. Microb. Cell Fact. 2022, 21, 19. [Google Scholar] [CrossRef]
- Dong, H.; Cui, Y.; Zhang, D. CRISPR/Cas Technologies and Their Applications in Escherichia coli. Front. Bioeng. Biotechnol. 2021, 9, 762676. [Google Scholar] [CrossRef]
- Brune, W.; Messerle, M.; Koszinowski, U.H. Forward with BACs: New tools for herpesvirus genomics. Trends Genet 2000, 16, 254–259. [Google Scholar] [CrossRef]
- Zhang, Y.; Buchholz, F.; Muyrers, J.P.; Stewart, A.F. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 1998, 20, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Kilby, N.J.; Snaith, M.R.; Murray, J.A. Site-specific recombinases: Tools for genome engineering. Trends Genet 1993, 9, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Zhao, B.; Kieff, E.; Aster, J.C.; Wang, F. EBNA-3B- and EBNA-3C-regulated cellular genes in Epstein-Barr virus-immortalized lymphoblastoid cell lines. J. Virol. 2006, 80, 10139–10150. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Tang, Y.; Zhou, M.; Wu, M.; Ouyang, J.; Gao, J.; Zhang, L.; Li, D.; Chen, Q.; Xiong, W.; et al. Gene modification in the genome of Epstein-Barr virus cloned as a bacterial artificial chromosome. Wei Sheng Wu Xue Bao 2008, 48, 385–390. [Google Scholar] [PubMed]
- Geiser, V.; Cahir-McFarland, E.; Kieff, E. Latent membrane protein 1 is dispensable for Epstein-Barr virus replication in human embryonic kidney 293 cells. PLoS ONE 2011, 6, e22929. [Google Scholar] [CrossRef]
- Raymond, C.S.; Soriano, P. High-efficiency FLP and PhiC31 site-specific recombination in mammalian cells. PLoS ONE 2007, 2, e162. [Google Scholar] [CrossRef] [PubMed]
- Kranz, A.; Fu, J.; Duerschke, K.; Weidlich, S.; Naumann, R.; Stewart, A.F.; Anastassiadis, K. An improved Flp deleter mouse in C57Bl/6 based on Flpo recombinase. Genesis 2010, 48, 512–520. [Google Scholar] [CrossRef]
- Osakada, F.; Mori, T.; Cetin, A.H.; Marshel, J.H.; Virgen, B.; Callaway, E.M. New rabies virus variants for monitoring and manipulating activity and gene expression in defined neural circuits. Neuron 2011, 71, 617–631. [Google Scholar] [CrossRef]
- Lee, E.C.; Yu, D.; Martinez de Velasco, J.; Tessarollo, L.; Swing, D.A.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 2001, 73, 56–65. [Google Scholar] [CrossRef]
- Jarosinski, K.W.; Osterrieder, N.; Nair, V.K.; Schat, K.A. Attenuation of Marek’s disease virus by deletion of open reading frame RLORF4 but not RLORF5a. J. Virol. 2005, 79, 11647–11659. [Google Scholar] [CrossRef]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [PubMed]
- Gupta, A.; Owens, S.M.; Oldenburg, D.G.; White, D.W.; Forrest, J.C. Lytic Replication and Reactivation from B Cells Is Not Required for Establishing or Maintaining Gammaherpesvirus Latency. J. Virol. 2022, 96, e0069022. [Google Scholar] [CrossRef] [PubMed]
- Sleman, S.; Najmuldeen, H.; Hao, H.; Jalal, P.; Saeed, N.; Othman, D.; Qian, Z. Human cytomegalovirus UL24 and UL43 products participate in SAMHD1 subcellular localization. Virus Disease 2022, 33, 383–396. [Google Scholar] [CrossRef]
- Benedyk, T.H.; Connor, V.; Caroe, E.R.; Shamin, M.; Svergun, D.I.; Deane, J.E.; Jeffries, C.M.; Crump, C.M.; Graham, S.C. Herpes simplex virus 1 protein pUL21 alters ceramide metabolism by activating the interorganelle transport protein CERT. J. Biol. Chem. 2022, 298, 102589. [Google Scholar] [CrossRef]
- Tang, J.; Brixel, R.; Brune, W. Copy-Paste Mutagenesis: A Method for Large-Scale Alteration of Viral Genomes. Int. J. Mol. Sci. 2019, 20, 913. [Google Scholar] [CrossRef] [PubMed]
- Heyn, I.; Bremer, L.; Zingler, P.; Fickenscher, H. Self-Repairing Herpesvirus Saimiri Deletion Variants. Viruses 2022, 14, 1525. [Google Scholar] [CrossRef]
- Gerlach, R.G.; Blank, K.; Wille, T. Site-Directed Mutagenesis Using Oligonucleotide-Based Recombineering. In Genetic Manipulation of DNA and Protein—Examples from Current Research; IntechOpen: London, UK, 2013. [Google Scholar]
- Warming, S.; Costantino, N.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. Simple and highly efficient BAC recombineering using galK selection. Nucleic. Acids. Res. 2005, 33, e36. [Google Scholar] [CrossRef]
- Ge, G.; Zhang, L.; Zhao, X.; Hu, X.; Li, Y. Optimization of the Method for Scarless Gene Knockout in Escherichia coli Genome. China Biotechnol. 2014, 34, 68–74. [Google Scholar] [CrossRef]
- Herring, C.D.; Glasner, J.D.; Blattner, F.R. Gene replacement without selection: Regulated suppression of amber mutations in Escherichia coli. Gene 2003, 311, 153–163. [Google Scholar] [CrossRef]
- Wang, W.; Pan, D.; Fu, W.; Ye, X.; Han, J.; Yang, L.; Jia, J.; Liu, J.; Zhu, R.; Zhang, Y.; et al. Development of a skin- and neuro-attenuated live vaccine for varicella. Nat. Commun. 2022, 13, 824. [Google Scholar] [CrossRef] [PubMed]
- Wass, A.B.; Krishna, B.A.; Herring, L.E.; Gilbert, T.S.K.; Nukui, M.; Groves, I.J.; Dooley, A.L.; Kulp, K.H.; Matthews, S.M.; Rotroff, D.M.; et al. Cytomegalovirus US28 regulates cellular EphA2 to maintain viral latency. Sci. Adv. 2022, 8, eadd1168. [Google Scholar] [CrossRef] [PubMed]
- Krauter, S.; Büscher, N.; Bräuchle, E.; Ortega Iannazzo, S.; Penner, I.; Krämer, N.; Gogesch, P.; Thomas, S.; Kreutz, M.; Dejung, M.; et al. An Attenuated Strain of Human Cytomegalovirus for the Establishment of a Subviral Particle Vaccine. Vaccines 2022, 10, 1326. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Y.; Leiby, M.; Zhu, J. A new positive/negative selection scheme for precise BAC recombineering. Mol. Biotechnol. 2009, 42, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Kasem, S.; Yu, M.H.H.; Yamada, S.; Kodaira, A.; Matsumura, T.; Tsujimura, K.; Madbouly, H.; Yamaguchi, T.; Ohya, K.; Fukushi, H. The ORF37 (UL24) is a neuropathogenicity determinant of equine herpesvirus 1 (EHV-1) in the mouse encephalitis model. Virology 2010, 400, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Lan, D.; Shi, X.; Wang, Y.; Liu, C.; Wang, M.; Cui, H.; Tian, G.; Li, J.; Tong, G. Construction of a recombinant HVT virus expressing the HA gene of avian influenza virus H5N1 via Rde/ET recombination system. Wei Sheng Wu Xue Bao 2009, 49, 78–84. [Google Scholar] [PubMed]
- Masud, H.M.A.A.; Yanagi, Y.; Watanabe, T.; Sato, Y.; Kimura, H.; Murata, T. Epstein-Barr Virus BBRF2 Is Required for Maximum Infectivity. Microorganisms 2019, 7, 705. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Hijioka, F.; Watanabe, T.; Yanagi, Y.; Okuno, Y.; Masud, H.M.A.A.; Sato, Y.; Murata, T.; Kimura, H. Role of Epstein-Barr Virus C Promoter Deletion in Diffuse Large B Cell Lymphoma. Cancers 2021, 13, 561. [Google Scholar] [CrossRef] [PubMed]
- Matsugo, H.; Kitamura-Kobayashi, T.; Kamiki, H.; Ishida, H.; Sekine, W.; Takenaka-Uema, A.; Nakagawa, T.; Murakami, S.; Horimoto, T. A potential bat adenovirus-based oncolytic virus targeting canine cancers. Sci. Rep. 2021, 11, 16706. [Google Scholar] [CrossRef]
- Matsugo, H.; Kobayashi-Kitamura, T.; Kamiki, H.; Ishida, H.; Takenaka-Uema, A.; Murakami, S.; Horimoto, T. Establishment of a Simple and Efficient Reverse Genetics System for Canine Adenoviruses Using Bacterial Artificial Chromosomes. Viruses 2020, 12, 767. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Damas, M.; Freise, A.; Hage, E.; Dhingra, A.; Rückert, J.; Gallo, A.; Kremmer, E.; Tegge, W.; Brönstrup, M.; et al. Kaposi’s sarcoma-associated herpesvirus vIRF2 protein utilizes an IFN-dependent pathway to regulate viral early gene expression. PLoS Pathog. 2019, 15, e1007743. [Google Scholar] [CrossRef]
- Bayer, C.N.; Sepulchro, A.G.V.; Rennig, M.; Nørholm, M.H.H. Efficient Bacterial Genome Engineering throughout the Central Dogma Using the Dual-Selection Marker. ACS Synth. Biol. 2022, 11, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Khetrapal, V.; Mehershahi, K.; Rafee, S.; Chen, S.; Lim, C.L.; Chen, S.L. A set of powerful negative selection systems for unmodified Enterobacteriaceae. Nucleic. Acids Res. 2015, 43, e83. [Google Scholar] [CrossRef]
- Sleman, S.; Hao, H.; Najmuldeen, H.; Jalal, P.; Saeed, N.; Othman, D.; Qian, Z. Human Cytomegalovirus UL24 and UL43 Cooperate to Modulate the Expression of Immunoregulatory UL16 Binding Protein 1. Viral. Immunol. 2022, 35, 529–544. [Google Scholar] [CrossRef]
- Neuhierl, B.; Delecluse, H.-J. The Epstein-Barr virus BMRF1 gene is essential for lytic virus replication. J. Virol. 2006, 80, 5078–5081. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, S.; Makvandi, M.; Abbasi, S.; Azadmanesh, K.; Teimoori, A. Developing oncolytic through UL39 knockout by CRISPR-Cas9. Iran. J. Basic. Med. Sci. 2020, 23, 937–944. [Google Scholar] [CrossRef]
- Ma, W.; He, H.; Wang, H. Oncolytic herpes simplex virus and immunotherapy. BMC Immunol. 2018, 19, 40. [Google Scholar] [CrossRef]
- Artusi, S.; Miyagawa, Y.; Goins, W.F.; Cohen, J.B.; Glorioso, J.C. Herpes Simplex Virus Vectors for Gene Transfer to the Central Nervous System. Diseases 2018, 6, 74. [Google Scholar] [CrossRef]
- Quinlivan, M.; Breuer, J. Clinical and molecular aspects of the live attenuated Oka varicella vaccine. Rev. Med. Virol. 2014, 24, 254–273. [Google Scholar] [CrossRef] [PubMed]
- Sadaoka, T.; Mori, Y. Vaccine Development for Varicella-Zoster Virus. Adv. Exp. Med. Biol. 2018, 1045, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Galea, S.A.; Sweet, A.; Beninger, P.; Steinberg, S.P.; Larussa, P.S.; Gershon, A.A.; Sharrar, R.G. The safety profile of varicella vaccine: A 10-year review. J. Infect. Dis. 2008, 197, S165–S169. [Google Scholar] [CrossRef]
- Heusel, E.H.; Grose, C. Twelve Children with Varicella Vaccine Meningitis: Neuropathogenesis of Reactivated Live Attenuated Varicella Vaccine Virus. Viruses 2020, 12, 1078. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-F.; Wang, W.; Jiang, X.; Zeng, W.-B.; Shen, Z.-Z.; Song, Y.-G.; Yang, H.; Liu, X.-J.; Dong, X.; Zhou, J.; et al. ORF7 of Varicella-Zoster Virus Is Required for Viral Cytoplasmic Envelopment in Differentiated Neuronal Cells. J. Virol. 2017, 91, e00127-17. [Google Scholar] [CrossRef]
- Zhang, Z.; Selariu, A.; Warden, C.; Huang, G.; Huang, Y.; Zaccheus, O.; Cheng, T.; Xia, N.; Zhu, H. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. PLoS Pathog. 2010, 6, e1000971. [Google Scholar] [CrossRef] [PubMed]
- Goulleret, N.; Mauvisseau, E.; Essevaz-Roulet, M.; Quinlivan, M.; Breuer, J. Safety profile of live varicella virus vaccine (Oka/Merck): Five-year results of the European Varicella Zoster Virus Identification Program (EU VZVIP). Vaccine 2010, 28, 5878–5882. [Google Scholar] [CrossRef] [PubMed]
- Gilden, D.H.; Kleinschmidt-DeMasters, B.K.; LaGuardia, J.J.; Mahalingam, R.; Cohrs, R.J. Neurologic complications of the reactivation of varicella-zoster virus. N. Engl. J. Med. 2000, 342, 635–645. [Google Scholar] [CrossRef]
- Mo, Z.-J.; Huang, S.-J.; Qiu, L.-X.; Li, C.-G.; Yu, X.-J.; Li, M.-Q.; Chen, Z.; Zhong, G.-H.; Pan, D.-Q.; Huang, L.-R.; et al. Safety and immunogenicity of a skin- and neuro-attenuated live vaccine for varicella: A randomized, double-blind, controlled, dose-escalation and age de-escalation phase 1 clinical trial. Lancet Reg. Health—West. Pac. 2023. [Google Scholar] [CrossRef]
- Goldstein, D.J.; Weller, S.K. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: Characterization of an ICP6 deletion mutant. Virology 1988, 166, 41–51. [Google Scholar] [CrossRef]
- Yoon, S.S.; Carroll, N.M.; Chiocca, E.A.; Tanabe, K.K. Cancer gene therapy using a replication-competent herpes simplex virus type 1 vector. Ann Surg 1998, 228, 366–374. [Google Scholar] [CrossRef]
- Miao, L.; Fraefel, C.; Sia, K.C.; Newman, J.P.; Mohamed-Bashir, S.A.; Ng, W.H.; Lam, P.Y.P. The potential application of a transcriptionally regulated oncolytic herpes simplex virus for human cancer therapy. Br. J. Cancer 2014, 110, 94–106. [Google Scholar] [CrossRef][Green Version]
- Neubauer, A.; Rudolph, J.; Brandmüller, C.; Just, F.T.; Osterrieder, N. The equine herpesvirus 1 UL34 gene product is involved in an early step in virus egress and can be efficiently replaced by a UL34-GFP fusion protein. Virology 2002, 300, 189–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hwang, S.; Wu, T.-T.; Tong, L.M.; Kim, K.S.; Martinez-Guzman, D.; Colantonio, A.D.; Uittenbogaart, C.H.; Sun, R. Persistent gammaherpesvirus replication and dynamic interaction with the host in vivo. J. Virol. 2008, 82, 12498–12509. [Google Scholar] [CrossRef] [PubMed]
- Reese, T.A.; Wakeman, B.S.; Choi, H.S.; Hufford, M.M.; Huang, S.C.; Zhang, X.; Buck, M.D.; Jezewski, A.; Kambal, A.; Liu, C.Y.; et al. Helminth infection reactivates latent γ-herpesvirus via cytokine competition at a viral promoter. Science 2014, 345, 573–577. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Editing Techniques | Length of Homologous Arms | Editing Efficiency | Requirement for Editing Site | Stability of BAC |
|---|---|---|---|---|
| RecA Recombination | 500 bp–3 k bp | 10−6 to 10−4 | None | Causes mutations in the BAC |
| λ-red Recombination | 30–50 bp | <1% | None | Maintained the stability of BAC |
| Base Editing | Not Required | Approaching 100% | PAM Sites & Base Editing Sites; Only Base Editing can be done | Maintained the stability of BAC |
| Screening Methods | Feasibility of Continuous Editing | Applicable E. coli Strain Type | Selection Efficiency | Auxiliary Proteins | Feasibility of Scarless Editing | Duration of Each Editing Cycle |
|---|---|---|---|---|---|---|
| Single selection | Difficult | All | >95% | None | No | 3 Days |
| combination site-specific recombinase recognition motif | Difficult | All | Approaching 100% | FLP/Cre | No | 7 Days |
| Combination I-sceI endonuclease recognition site | Feasible | All | Negtive: 1–15% | I-sceI | Yes | 7 Days |
| Positive and negative (dual) selection cassettes | Feasible | rpsL; StrR galK; ΔgalK | Negative: rpsL-7.8% galK-16% | None | Yes | rpsL-Kana: 7 Days galK: 12 Days galK-Kana: 9 Days |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, M.; Tang, J.; Ge, S.; Li, T.; Xia, N. Bacterial-Artificial-Chromosome-Based Genome Editing Methods and the Applications in Herpesvirus Research. Microorganisms 2023, 11, 589. https://doi.org/10.3390/microorganisms11030589
Hao M, Tang J, Ge S, Li T, Xia N. Bacterial-Artificial-Chromosome-Based Genome Editing Methods and the Applications in Herpesvirus Research. Microorganisms. 2023; 11(3):589. https://doi.org/10.3390/microorganisms11030589
Chicago/Turabian StyleHao, Mengling, Jiabao Tang, Shengxiang Ge, Tingdong Li, and Ningshao Xia. 2023. "Bacterial-Artificial-Chromosome-Based Genome Editing Methods and the Applications in Herpesvirus Research" Microorganisms 11, no. 3: 589. https://doi.org/10.3390/microorganisms11030589
APA StyleHao, M., Tang, J., Ge, S., Li, T., & Xia, N. (2023). Bacterial-Artificial-Chromosome-Based Genome Editing Methods and the Applications in Herpesvirus Research. Microorganisms, 11(3), 589. https://doi.org/10.3390/microorganisms11030589