
Identifying New Clusterons: Application of TBEV Analyzer 3.0
 , , , , and
, , , , and
Abstract
1. Introduction
- Theoretical improvement:
- −
- Performing whole GenBank analysis and introducing new clusterons, followed by updating of the model of clusteron structure obtained by the clusteron approach.
- Practical improvements leading to enhancement of platform functionality:
- −
- Automatic monitoring of GenBank for emerging novel strains.
- −
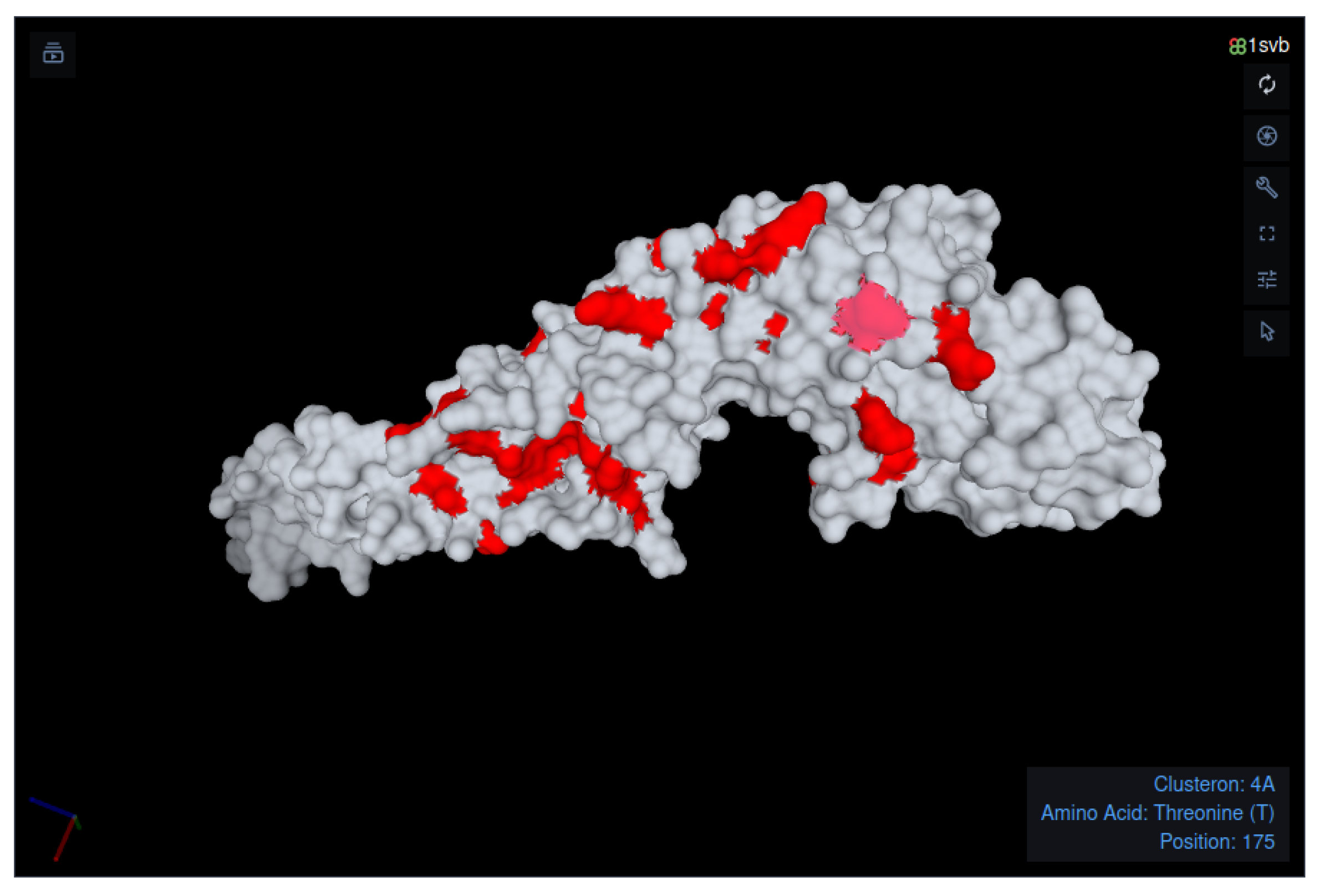
- Identification of the query’s amino acid signature and its visualization on the E protein surface.
- −
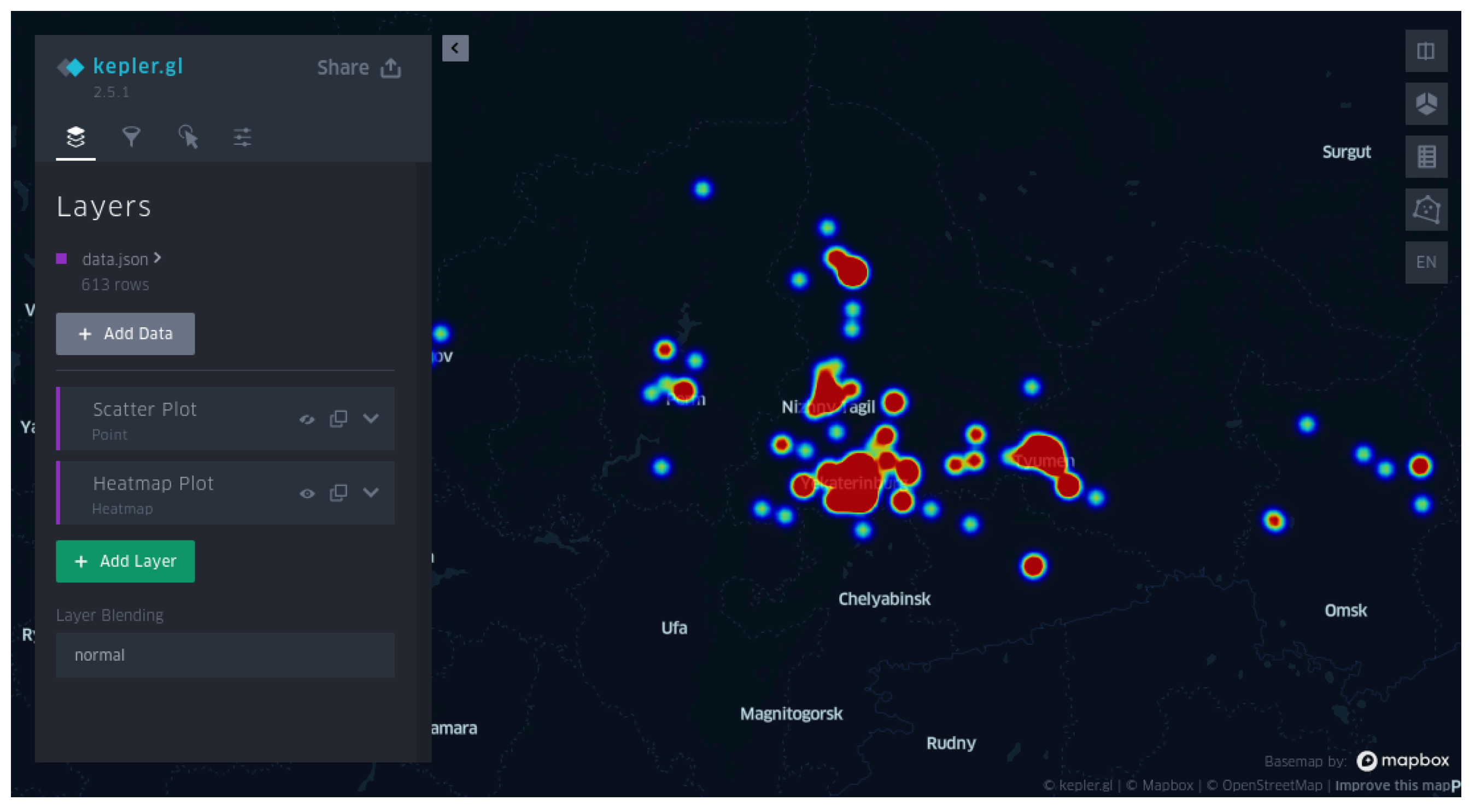
- Provision of high-quality visualization of clusteron spatial distributions and visualization of a query on a geographical map via its latitude and longitude.
- −
- Interactive visualization of the clusteron structure.
- −
- Equipping the platform with an Application Programming Interface (API).
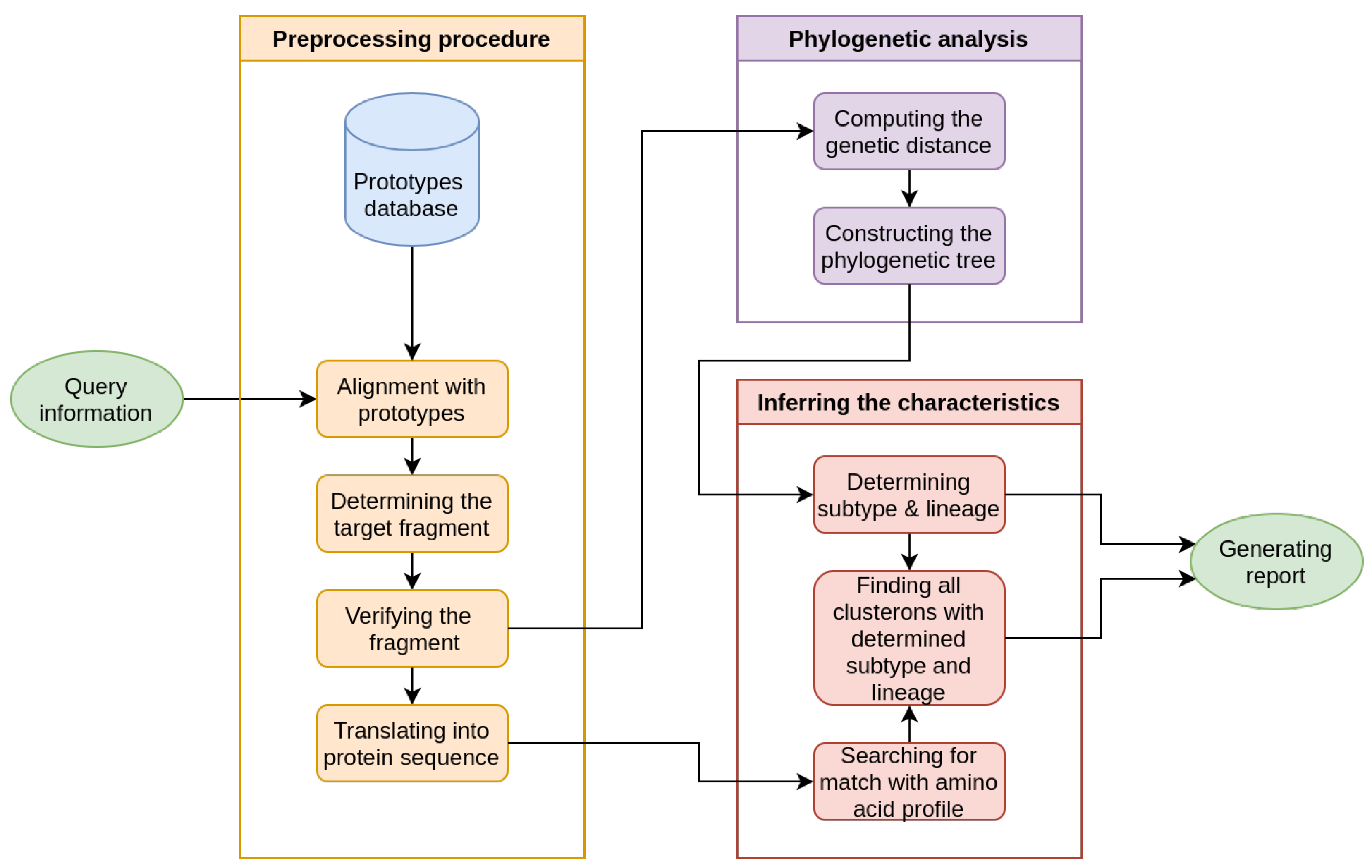
2. Materials and Methods
- Constructing the CS via phylogenetic network analysis.
- Application of the CS to identify the hierarchical three-fold phylogenetic characteristics of a query strain.
- General Report
- Clusteron Structure
- Phylogenetic Tree
- Nucleotide Alignment Table
- Protein Alignment Table
- Amino Acid Signature
- Geographical Map
- Supplementary Files
- Phylogenetic characteristics
- Specific amino acid signature
- E protein fragment coding sequence (prototype)
3. Experiment and Results
- Determining the three-fold phylogenitic characteristics for all new strains
- Reconsideration of the set of strains identified as unique, for finding any new clusterons
4. Discussion
- Performing CA analysis
- Monitoring the geographical distribution of clusterons
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TBEV | Tick-Borne Encephalitis Virus |
| TBEV-FE | Tick-Borne Encephalitis Virus—Far Eastern (subtype) |
| TBEV-Sib | Tick-Borne Encephalitis Virus—Siberian (subtype) |
| TBEV-Eu | Tick-Borne Encephalitis Virus—European (subtype) |
| TBEV-Bkl | Tick-Borne Encephalitis Virus—Baikalian (subtype) |
| TBEV-Him | Tick-Borne Encephalitis Virus—Himalayan (subtype) |
| TBEV-Sib-Balt | Tick-Borne Encephalitis Virus—Siberian (subtype)—Baltic (lineage) |
| TBEV-Sib-Asia | Tick-Borne Encephalitis Virus—Siberian (subtype)—Asian (lineage) |
| TBEV-Sib-S.-Sib | Tick-Borne Encephalitis Virus—Siberian (subtype)—South-Siberian (lineage) |
| TBEV-Sib-E.-Sib | Tick-Borne Encephalitis Virus—Siberian (subtype)—East-Siberian (lineage) |
| CA | Clusteron Approach |
| CS | Clusteron Structure (subtype) |
| API | Application Programming Interface (subtype) |
| PDB | Protein Data Bank |
References
- Amato-Gauci, A.; Zeller, H. Tick-borne encephalitis joins the diseases under surveillance in the European Union. Eurosurveillance 2012, 17, 20299. [Google Scholar] [CrossRef]
- Rospotrebnadzor. Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing. 2018. Available online: www.rospotrebnadzor.ru/activities/statistical-materials/ (accessed on 15 August 2021).
- Fares, W.; Dachraoui, K.; Cherni, S.; Barhoumi, W.; Slimane, T.B.; Younsi, H.; Zhioua, E. Tick-borne encephalitis virus in Ixodes ricinus (Acari: Ixodidae) ticks, Tunisia. Ticks Tick-Borne Dis. 2021, 12, 101606. [Google Scholar] [CrossRef]
- Holding, M.; Dowall, S.D.; Medlock, J.M.; Carter, D.P.; McGinley, L.; Curran-French, M.; Pullan, S.T.; Chamberlain, J.; Hansford, K.M.; Baylis, M.; et al. Detection of new endemic focus of tick-borne encephalitis virus (TBEV), Hampshire/Dorset border, England, September 2019. Eurosurveillance 2019, 24, 1900658. [Google Scholar] [CrossRef]
- Holding, M.; Dowall, S.D.; Medlock, J.M.; Carter, D.P.; Pullan, S.T.; Lewis, J.; Vipond, R.; Rocchi, M.S.; Baylis, M.; Hewson, R. Tick-borne encephalitis virus, United Kingdom. Emerg. Infect. Dis. 2020, 26, 90. [Google Scholar] [CrossRef]
- Kovalev, S.; Mukhacheva, T. Reconsidering the classification of tick-borne encephalitis virus within the Siberian subtype gives new insights into its evolutionary history. Infect. Genet. Evol. 2017, 55, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Shang, G.; Lu, S.; Yang, J.; Xu, J. A new subtype of eastern tick-borne encephalitis virus discovered in Qinghai-Tibet Plateau, China. Emerg. Microbes Infect. 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Choklikitumnuey, P. Species Identification of Ticks Collected in Skåne County and Their Role in the Epidemiology of Tick-Borne Encephalitis (TBEV). Master’s Thesis, Swedish University of Agricultural Sciences, Uppsala, Sweden, 2019. [Google Scholar]
- Wallenhammar, A.; Lindqvist, R.; Asghar, N.; Gunaltay, S.; Fredlund, H.; Davidsson, Å.; Andersson, S.; Överby, A.K.; Johansson, M. Revealing new tick-borne encephalitis virus foci by screening antibodies in sheep milk. Parasites Vectors 2020, 13, 1–12. [Google Scholar] [CrossRef]
- Gritsun, T.; Lashkevich, V.; Gould, E. Tick-borne encephalitis. Antivir. Res. 2003, 57, 129–146. [Google Scholar] [CrossRef]
- Dobler, G.; Erber, W.; Bröker, M.; Schmitt, H.J. The TBE Book; Global Health Press Pte Ltd.: Singapore, 2020. [Google Scholar]
- Liebig, K.; Boelke, M.; Grund, D.; Schicht, S.; Bestehorn-Willmann, M.; Chitimia-Dobler, L.; Dobler, G.; Jung, K.; Becker, S.C. The Stable Matching Problem in TBEV Enzootic Circulation: How Important Is the Perfect Tick-Virus Match? Microorganisms 2021, 9, 196. [Google Scholar] [CrossRef]
- Heinz, F.X. Epitope mapping of flavivirus glycoproteins. Adv. Virus Res. 1986, 31, 103–168. [Google Scholar] [CrossRef]
- Michelitsch, A.; Wernike, K.; Klaus, C.; Dobler, G.; Beer, M. Exploring the reservoir hosts of tick-borne encephalitis virus. Viruses 2019, 11, 669. [Google Scholar] [CrossRef]
- Süss, J.; Klaus, C.; Gerstengarbe, F.W.; Werner, P.C. What makes ticks tick? Climate change, ticks, and tick-borne diseases. J. Travel Med. 2008, 15, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Ruzhnikov, G.; Danchinova, G.; Fedorov, R.; Khasnatinov, M.; Paramonov, V.; Lyapunov, A. Modern technologies for informational and analytical evaluation of activity and forecast of spatial distribution of ixodid ticks by the example of Irkutsk City. Bull. Sib. Branch Russ. Acad. Med. Sci. 2012, 32, 55–59. [Google Scholar]
- Molorodov, Y.; Khodorchenko, D. Development of the information system for tick-hazard on the basis of the objective ontology (in Russian). Vestn. NSU Ser. Inf. Technol. 2018, 16, 107–114. [Google Scholar] [CrossRef]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Forghani, M.; Kovalev, S.; Vasev, P.; Bolkov, M. TBEV Analyzer: A Platform for Evolutionary Analysis of Tick-borne Encephalitis Virus. In Proceedings of the 2019 International Multi-Conference on Engineering, Computer and Information Sciences (SIBIRCON), Novosibirsk, Russia, 21–27 October 2019; pp. 397–402. [Google Scholar] [CrossRef]
- Kovalev, S.Y.; Mukhacheva, T.A. Clusteron structure of tick-borne encephalitis virus populations. Infect. Genet. Evol. 2013, 14, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Ostell, J.; Pruitt, K.D.; Sayers, E.W. GenBank. Nucleic Acids Res. 2018, 46, D41–D47. [Google Scholar] [CrossRef]
- Kovalev, S.Y.; Mukhacheva, T.A. Clusterons as a tool for monitoring populations of tick-borne encephalitis virus. J. Med. Virol. 2014, 86, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Forghani, M.; Kovalev, S.; Bolkov, M.; Khachay, M.; Vasev, P. TBEV analyzer platform for evolutionary analysis and monitoring tick-borne encephalitis virus: 2020 update. Biostat. Epidemiol. 2022, 6, 57–73. [Google Scholar] [CrossRef]
- Kovalev, S.Y.; Mukhacheva, T.A. Tick-borne encephalitis virus subtypes emerged through rapid vector switches rather than gradual evolution. Ecol. Evol. 2014, 4, 4307–4316. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.A.; Heinz, F.X.; Mandl, C.; Kunz, C.; Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature 1995, 375, 291–298. [Google Scholar] [CrossRef]
- Sehnal, D.; Rose, A.S.; Koca, J.; Burley, S.K.; Velankar, S. Mol*: Towards a Common Library and Tools for Web Molecular Graphics. In Proceedings of the MolVa@ EuroVis, Brno, Czech, 4–8 June 2018; pp. 29–33. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- PDBe Molstar. 2020. Available online: https://github.com/PDBeurope/pdbe-molstar (accessed on 15 August 2021).
- Uber. Uber(2018).Kepler.gl. 2018. Available online: http://kepler.gl (accessed on 15 August 2021).
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.P.; Vaz, C.; Monteiro, P.T.; Melo-Cristino, J.; Ramirez, M.; Carriço, J.A. PHYLOViZ: Phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinform. 2012, 13, 87. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
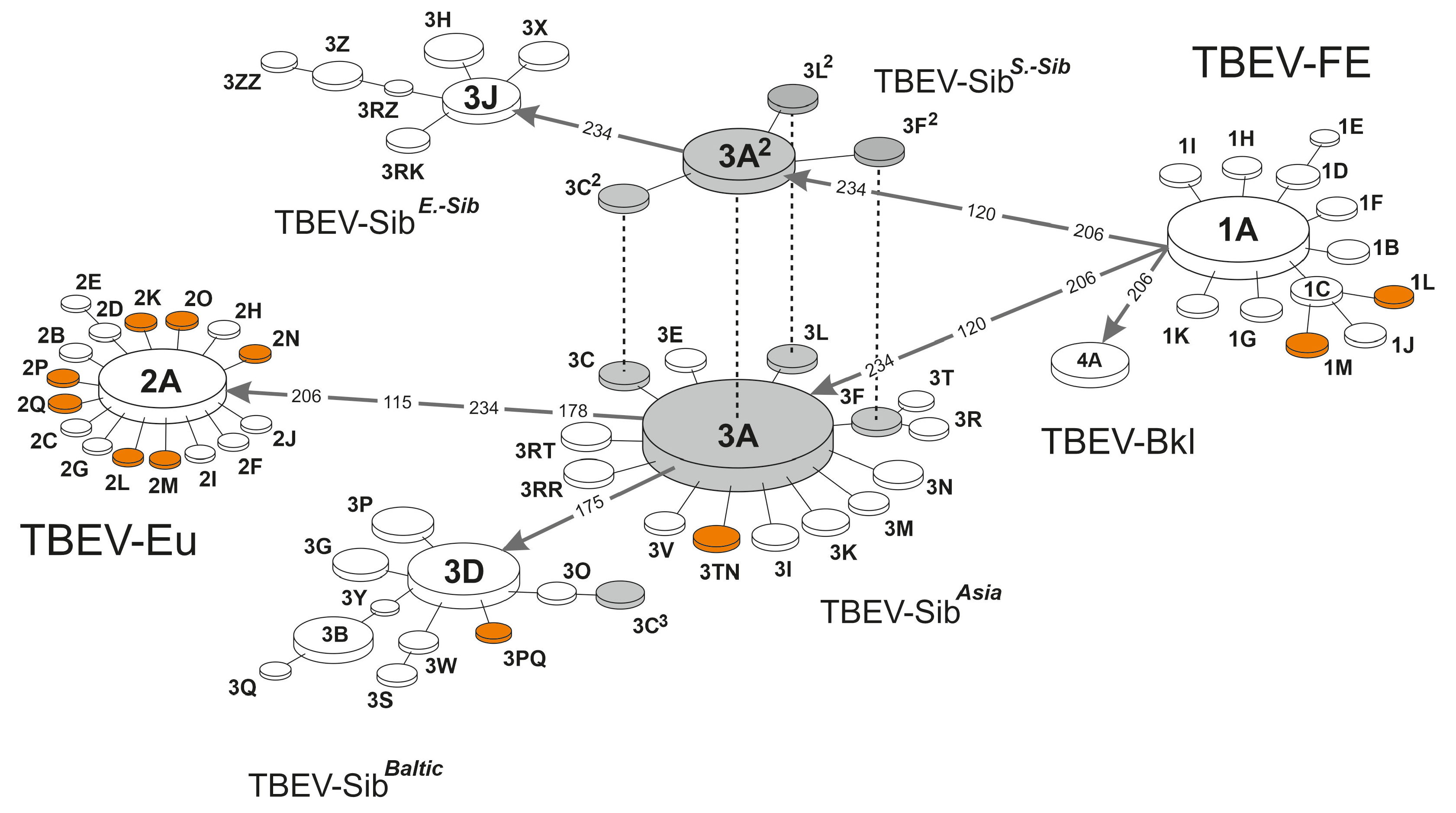
| Clusteron | Prototype Strains GenBank Acc. No. | Freq. (%) | Clusteron-Specific Amino Acid Signature | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 115 | 119 | 120 | 122 | 126 | 128 | 130 | 136 | 137 | 148 | 151 | 153 | 158 | 163 | 165 | 167 | 169 | 171 | 175 | 178 | 188 | 189 | 192 | 195 | 201 | 204 | 206 | 221 | 228 | 232 | 234 | 237 | 239 | 246 | ||||
| TBEV-FE | |||||||||||||||||||||||||||||||||||||
| 1A | FJ402886 | 100 (6.83) | T | A | S | E | K | T | H | K | I | G | V | A | S | A | F | V | S | K | T | D | V | A | V | A | E | K | S | N | K | A | N | N | E | A | |
| 1B | FJ214111 | 16 (1.09) | . | . | . | . | . | . | . | . | . | . | . | V | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | |
| 1C | DQ989336 | 14 (0.96) | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | R | . | . | . | . | . | |
| 1D | AF091013 | 3 (0.2) | . | . | . | . | . | . | Y | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | |
| 1E | HM008981 | 6 (0.41) | . | . | . | . | . | . | Y | . | . | . | . | . | . | V | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | |
| 1F | HM008978 | 3 (0.2) | . | . | . | . | . | . | . | V | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | |
| 1G | AB237185 | 2 (0.14) | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | |
| 1H | AF091008 | 8 (0.55) | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | V | |
| 1I | AY178833 | 5 (0.34) | . | . | . | G | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | |
| 1J | FJ214120 | 3 (0.2) | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | R | . | . | E | . | . | |
| 1K | X07755 | 14 (0.96) | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | R | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | |
| 1L * | KM019546 | 3 (0.2) | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | R | . | . | . | . | . | . | . | . | . | . | R | . | . | . | . | . | |
| 1M * | KF880804 | 3 (0.2) | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | R | . | T | . | . | . | |
| TBEV-Eu | |||||||||||||||||||||||||||||||||||||
| 2A | EF113081 | 290 (19.8) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| 2B | HM468162 | 23 (1.57) | A | . | A | . | . | . | Y | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| 2C | GQ266392 | 8 (0.55) | A | . | A | . | . | I | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| 2D | AJ414703 | 2 (0.14) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | A | . | . | . | . | . | . | . | |
| 2E | HM468153 | 2 (0.14) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | T | . | . | . | . | A | . | . | . | . | . | . | . | |
| 2F | AF091012 | 9 (0.61) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | T | . | . | . | |
| 2G | EF113085 | 3 (0.2) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| 2H | TEU27495 | 4 (0.27) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | I | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| 2I | JF501438 | 11 (0.75) | A | . | A | G | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| 2J | AJ319583 | 2 (0.14) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | K | . | |
| 2K * | KC292217 | 9 (0.61) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | V | |
| 2L * | DQ393776 | 3 (0.2) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | A | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| 2M * | KC154196 | 2 (0.14) | A | . | A | . | . | . | . | . | . | R | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| 2N * | JQ654653 | 4 (0.27) | A | . | A | . | . | . | . | . | M | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| 2O * | KF991107 | 5 (0.34) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | R | . | . | . | . | . | |
| 2P * | MG210946 | 4 (0.27) | A | . | A | . | . | . | . | . | . | . | . | . | N | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| 2Q * | LC171402 | 2 (0.14) | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | V | . | . | . | . | . | . | . | |
| TBEV-Sib-Asia | |||||||||||||||||||||||||||||||||||||
| 3A | AF527415 | 404 (27.58) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3C | GU444159 | 14 (0.96) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | R | L | . | . | . | H | . | . | . | |
| 3E | GU444279 | 5 (0.34) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | V | H | . | . | . | |
| 3F | GU444225 | 64 (4.37) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | R | . | H | . | . | . | |
| 3I | GU444204 | 12 (0.82) | . | . | A | . | . | . | . | . | . | . | I | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3K | EU443275 | 8 (0.55) | . | . | A | . | . | . | . | R | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3L | GU444211 | 2 (0.14) | . | . | A | . | . | I | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3M | GU143822 | 4 (0.27) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | I | . | . | . | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3N | AB049349 | 3 (0.2) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | S | . | . | H | . | . | . | |
| 3R | KT748745 | 4 (0.27) | . | . | A | . | . | S | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | R | . | H | . | . | . | |
| 3RR | GU444227 | 3 (0.2) | . | . | A | . | R | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3RT | GU444136 | 4 (0.27) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3T | KT749641 | 4 (0.27) | . | . | A | . | . | . | . | R | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | R | . | H | . | . | . | |
| 3TN * | MG675054 | 7 (0.48) | A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3V | FJ214137 | 13 (0.89) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | Y | . | . | . | |
| TBEV-Sib-S.-Siberian | |||||||||||||||||||||||||||||||||||||
| 3A | GQ845432 | 50 (3.41) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3C | EF566817 | 2 (0.14) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | R | L | . | . | . | H | . | . | . | |
| 3F | GQ845430 | 3 (0.2) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | R | . | H | . | . | . | |
| 3L | EU443268 | 7 (0.48) | . | . | A | . | . | I | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3C | JX315786 | 2 (0.14) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | R | L | . | . | . | H | . | . | . | |
| TBEV-Sib-Baltic | |||||||||||||||||||||||||||||||||||||
| 3D | DQ393773 | 73 (4.98) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3B | GU444253 | 36 (2.46) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | . | . | . | . | . | L | . | . | G | Q | . | . | . | |
| 3G | DQ451295 | 10 (0.68) | . | V | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3O | GU444224 | 4 (0.27) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | . | . | . | . | R | L | . | . | . | H | . | . | . | |
| 3P | FJ214139 | 17 (1.16) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | . | . | . | . | . | L | . | . | . | Y | . | . | . | |
| 3Q | JX315727 | 5 (0.34) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | S | . | . | . | N | . | . | . | . | . | . | . | L | . | . | G | Q | . | . | . | |
| 3PQ * | JX315894 | 4 (0.27) | . | . | A | . | . | I | . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | . | . | . | . | . | L | . | . | . | H | . | . | . | |
| 3S | KT749572 | 6 (0.41) | . | . | A | . | . | . | . | R | . | . | . | . | . | . | . | . | . | . | N | . | . | . | . | . | . | . | L | . | R | . | H | . | . | . | |
| 3W | DQ486861 | 7 (0.48) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | . | . | . | . | . | L | . | R | . | H | . | . | . | |
| 3Y | FJ214136 | 1 (0.07) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | . | . | . | . | . | L | . | . | . | Q | . | . | . | |
| TBEV-Sib-E.-Siberian | |||||||||||||||||||||||||||||||||||||
| 3J | AB049348 | 24 (1.64) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | Q | . | . | . | |
| 3H | AF091006 | 42 (2.87) | . | V | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | Q | . | . | . | |
| 3RK | KT749636 | 3 (0.2) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . | . | . | L | . | . | . | Q | . | . | . | |
| 3X | LC017692 | 7 (0.48) | . | . | A | G | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | Q | . | . | . | |
| 3RZ | KT321407 | 1 (0.07) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | I | . | . | . | L | . | . | . | Q | . | . | . | |
| 3Z | KT321372 | 22 (1.5) | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | I | S | . | . | L | . | . | . | Q | . | . | . | |
| 3ZZ | KC417474 | 8 (0.55) | . | . | A | . | . | . | . | . | . | . | I | . | . | . | . | . | . | . | . | . | . | . | I | S | . | . | L | . | . | . | Q | . | . | . | |
| TBEV-Baikalian | |||||||||||||||||||||||||||||||||||||
| 4A | EF469662 | 17 (1.16) | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | . | . | . | . | |
| Subtype | Lineage | Clusteron | Strains | Region |
|---|---|---|---|---|
| Far-Eastern | – | 1L | KJ914682 KM019546 KJ739729 | Tomsk and Novosibirsk regions (Russia) |
| – | 1M | KP869172 KF880804 KT001073 | Russia: Far East, Khabarovsk territory, Nikolaevsk and Lazo regions | |
| Siberian | Asian | 3TN | JX315908 JX315996 MF161158 MG675054 MT113363 MT113364 MT113367 | Perm, Sverdlovsk and Irkutsk regions (Russia) |
| Baltic | 3PQ | JX315894 KT748739 MT889225 MT424734 | Hizhnyi Tagil in Sverdlovsk region, Petrozavodsk and Valaam Island in Republic of Karelia, and Omsk region (Russia) | |
| European | – | 2K | KY069126 KY069125 KY069124 KT895102 KT895101 KT895100 KT895099 KT895098 KC292217 | Altai region (Russia) and Baden-Wuerttemberg (Germany) |
| – | 2L | GU183383 DQ393776 GU183381 | Estonia and Finland | |
| – | 2M | KC154196 KC154197 | Heselbach in South-East Germany | |
| – | 2N | JQ654653 MK903681 MK903683 MK922617 | (Slovenia) and Lower Saxony (Germany) | |
| – | 2O | MN735988 MN128700 KF991107 MN735989 MN735990 | Northern Zealand (Denmark), UK-Thetford Forest (United Kingdom), and Mandal (Norway) | |
| – | 2P | AF091011 MG210946 MG210948 MG210947 | Tatabanya (Hungary) | |
| – | 2Q | MN661145 LC171402 | UK-Hampshire (United Kingdom) and Netherlands |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forghani, M.; Kovalev, S.; Khachay, M.; Ramsay, E.; Bolkov, M.; Vasev, P. Identifying New Clusterons: Application of TBEV Analyzer 3.0. Microorganisms 2023, 11, 324. https://doi.org/10.3390/microorganisms11020324
Forghani M, Kovalev S, Khachay M, Ramsay E, Bolkov M, Vasev P. Identifying New Clusterons: Application of TBEV Analyzer 3.0. Microorganisms. 2023; 11(2):324. https://doi.org/10.3390/microorganisms11020324
Chicago/Turabian StyleForghani, Majid, Sergey Kovalev, Michael Khachay, Edward Ramsay, Mikhail Bolkov, and Pavel Vasev. 2023. "Identifying New Clusterons: Application of TBEV Analyzer 3.0" Microorganisms 11, no. 2: 324. https://doi.org/10.3390/microorganisms11020324
APA StyleForghani, M., Kovalev, S., Khachay, M., Ramsay, E., Bolkov, M., & Vasev, P. (2023). Identifying New Clusterons: Application of TBEV Analyzer 3.0. Microorganisms, 11(2), 324. https://doi.org/10.3390/microorganisms11020324