

State-of the-Art Constraint-Based Modeling of Microbial Metabolism: From Basics to Context-Specific Models with a Focus on Methanotrophs
 ,
,
Abstract
1. Introduction
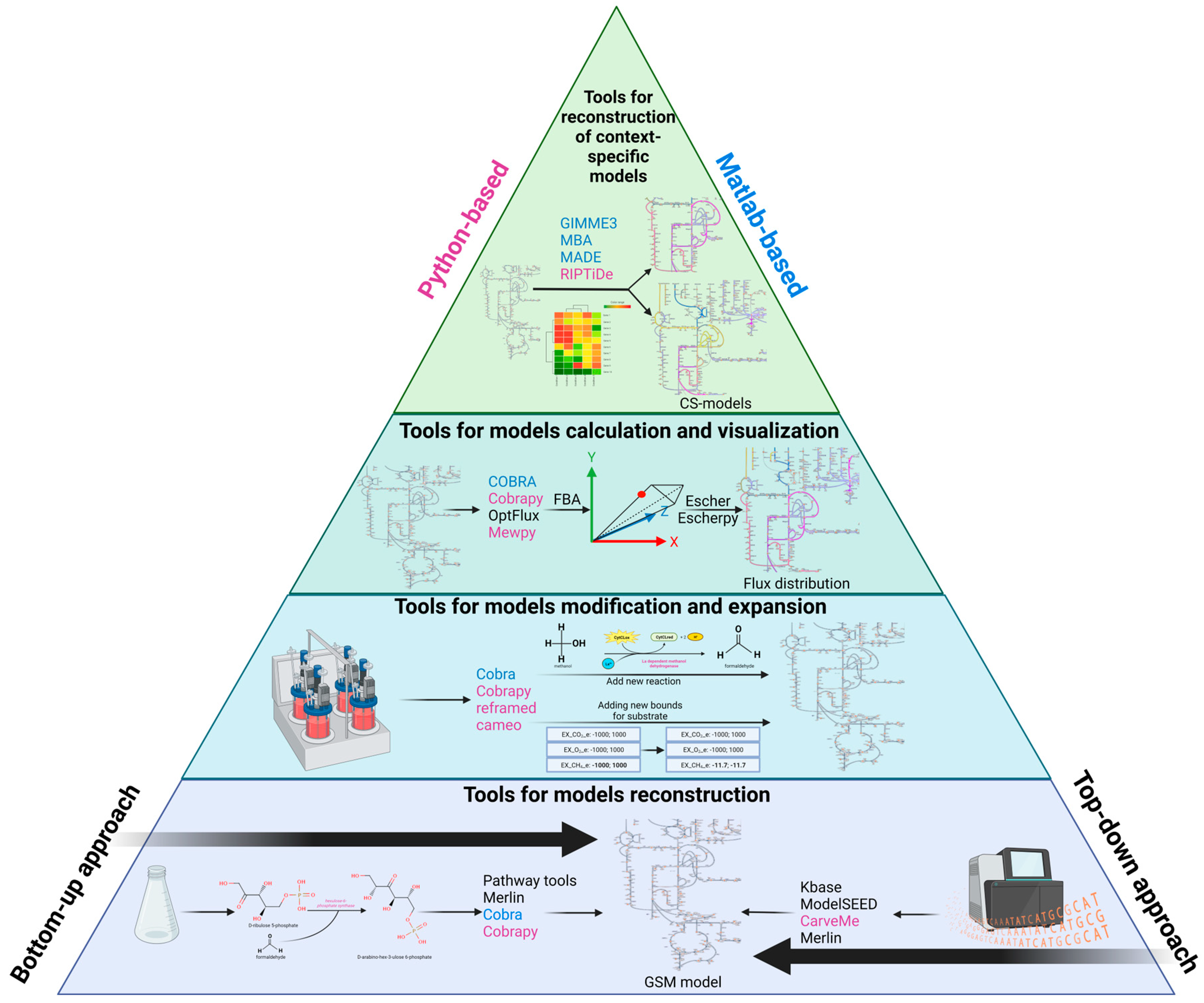
2. Reconstruction and Analysis of Genome-Scale Metabolic Models
2.1. The Stages of Metabolic Model Reconstruction
2.2. Databases of the Microorganisms’ Genomes
2.3. GSM Models for C1-Utilizing Bacteria
2.4. Web Resources and Tools for Automatic Reconstruction of GSM Models
2.4.1. Web Resources

{kind=link}
| Program | Tool Type | Type of Reconstruction | Databases for Reaction Information | Programs Availability | Reference |
|---|---|---|---|---|---|
| Kbase http://kbase.us | Web-service | automatic | ModelSEED | Available | [18] |
| ModelSEED http://www.theseed.org/models/ | Web-service | automatic | ModelSEED | Available | [66,67] |
| FAME http://f-a-m-e.org | Web-service | automatic | KEGG | Not available | [68] |
| Pathway tools ttp://pathwaytools.com | GUI based program | MetaCyc Template models | Available, but works with BioCyc license | [34] | |
| GEMSiRV http://sb.nhri.org.tw/GEMSiRV | GUI based program | semi-automatic | Template models | Not available | [70] |
| AuReMe http://aureme.genouest.org | Command line program | automatic | MetaCyc, BiGG, ModelSEED | Available | [71] |
| Merlin v.4 https://www.merlin-sysbio.org/ | GUI based program | semi-automatic | KEGG, BiGG | Available | [72] |
| Gapseq https://github.com/jotech/gapseq | Command line program, R package | automatic | MNXref, KEGG, BiGG, MetaCyc, ModelSEED | Available | [73] |
| AutoKEGGRec https://www.ntnu.edu/almaaslab and https://github.com/emikar/AutoKEGGRec | Matlab package | automatic | KEGG | Available but needed Matlab. Last update more than 5 years ago | [74] |
| RAVEN v2 https://github.com/SysBioChalmers/RAVEN | Matlab package | semi-automatic | KEGG, MetaCyc Template models | Available, but needed Matlab | [75] |
| MicrobesFlux http://tanglab.engineering.wustl.edu/static/MicrobesFlux.html | Web-service | automatic | KEGG | Not available | [69] |
| ScrumPy https://mudshark.brookes.ac.uk/ScrumPy | Python package | semi-automatic | BioCyc | Available | [76] |
| CarveMe github.com/cdanielmachado/carveme | Command line program, Python package | automatic | BiGG | Available, but needed commercial solvers (IBM CPLEX or Gurobi) | [77] |
| PADMet (AuReMe) https://pypi.python.org/pypi/padmet and https://gitlab.inria.fr/maite/padmet | Python package | MetaCyc, BiGG | Available | [71] | |
| MetaDraft https://systemsbioinformatics.github.io/cbmpy-metadraft/ | GUI based program | semi-automatic | Template models | Available | [78] |
| moped https://gitlab.com/marvin.vanaalst/moped-publication-2021 | Python package | semi-automatic | MetaCyc, BioCyc | Available | [79] |
| Reconstructor http://github.com/emmamglass/reconstructor | Command line program, Python package | automatic | KEGG ModelSEED | Available | [80] |
| Bactabolize github.com/kelwyres/Bactabolize | Command line program, Python package | automatic | BiGG | Available | [81] |
| AuCoMe https://github.com/AuReMe/aucome | Command line program, Python package | automatic | MetaCyc | Free access, but needed Pathway tools | [82] |
2.4.2. GUI-Based Desktop Programs
2.4.3. Packages and Command Line Programs
2.5. Web-Resources and Tools for Analysis of GSM Models
2.6. Tools for the Integration of Omics-Data into GSM Models
- The GIMME-like group, where most of the methods of this group conduct reconstruction of metabolic models in two steps: the first step is the maximization of a required metabolic functionality (RMF) based on the FBA (or similar) algorithm. The second step is to minimize the penalty function describing the discrepancy between the obtained reaction fluxes and the experimental data while maintaining the flux through the RMF above the given flux fraction. As a rule, the pseudo-reaction of the biomass equation is chosen as the RMF [14]. GIMME-like algorithms include GIMME [13], GIMMEp [118], GIM3E [110] and RIPTiDe [119];
- The iMAT-like family of methods, in contrast to the group above, does not require a precise definition of RMF. This group of algorithms is based on the classification of reactions in the reference model as active or inactive in accordance with the corresponding states in the experimental data, on the basis of which the GSM model is reconstructed. As a consequence, this approach requires that the experimental data be categorized into two or more groups describing different states of the data (e.g., low-expressed and high-expressed in the context of transcriptomics data) [14]. The algorithms of the iMAT-like group include: iMAT [106], INIT [108], ftINIT [112], Lee [120] and RegrEx [121];
- The MADE-like methods rely on differential expression data in the process of GSM models reconstruction. The last ones describe differences in metabolic fluxes between two contexts/conditions. Similar to the GIMME-like group, the preservation of the minimum flux value required for RMF is also taken into account in these algorithms [14]. Algorithms of the MADE-like group include MADE [107], RMetD2 [122] and deltaFBA [117];
- The MBA-like algorithms are based on the identification of key reactions and the subsequent removal of reactions that are not part of the core set. Similar to the iMAT-like group, MBA-like algorithms do not have the choice of selecting an RMF, nor do they have the choice of maintaining the flux through it [14]. The MBA-like algorithms include MBA [123] and mCADRE [114], as well as pymCADRE [124], the FASTCORE algorithm group [116] and CORDA [115].
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, W.J.; Kim, H.U.; Lee, S.Y. Current state and applications of microbial genome-scale metabolic models. Curr. Opin. Syst. Biol. 2017, 2, 10–18. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef]
- Walakira, A.; Rozman, D.; Režen, T.; Mraz, M.; Moškon, M. Guided extraction of genome-scale metabolic models for the integration and analysis of omics data. Comput. Struct. Biotechnol. J. 2021, 19, 3521–3530. [Google Scholar] [CrossRef] [PubMed]
- Richelle, A.; Kellman, B.P.; Wenzel, A.T.; Chiang, A.W.T.; Reagan, T.; Gutierrez, J.M.; Joshi, C.; Li, S.; Liu, J.K.; Masson, H.; et al. Model-based assessment of mammalian cell metabolic functionalities using omics data. Cell Rep. Methods 2021, 1, 100040. [Google Scholar] [CrossRef] [PubMed]
- Nogales, J.; Garmendia, J. Bacterial metabolism and pathogenesis intimate intertwining: Time for metabolic modelling to come into action. Microb. Biotechnol. 2022, 15, 95–102. [Google Scholar] [CrossRef]
- Ye, C.; Wei, X.; Shi, T.; Sun, X.; Xu, N.; Gao, C.; Zou, W. Genome-scale metabolic network models: From first-generation to next-generation. Appl. Microbiol. Biotechnol. 2022, 106, 4907–4920. [Google Scholar] [CrossRef]
- Chubukov, V.; Mukhopadhyay, A.; Petzold, C.J.; Keasling, J.D.; Martín, H.G. Synthetic and systems biology for microbial production of commodity chemicals. npj Syst. Biol. Appl. 2018, 2, 16009. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.L.; Kim, T.Y.; Kim, H.U.; Palsson, B.; Lee, S.Y. Systems biology as a foundation for genome-scale synthetic biology. Curr. Opin. Biotechnol. 2006, 17, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Esvelt, K.M.; Wang, H.H. Genome-scale engineering for systems and synthetic biology. Mol. Syst. Biol. 2013, 9, 641. [Google Scholar] [CrossRef] [PubMed]
- Akberdin, I.R.; Thompson, M.; Kalyuzhnaya, M.G. Systems biology and metabolic modeling of C1-metabolism. In Methane Biocatalysis: Paving the Way to Sustainability; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Kabimoldayev, I.; Nguyen, A.D.; Yang, L.; Park, S.; Lee, E.Y.; Kim, D. Basics of genome-scale metabolic modeling and applications on C1-utilization. FEMS Microbiol. Lett. 2018, 365, fny241. [Google Scholar] [CrossRef]
- Hwangbo, M.; Shao, Y.; Hatzinger, P.B.; Chu, K.H. Acidophilic methanotrophs: Occurrence, diversity, and possible bioremediation applications. Environ. Microbiol. Rep. 2023, 15, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.A.; Palsson, B.O. Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef]
- Moškon, M.; Režen, T. Context-Specific Genome-Scale Metabolic Modelling and Its Application to the Analysis of COVID-19 Metabolic Signatures. Metabolites 2023, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Feist, A.M.; Herrgård, M.J.; Thiele, I.; Reed, J.L.; Palsson, B. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 2009, 7, 129–143. [Google Scholar] [CrossRef]
- Haggart, C.R.; Bartell, J.A.; Saucerman, J.J.; Papin, J.A. Whole-genome metabolic network reconstruction and constraint-based modeling. Methods Enzymol. 2011, 500, 411–433. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.J.; Sapra, R.; Joyner, D.; Hazen, T.C.; Myers, S.; Reichmuth, D.; Blanch, H.; Keasling, J.D. Analysis of metabolic pathways and fluxes in a newly discovered thermophilic and ethanol-tolerant Geobacillus strain. Biotechnol. Bioeng. 2009, 102, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States department of energy systems biology knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Edwards, J.S.; Covert, M.; Palsson, B. Metabolic modelling of microbes: The flux-balance approach. Environ. Microbiol. 2002, 4, 133–140. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Bordbar, A.; Monk, J.M.; King, Z.A.; Palsson, B.O. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 2014, 15, 107–120. [Google Scholar] [CrossRef]
- Cordova, L.T.; Long, C.P.; Venkataramanan, K.P.; Antoniewicz, M.R. Complete genome sequence, metabolic model construction and phenotypic characterization of Geobacillus LC300, an extremely thermophilic, fast growing, xylose-utilizing bacterium. Metab. Eng. 2015, 32, 74–81. [Google Scholar] [CrossRef]
- Akberdin, I.R.; Thompson, M.; Hamilton, R.; Desai, N.; Alexander, D.; Henard, C.A.; Guarnieri, M.T.; Kalyuzhnaya, M.G. Methane utilization in Methylomicrobium alcaliphilum 20ZR: A systems approach. Sci. Rep. 2018, 8, 2512. [Google Scholar] [CrossRef]
- Henard, C.A.; Akberdin, I.R.; Kalyuzhnaya, M.G.; Guarnieri, M.T. Muconic acid production from methane using rationally-engineered methanotrophic biocatalysts. Green Chem. 2019, 21, 6731–6737. [Google Scholar] [CrossRef]
- Gupta, A.; Ahmad, A.; Chothwe, D.; Madhu, M.K.; Srivastava, S.; Sharma, V.K. Genome-scale metabolic reconstruction and metabolic versatility of an obligate methanotroph Methylococcus capsulatus str. Bath. PeerJ 2019, 7, e6685. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2018, 20, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.D.; Allen, J.; Amode, R.M.; Azov, A.G.; Barba, M.; Becerra, A.; Bhai, J.; Campbell, L.I.; Carbajo Martinez, M.; Chakiachvili, M.; et al. Ensembl Genomes 2022: An expanding genome resource for non-vertebrates. Nucleic Acids Res. 2022, 50, D996–D1003. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef] [PubMed]
- Vallenet, D.; Calteau, A.; Dubois, M.; Amours, P.; Bazin, A.; Beuvin, M.; Burlot, L.; Bussell, X.; Fouteau, S.; Gautreau, G.; et al. MicroScope: An integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis. Nucleic Acids Res. 2020, 48, D579–D589. [Google Scholar] [CrossRef]
- Chen, I.M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.J.; Webb, C.; Wu, D.; et al. The IMG/M data management and analysis system v.7: Content updates and new features. Nucleic Acids Res. 2023, 51, D723–D732. [Google Scholar] [CrossRef]
- Karp, P.D.; Midford, P.E.; Billington, R.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Ong, W.K.; Subhraveti, P.; Caspi, R.; Fulcher, C.; et al. Pathway Tools version 23.0 update: Software for pathway/genome informatics and systems biology. Brief Bioinform. 2021, 22, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Keseler, I.M.; Gama-Castro, S.; Mackie, A.; Billington, R.; Bonavides-Martínez, C.; Caspi, R.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Muñiz-Rascado, L.; et al. The EcoCyc Database in 2021. Front. Microbiol. 2021, 12, 711077. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; Cukura, A.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef] [PubMed]
- Wittig, U.; Rey, M.; Weidemann, A.; Kania, R.; Müller, W. SABIO-RK: An updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Res. 2018, 46, D656–D660. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Sherry, S.T.; Yankie, L.; Karsch-Mizrachi, I. GenBank 2023 update. Nucleic Acids Res. 2023, 51, D141–D144. [Google Scholar] [CrossRef] [PubMed]
- Kersey, P.J.; Allen, J.E.; Allot, A.; Barba, M.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Grabmueller, C.; et al. Ensembl Genomes 2018: An integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 2018, 46, D802–D808. [Google Scholar] [CrossRef]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef]
- Wattam, A.R.; Brettin, T.; Davis, J.J.; Gerdes, S.; Kenyon, R.; Machi, D.; Mao, C.; Olson, R.; Overbeek, R.; Pusch, G.D.; et al. Assembly, annotation, and comparative genomics in PATRIC, the all bacterial bioinformatics resource center. Methods Mol. Biol. 2018, 1704, 79–101. [Google Scholar]
- Chen, I.M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.; Varghese, N.; Seshadri, R.; et al. The IMG/M data management and analysis system v.6.0: New tools and advanced capabilities. Nucleic Acids Res. 2021, 49, D751–D763. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.D.; Lee, E.Y. Engineered Methanotrophy: A Sustainable Solution for Methane-Based Industrial Biomanufacturing. Trends Biotechnol. 2021, 39, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Nguyen, D.T.N.; Chau, T.H.T.; Fei, Q.; Lee, E.Y. Systems Metabolic Engineering of Methanotrophic Bacteria for Biological Conversion of Methane to Value-Added Compounds. Adv. Biochem. Eng./Biotechnol. 2022, 180, 91–126. [Google Scholar]
- Van Dien, S.J.; Lidstrom, M.E. Stoichiometric model for evaluating the metabolic capabilities of the facultative methylotroph Methylobacterium extorquens AM1, with application to reconstruction of C3 and C4 metabolism. Biotechnol. Bioeng. 2002, 78, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, R.; Schneider, K.; Kiefer, P.; Massou, S.; Vorholt, J.A.; Portais, J.C. Genome-scale reconstruction and system level investigation of the metabolic network of Methylobacterium extorquens AM1. BMC Syst. Biol. 2011, 5, 189. [Google Scholar] [CrossRef] [PubMed]
- Torre, A.; Metivier, A.; Chu, F.; Laurens, L.M.L.; Beck, D.A.C.; Pienkos, P.T.; Lidstrom, M.E.; Kalyuzhnaya, M.G. Genome-scale metabolic reconstructions and theoretical investigation of methane conversion in Methylomicrobium buryatense strain 5G(B1). Microb. Cell Fact. 2015, 14, 188. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, A.; Akberdin, I.R.; Allemann, M.; Allen, E.E.; Kalyuzhnaya, M.G. Fatty acid biosynthesis pathways in Methylomicrobium buryatense 5G(B1). Front. Microbiol. 2017, 7, 2167. [Google Scholar] [CrossRef]
- Akberdin, I.R.; Collins, D.A.; Hamilton, R.; Oshchepkov, D.Y.; Shukla, A.K.; Nicora, C.D.; Nakayasu, E.S.; Adkins, J.N.; Kalyuzhnaya, M.G. Rare Earth Elements alter redox balance in Methylomicrobium alcaliphilum 20ZR. Front. Microbiol. 2018, 9, 2735. [Google Scholar] [CrossRef]
- Lieven, C.; Petersen, L.A.H.; Jørgensen, S.B.; Gernaey, K.V.; Herrgard, M.J.; Sonnenschein, N. A Genome-Scale Metabolic Model for Methylococcus capsulatus (Bath) Suggests Reduced Efficiency Electron Transfer to the Particulate Methane Monooxygenase. Front. Microbiol. 2018, 9, 2947. [Google Scholar] [CrossRef]
- Bordel, S.; Rodríguez, Y.; Hakobyan, A.; Rodríguez, E.; Lebrero, R.; Muñoz, R. Genome scale metabolic modeling reveals the metabolic potential of three Type II methanotrophs of the genus Methylocystis. Metab. Eng. 2019, 54, 191–199. [Google Scholar] [CrossRef]
- Bordel, S.; Rojas, A.; Muñoz, R. Reconstruction of a Genome Scale Metabolic Model of the polyhydroxybutyrate producing methanotroph Methylocystis parvus OBBP. Microb. Cell Fact. 2019, 18, 104. [Google Scholar] [CrossRef] [PubMed]
- Naizabekov, S.; Lee, E.Y. Genome-scale metabolic model reconstruction and in silico investigations of methane metabolism in Methylosinus trichosporium ob3b. Microorganisms 2020, 8, 437. [Google Scholar] [CrossRef] [PubMed]
- Bordel, S.; Crombie, A.T.; Muñoz, R.; Murrell, J.C. Genome Scale Metabolic Model of the versatile methanotroph Methylocella silvestris. Microb. Cell Fact. 2020, 19, 144. [Google Scholar] [CrossRef] [PubMed]
- Desouki, A.A.; Jarre, F.; Gelius-Dietrich, G.; Lercher, M.J. CycleFreeFlux: Efficient removal of thermodynamically infeasible loops from flux distributions. Bioinformatics 2015, 31, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Villada, J.C.; Duran, M.F.; Lim, C.K.; Stein, L.Y.; Lee, P.K.H. Integrative Genome-Scale Metabolic Modeling Reveals Versatile Metabolic Strategies for Methane Utilization in Methylomicrobium album BG8. mSystems 2022, 7, e0007322. [Google Scholar] [CrossRef]
- Kalyuzhnaya, M.G.; Yang, S.; Rozova, O.N.; Smalley, N.E.; Clubb, J.; Lamb, A.; Gowda, G.A.N.; Raftery, D.; Fu, Y.; Bringel, F.; et al. Highly efficient methane biocatalysis revealed in a methanotrophic bacterium. Nat. Commun. 2013, 4, 2785. [Google Scholar] [CrossRef]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020, 38, 504. [Google Scholar] [CrossRef]
- He, B.; Cai, C.; McCubbin, T.; Muriel, J.C.; Sonnenschein, N.; Hu, S.; Yuan, Z.; Marcellin, E. A Genome-Scale Metabolic Model of Methanoperedens nitroreducens: Assessing Bioenergetics and Thermodynamic Feasibility. Metabolites 2022, 12, 314. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, S.N.; Olivier, B.G.; Molenaar, D.; Teusink, B. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 2019, 20, 158. [Google Scholar] [CrossRef]
- King, Z.A.; Dräger, A.; Ebrahim, A.; Sonnenschein, N.; Lewis, N.E.; Palsson, B.O. Escher: A Web Application for Building, Sharing, and Embedding Data-Rich Visualizations of Biological Pathways. PLoS Comput. Biol. 2015, 11, e1004321. [Google Scholar] [CrossRef] [PubMed]
- Devoid, S.; Overbeek, R.; DeJongh, M.; Vonstein, V.; Best, A.A.; Henry, C. Automated genome annotation and metabolic model reconstruction in the SEED and model SEED. Methods Mol. Biol. 2013, 985, 17–45. [Google Scholar] [CrossRef] [PubMed]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef] [PubMed]
- Faria, J.P.; Liu, F.; Edirisinghe, J.N.; Gupta, N.; Seaver, S.M.D.; Freiburger, A.P.; Zhang, Q.; Weisenhorn, P.; Conrad, N.; Zarecki, R.; et al. ModelSEED v2: High-throughput genome-scale metabolic model reconstruction with enhanced energy biosynthesis pathway prediction. bioRxiv 2023. [Google Scholar] [CrossRef]
- Boele, J.; Olivier, B.G.; Teusink, B. FAME, the Flux Analysis and Modeling Environment. BMC Syst. Biol. 2012, 6, 8. [Google Scholar] [CrossRef]
- Feng, X.; Xu, Y.; Chen, Y.; Tang, Y.J. MicrobesFlux: A web platform for drafting metabolic models from the KEGG database. BMC Syst. Biol. 2012, 6, 94. [Google Scholar] [CrossRef]
- Liao, Y.C.; Tsai, M.H.; Chen, F.C.; Hsiung, C.A. GEMSiRV: A software platform for GEnome-scale metabolic model simulation, reconstruction and visualization. Bioinformatics 2012, 28, 1752–1758. [Google Scholar] [CrossRef]
- Aite, M.; Chevallier, M.; Frioux, C.; Trottier, C.; Got, J.; Cortés, M.P.; Mendoza, S.N.; Carrier, G.; Dameron, O.; Guillaudeux, N.; et al. Traceability, reproducibility and wiki-exploration for “à-la-carte” reconstructions of genome-scale metabolic models. PLoS Comput. Biol. 2018, 14, e1006146. [Google Scholar] [CrossRef]
- Capela, J.; Lagoa, D.; Rodrigues, R.; Cunha, E.; Cruz, F.; Barbosa, A.; Bastos, J.; Lima, D.; Ferreira, E.C.; Rocha, M.; et al. merlin, an improved framework for the reconstruction of high-quality genome-scale metabolic models. Nucleic Acids Res. 2022, 50, 6052–6066. [Google Scholar] [CrossRef]
- Zimmermann, J.; Kaleta, C.; Waschina, S. gapseq: Informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. Genome Biol. 2021, 22, 81. [Google Scholar] [CrossRef]
- Karlsen, E.; Schulz, C.; Almaas, E. Automated generation of genome-scale metabolic draft reconstructions based on KEGG. BMC Bioinform. 2018, 19, 467. [Google Scholar] [CrossRef]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef] [PubMed]
- Poolman, M.G. ScrumPy: Metabolic modelling with Python. IEE Proc. Syst. Biol. 2006, 153, 375–378. [Google Scholar] [CrossRef]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef]
- Olivier, B.G.; Mendoza, S.; Molenaar, D.; Teusink, B. MetaDraft Release: 0.9.5 2020. Available online: https://zenodo.org/records/4291058 (accessed on 12 December 2023).
- Saadat, N.P.; van Aalst, M.; Ebenhöh, O. Network Reconstruction and Modelling Made Reproducible with moped. Metabolites 2022, 12, 275. [Google Scholar] [CrossRef]
- Jenior, M.L.; Glass, E.M.; Papin, J.A. Reconstructor: A COBRApy compatible tool for automated genome-scale metabolic network reconstruction with parsimonious flux-based gap-filling. Bioinformatics 2023, 39, btad367. [Google Scholar] [CrossRef]
- Vezina, B.; Watts, S.C.; Hawkey, J.; Cooper, H.B.; Judd, L.M.; Jenney, A.; Monk, J.M.; Holt, K.E.; Wyres, K.L. Bactabolize: A tool for high-throughput generation of bacterial strain-specific metabolic models. bioRxiv 2023. [Google Scholar] [CrossRef]
- Belcour, A.; Got, J.; Aite, M.; Delage, L.; Collén, J.; Frioux, C.; Leblanc, C.; Dittami, S.M.; Blanquart, S.; Markov, G.V.; et al. Inferring and comparing metabolism across heterogeneous sets of annotated genomes using AuCoMe. Genome Res. 2023, 33, 972–987. [Google Scholar] [CrossRef]
- Norsigian, C.J.; Pusarla, N.; McConn, J.L.; Yurkovich, J.T.; Dräger, A.; Palsson, B.O.; King, Z. BiGG Models 2020: Multi-strain genome-scale models and expansion across the phylogenetic tree. Nucleic Acids Res. 2020, 48, D402–D406. [Google Scholar] [CrossRef] [PubMed]
- Hucka, M.; Bergmann, F.T.; Dräger, A.; Hoops, S.; Keating, S.M.; Le Novère, N.; Myers, C.J.; Olivier, B.G.; Sahle, S.; Schaff, J.C.; et al. The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 2 Core. J. Integr. Bioinform. 2018, 15, 20170081. [Google Scholar] [CrossRef]
- Prigent, S.; Frioux, C.; Dittami, S.M.; Thiele, S.; Larhlimi, A.; Collet, G.; Gutknecht, F.; Got, J.; Eveillard, D.; Bourdon, J.; et al. Meneco, a Topology-Based Gap-Filling Tool Applicable to Degraded Genome-Wide Metabolic Networks. PLoS Comput. Biol. 2017, 13, e1005276. [Google Scholar] [CrossRef] [PubMed]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef]
- Rocha, I.; Maia, P.; Evangelista, P.; Vilaça, P.; Soares, S.; Pinto, J.P.; Nielsen, J.; Patil, K.R.; Ferreira, E.C.; Rocha, M. OptFlux: An open-source software platform for in silico metabolic engineering. BMC Syst. Biol. 2010, 4, 45. [Google Scholar] [CrossRef]
- Kelley, J.J.; Lane, A.; Li, X.; Mutthoju, B.; Maor, S.; Egen, D.; Lun, D.S. MOST: A software environment for constraint-based metabolic modeling and strain design. Bioinformatics 2015, 31, 610–611. [Google Scholar] [CrossRef][Green Version]
- Kelley, J.J.; Maor, S.; Kim, M.K.; Lane, A.; Lun, D.S. MOST-Visualization: Software for producing automated textbook-style maps of genome-scale metabolic networks. Bioinformatics 2017, 33, 2596–2597. [Google Scholar] [CrossRef][Green Version]
- Hari, A.; Lobo, D. Fluxer: A web application to compute, analyze and visualize genome-scale metabolic flux networks. Nucleic Acids Res. 2020, 48, W427–W435. [Google Scholar] [CrossRef]
- Mao, Z.; Yuan, Q.; Li, H.; Hang, Y.; Huang, Y.; Yang, C.; Wang, R.; Yang, Y.; Wu, Y.; Yang, S.; et al. CAVE: A cloud-based platform for analysis and visualization of metabolic pathways. Nucleic Acids Res. 2023, 51, W70–W77. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef]
- Cardoso, J.G.R.; Jensen, K.; Lieven, C.; Hansen, A.S.L.; Galkina, S.; Beber, M.; Özdemir, E.; Herrgård, M.J.; Redestig, H.; Sonnenschein, N. Cameo: A Python Library for Computer Aided Metabolic Engineering and Optimization of Cell Factories. ACS Synth. Biol. 2018, 7, 1163–1166. [Google Scholar] [CrossRef]
- Machado, D.; Ochsner, N.; Colpo, R.A. cdanielmachado/reframed: 1.4.0 2023. Available online: https://zenodo.org/records/7778365 (accessed on 12 December 2023).
- Pereira, V.; Cruz, F.; Rocha, M. MEWpy: A computational strain optimization workbench in Python. Bioinformatics 2021, 37, 2494–2496. [Google Scholar] [CrossRef]
- Olivier, B.; Gottstein, W.; Molenaar, D.; Teusink, B. CBMPy release 0.8.8 2023. Available online: https://zenodo.org/records/10067017 (accessed on 12 December 2023).
- Klamt, S.; Saez-Rodriguez, J.; Gilles, E.D. Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst. Biol. 2007, 1, 2. [Google Scholar] [CrossRef]
- von Kamp, A.; Thiele, S.; Hädicke, O.; Klamt, S. Use of CellNetAnalyzer in biotechnology and metabolic engineering. J. Biotechnol. 2017, 261, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Thiele, S.; Von Kamp, A.; Bekiaris, P.S.; Schneider, P.; Klamt, S. CNApy: A CellNetAnalyzer GUI in Python for analyzing and designing metabolic networks. Bioinformatics 2022, 38, 1467–1469. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Bekiaris, P.S.; von Kamp, A.; Klamt, S. StrainDesign: A comprehensive Python package for computational design of metabolic networks. Bioinformatics 2022, 38, 4981–4983. [Google Scholar] [CrossRef] [PubMed]
- Vilaça, P.; Maia, P.; Giesteira, H.; Rocha, I.; Rocha, M. Analyzing and designing cell factories with OptFlux. Methods Mol. Biol. 2018, 1716, 37–76. [Google Scholar] [PubMed]
- Sánchez, B.J.; Zhang, C.; Nilsson, A.; Lahtvee, P.; Kerkhoven, E.J.; Nielsen, J. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol. Syst. Biol. 2017, 13, 935. [Google Scholar] [CrossRef]
- Shen, F.; Sun, R.; Yao, J.; Li, J.; Liu, Q.; Price, N.D.; Liu, C.; Wang, Z. Optram: In-silico strain design via integrative regulatory-metabolic network modeling. PLoS Comput. Biol. 2019, 15, e1006835. [Google Scholar] [CrossRef]
- Kim, M.K.; Lane, A.; Kelley, J.J.; Lun, D.S. E-Flux2 and sPOT: Validated methods for inferring intracellular metabolic flux distributions from transcriptomic data. PLoS ONE 2016, 11, e0157101. [Google Scholar] [CrossRef]
- Åkesson, M.; Förster, J.; Nielsen, J. Integration of gene expression data into genome-scale metabolic models. Metab. Eng. 2004, 6, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Zur, H.; Ruppin, E.; Shlomi, T. iMAT: An integrative metabolic analysis tool. Bioinformatics 2010, 26, 3140–3142. [Google Scholar] [CrossRef]
- Jensen, P.A.; Papin, J.A. Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 2011, 27, 541–547. [Google Scholar] [CrossRef]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef]
- Machado, D.; Herrgård, M. Systematic Evaluation of Methods for Integration of Transcriptomic Data into Constraint-Based Models of Metabolism. PLoS Comput. Biol. 2014, 10, e1003580. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.J.; Ebrahim, A.; Metz, T.O.; Adkins, J.N.; Palsson, B.; Hyduke, D.R. GIM3E: Condition-specific models of cellular metabolism developed from metabolomics and expression data. Bioinformatics 2013, 29, 2900–2908. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 2014, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, J.; Anton, M.; Roshanzamir, F.; Jörnsten, R.; Kerkhoven, E.J.; Robinson, J.L.; Nielsen, J. Generation and analysis of context-specific genome-scale metabolic models derived from single-cell RNA-Seq data. Proc. Natl. Acad. Sci. USA 2023, 120, e2217868120. [Google Scholar] [CrossRef]
- Jensen, P.A.; Lutz, K.A.; Papin, J.A. TIGER: Toolbox for integrating genome-scale metabolic models, expression data, and transcriptional regulatory networks. BMC Syst. Biol. 2011, 5, 147. [Google Scholar] [CrossRef]
- Wang, Y.; Eddy, J.A.; Price, N.D. Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst. Biol. 2012, 6, 153. [Google Scholar] [CrossRef]
- Schultz, A.; Qutub, A.A. Reconstruction of Tissue-Specific Metabolic Networks Using CORDA. PLoS Comput. Biol. 2016, 12, e1004808. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.P.; Sauter, T. The FASTCORE family: For the fast reconstruction of compact context-specific metabolic networks models. Methods Mol. Biol. 2018, 1716, 101–110. [Google Scholar] [PubMed]
- Ravi, S.; Gunawan, R. ΔFBA-Predicting metabolic flux alterations using genome-scale metabolic models and differential transcriptomic data. PLoS Comput. Biol. 2021, 17, e1009589. [Google Scholar] [CrossRef] [PubMed]
- Bordbar, A.; Mo, M.L.; Nakayasu, E.S.; Schrimpe-Rutledge, A.C.; Kim, Y.M.; Metz, T.O.; Jones, M.B.; Frank, B.C.; Smith, R.D.; Peterson, S.N.; et al. Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol. Syst. Biol. 2012, 8, 558. [Google Scholar] [CrossRef] [PubMed]
- Jenior, M.L.; Moutinho, T.J.; Dougherty, B.V.; Papin, J.A. Transcriptome-guided parsimonious flux analysis improves predictions with metabolic networks in complex environments. PLoS Comput. Biol. 2020, 16, e1007099. [Google Scholar] [CrossRef]
- Lee, D.; Smallbone, K.; Dunn, W.B.; Murabito, E.; Winder, C.L.; Kell, D.B.; Mendes, P.; Swainston, N. Improving metabolic flux predictions using absolute gene expression data. BMC Syst. Biol. 2012, 6, 73. [Google Scholar] [CrossRef]
- Estévez, S.R.; Nikoloski, Z. Context-specific metabolic model extraction based on regularized least squares optimization. PLoS ONE 2015, 10, e0131875. [Google Scholar] [CrossRef]
- Zhang, C.; Lee, S.; Bidkhori, G.; Benfeitas, R.; Lovric, A.; Chen, S.; Uhlen, M.; Nielsen, J.; Mardinoglu, A. RMetD2: A tool for integration of relative transcriptomics data into Genome-scale metabolic models. bioRxiv 2019. [Google Scholar] [CrossRef]
- Jerby, L.; Shlomi, T.; Ruppin, E. Computational reconstruction of tissue-specific metabolic models: Application to human liver metabolism. Mol. Syst. Biol. 2010, 6, 401. [Google Scholar] [CrossRef]
- Leonidou, N.; Renz, A.; Mostolizadeh, R.; Dräger, A. New workflow predicts drug targets against SARS-CoV-2 via metabolic changes in infected cells. PLoS Comput. Biol. 2023, 19, e1010903. [Google Scholar] [CrossRef] [PubMed]
- Grausa, K.; Mozga, I.; Pleiko, K.; Pentjuss, A. Integrative Gene Expression and Metabolic Analysis Tool IgemRNA. Biomolecules 2022, 12, 586. [Google Scholar] [CrossRef] [PubMed]
- Motamedian, E.; Mohammadi, M.; Shojaosadati, S.A.; Heydari, M. TRFBA: An algorithm to integrate genome-scale metabolic and transcriptional regulatory networks with incorporation of expression data. Bioinformatics 2017, 33, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S. A protocol for the construction and curation of genome-scale integrated metabolic and regulatory network models. Methods Mol. Biol. 2019, 1927, 203–214. [Google Scholar] [PubMed]
- Yao, H.; Dahal, S.; Yang, L. Novel context-specific genome-scale modelling explores the potential of triacylglycerol production by Chlamydomonas reinhardtii. Microb. Cell Fact. 2023, 22, 13. [Google Scholar] [CrossRef]
- Muriel, J.C.; Long, C.P.; Sonnenschein, N. Geckopy 3.0: Enzyme constraints, thermodynamics constraints and omics integration in python. bioRxiv 2023. [Google Scholar] [CrossRef]
- Jalili, M.; Scharm, M.; Wolkenhauer, O.; Salehzadeh-Yazdi, A. Metabolic function-based normalization improves transcriptome data-driven reduction of genome-scale metabolic models. npj Syst. Biol. Appl. 2023, 9, 15. [Google Scholar] [CrossRef]
- Powers, D.A.; Jenior, M.L.; Kolling, G.L.; Papin, J.A. Network analysis of toxin production in Clostridioides difficile identifies key metabolic dependencies. PLoS Comput. Biol. 2023, 19, e1011076. [Google Scholar] [CrossRef]
- Leonidou, N. pymCADRE: Tissue-Specific Model Reconstruction. 2021. Available online: https://zenodo.org/records/5566815 (accessed on 12 December 2023).
- Ferreira, J.; Vieira, V.; Gomes, J.; Correia, S.; Rocha, M. Troppo-A Python Framework for the Reconstruction of Context-Specific Metabolic Models. In Practical Applications of Computational Biology and Bioinformatics, 13th International Conference. PACBB 2019; Springer: Cham, Switzerland, 2020; Volume 1005. [Google Scholar]
- Vieira, V.; Ferreira, J.; Rocha, M. A pipeline for the reconstruction and evaluation of context-specific human metabolic models at a large-scale. PLoS Comput. Biol. 2022, 18, e1009294. [Google Scholar] [CrossRef] [PubMed]
- Troitiño-Jordedo, D.; Carvalho, L.; Henriques, D.; Pereira, V.; Rocha, M.; Balsa-Canto, E. A New GIMME–Based Heuristic for Compartmentalised Transcriptomics Data Integration. In Practical Applications of Computational Biology and Bioinformatics, 17th International Conference (PACBB 2023); Springer: Cham, Switzerland, 2023; Volume 743. [Google Scholar]
- Sugden, S.; Lazic, M.; Sauvageau, D.; Stein, L.Y. Transcriptomic and Metabolomic Responses to Carbon and Nitrogen Sources in Methylomicrobium album BG8. Appl. Environ. Microbiol. 2021, 87, e0038521. [Google Scholar] [CrossRef]
- Pei, S.; Liu, P.; Parker, D.A.; Mackie, R.I.; Rao, C.V. Systems analysis of the effect of hydrogen sulfide on the growth of Methylococcus capsulatus Bath. Appl. Microbiol. Biotechnol. 2022, 106, 7879–7890. [Google Scholar] [CrossRef]
- Schmitz, R.A.; Peeters, S.H.; Mohammadi, S.S.; Berben, T.; van Erven, T.; Iosif, C.A.; van Alen, T.; Versantvoort, W.; Jetten, M.S.M.; Op den Camp, H.J.M.; et al. Simultaneous sulfide and methane oxidation by an extremophile. Nat. Commun. 2023, 14, 2974. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Groom, J.D.; Wilson, E.H.; Fernandez, J.; Konopka, M.C.; Beck, D.A.C.; Lidstrom, M.E. A methanotrophic bacterium to enable methane removal for climate mitigation. Proc. Natl. Acad. Sci. USA 2023, 120, e2310046120. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Yang, Y.; Yan, X.; Zhang, T.; Xiang, J.; Gao, Z.; Chen, Y.; Yang, S.; Fei, Q. Molecular Mechanism Associated With the Impact of Methane/Oxygen Gas Supply Ratios on Cell Growth of Methylomicrobium buryatense 5GB1 Through RNA-Seq. Front. Bioeng. Biotechnol. 2020, 8, 263. [Google Scholar] [CrossRef]
- Gilman, A.; Fu, Y.; Hendershott, M.; Chu, F.; Puri, A.W.; Smith, A.L.; Pesesky, M.; Lieberman, R.; Beck, D.A.C.; Lidstrom, M.E. Oxygen-limited metabolism in the methanotroph Methylomicrobium buryatense 5GB1C. PeerJ 2017, 5, e3945. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; He, L.; Reeve, J.; Beck, D.A.C.; Lidstrom, M.E. Core metabolism shifts during growth on methanol versus methane in the methanotroph Methylomicrobium buryatense 5GB1. mBio 2019, 10, e00406-19. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.D.; Ford, S.M.; Pesesky, M.W.; Lidstrom, M.E. A mutagenic screen identifies a TonB-dependent receptor required for the lanthanide metal switch in the type I methanotroph “Methylotuvimicrobium buryatense” 5GB1C. J. Bacteriol. 2021, 201, e00120-19. [Google Scholar] [CrossRef]
- Nguyen, A.D.; Kim, D.; Lee, E.Y. A comparative transcriptome analysis of the novel obligate methanotroph Methylomonas sp. DH-1 reveals key differences in transcriptional responses in C1 and secondary metabolite pathways during growth on methane and methanol. BMC Genom. 2019, 20, 130. [Google Scholar] [CrossRef]
- Puri, A.W.; Liu, D.; Schaefer, A.L.; Yu, Z.; Pesesky, M.W.; Peter Greenberg, E.; Lidstrom, M.E. Interspecies chemical signaling in a methane-oxidizing bacterial community. Appl. Environ. Microbiol. 2019, 85, e02702-18. [Google Scholar] [CrossRef]
- Picone, N.; Mohammadi, S.S.; Waajen, A.C.; van Alen, T.A.; Jetten, M.S.M.; Pol, A.; Op den Camp, H.J.M. More Than a Methanotroph: A Broader Substrate Spectrum for Methylacidiphilum fumariolicum SolV. Front. Microbiol. 2020, 11, 604485. [Google Scholar] [CrossRef]
- Yu, Z.; Pesesky, M.; Zhang, L.; Huang, J.; Winkler, M.; Chistoserdova, L. A Complex Interplay between Nitric Oxide, Quorum Sensing, and the Unique Secondary Metabolite Tundrenone Constitutes the Hypoxia Response in Methylobacter. mSystems 2020, 5, e00770-19. [Google Scholar] [CrossRef]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S.; et al. Jupyter Notebooks: A publishing format for reproducible computational workflows. In Proceedings of the Positioning and Power in Academic Publishing: Players, Agents and Agendas, 20th International Conference on Electronic Publishing, Göttingen, Germany, 7–9 June 2016; IOS Press: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Perkel, J.M. Why Jupyter is data scientists’ computational notebook of choice. Nature 2018, 563, 145–146. [Google Scholar] [CrossRef] [PubMed]

| Organism | ID * | Genes | Reactions | Metabolites | Tools & Databases ** | References | Memote Score *** | Cobrapy Model Consistency # |
|---|---|---|---|---|---|---|---|---|
| Methylobacterium extorquens AM1 | – | 67 | 65 | [48] | ||||
| iRP911 | 911 | 1139 | 977 | CellNet Analyser MicroScope MetaCyc KEGG | [49] | - | - | |
| Methylotuvimicrobium buryatense 5G | iMb5G (B1) | – | 841 | – | Pathway-Tools MicroScope | [50] | ||
| iMb5GB1 update | 314 | 402 | 403 | COBRA Toolbox | [51] | 42% | 98% | |
| Methylotuvimicrobium alcaliphilum 20ZR | iIA409 | 409 | 436 | 423 | COBRA toolbox KEGG, BiGG, BioCyc | [24,52] | 25% | 94.5% |
| Methylococcus capsulatus | iMcBath | 730 | 898 | 877 | Cobrapy KEGG, BiGG MetaCyc | [53] | 54% | 66.3% |
| iMC535 | 535 | 899 | 865 | ModelSEED COBRA Toolbox KEGG, MetaCyc | [26] | 21% | 60.1% | |
| Methylocystis hirsuta CSC1 | 2478 | 1399 | 1460 | ModelSEED Cobrapy KEGG | [54] | 19% | 50.75% | |
| Methylocystis sp. SC2 | 2251 | 1449 | 1434 | ModelSEED Cobrapy KEGG | [54] | 19% | 49.82% | |
| Methylocystis sp. SB2 | 2281 | 1380 | 1453 | ModelSEED Cobrapy KEGG | [54] | 19% | 50.86% | |
| Methylocystis parvus OBBP | 2795 | 1326 | 1399 | ModelSEED Cobrapy | [55] | 19% | 53.1% | |
| Methylosinus trichosporium OB3b | iMsOB3b | 683 | 1043 | 1020 | Cobrapy KEGG | [56] | 23% | 67.24% |
| Methylocella silvestris BL2 | 681 | 1436 | 1474 | ModelSEED Cobrapy | [57] | 19% | 48% | |
| Methylomicrobium album BG8 | iJV806 | 803 | 1358 | 1367 | KBase COBRA Toolbox Cobrapy KEGG CycleFreeFlux [58] | [59] | 27% | 53.52% |
| Program | Tool Type | Algorithms for Optimization | Programs Availability | Reference |
|---|---|---|---|---|
| COBRA Toolbox 3.0 https://github.com/opencobra/cobratoolbox | Matlab package | FBA, pFBA, dFBA, dynamic rFBA, geometricFBA, relaxed FBA, FVA, MOMA, ROOM, FASTCORE, thermo FBA, looples FBA | Available, but needed Matlab | [86] |
| OptFlux http://www.optflux.org | GUI based program | FBA, pFBA, FVA, MOMA, LMOMA, ROOM, MiMBL, OptRAM, OptGene, OptKnock. | Available | [87] |
| MOST http://most.ccib.rutgers.edu/ | GUI based program | FBA, FVA, E-Flux2, SPOT | Available, but last update 5 years ago | [88,89] |
| In silico discovery https://www.insilico-biotechnology.com/ | GUI based program | FBA, FVA | Commercial | |
| Fluxer https://fluxer.umbc.edu/ | Web-service | FBA | Available | [90] |
| CAVE https://cave.biodesign.ac.cn/ | Web-service | FBA, FVA | Available | [91] |
| Cobrapy http://opencobra.sourceforge.net/ | Python package | FBA, pFBA, dFBA, geometric FBA, relaxed FBA, FVA, MOMA, ROOM, FASTCORE, thermodynamic FBA, looples FBA | Available | [92] |
| cameo http://cameo.bio. http://try.cameo.bio | Python package | FBA, FVA, OptKnock, OptGene | Available | [93] |
| ReFramed https://github.com/cdanielmachado/reframed | Python package | FBA, FVA, pFBA, FBrAtio, CAFBA, MOMA, lMOMA, ROOM, looples FBA, thermodynamic FBA, TVA, NET, GIMME, E-Flux, SteadyCom | Available | [94] |
| Mewpy https://github.com/BioSystemsUM/mewpy | Python package | FBA, pFBA, FVA, MOMA, LMOMA, ROOM, MiMBL, OptRAM, OptGene, OptKnock | Available | [95] |
| PySCeS CBMPy https://cbmpy.sourceforge.net/ | Python package | FBA, FVA | Available | [96] |
| CellNetAnalyzer (CNA) https://www2.mpi-magdeburg.mpg.de/projects/cna/cna.html | MATLAB toolbox | MFA, FBA, FVA, EFM, Yield analysis, Strain optimization (CASOP) | Available | [97,98] |
| CNApy https://github.com/cnapy-org/CNApy | Python package | FBA, pFBA, FVA, EFM, Yield optimization, Computational strain design (OptKnock, RobustKnock, OptCouple and advanced Minimal Cut Sets), OptMDFpathway, thermodynamic FBA, phase plane analysis | Available | [99] |
| StrainDesign https://github.com/klamt-lab/straindesign | Python package | FBA, pFBA, FVA, OptKnock, RobustKnock, OptCouple, general minimal cut set (MCS) approach, cRegMCS, FOCAL, ModCell2 | Available | [100] |
| Program | Data Type | Requirements | Examples of Use |
|---|---|---|---|
| RIPTiDe https://github.com/mjenior/riptide | Transcriptomic | GSM model, transcriptomics data file | [119,131] |
| pymCADRE https://github.com/draeger-lab/pymCADRE/ | Transcriptomic Metabolomic | GSM model, list of precursor metabolites, confidence scores, list of gene IDs for all genes in model, list of ubiquity scores calculated for all genes in model | [124,132] |
| Troppo https://github.com/BioSystemsUM/troppo | Transcriptomic | GSM or enzyme-constrained model, multi-omics datasets | [133,134] |
| Geckopy3.0 https://doi.org/10.1101/2023.03.20.533446 | Proteomic | Enzyme-constrained model, kinetics and omics data | [129] |
| A new GIMME–Based method | Transcriptomic | GSM model, transcriptomics data file | [135] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulyashov, M.A.; Kolmykov, S.K.; Khlebodarova, T.M.; Akberdin, I.R. State-of the-Art Constraint-Based Modeling of Microbial Metabolism: From Basics to Context-Specific Models with a Focus on Methanotrophs. Microorganisms 2023, 11, 2987. https://doi.org/10.3390/microorganisms11122987
Kulyashov MA, Kolmykov SK, Khlebodarova TM, Akberdin IR. State-of the-Art Constraint-Based Modeling of Microbial Metabolism: From Basics to Context-Specific Models with a Focus on Methanotrophs. Microorganisms. 2023; 11(12):2987. https://doi.org/10.3390/microorganisms11122987
Chicago/Turabian StyleKulyashov, Mikhail A., Semyon K. Kolmykov, Tamara M. Khlebodarova, and Ilya R. Akberdin. 2023. "State-of the-Art Constraint-Based Modeling of Microbial Metabolism: From Basics to Context-Specific Models with a Focus on Methanotrophs" Microorganisms 11, no. 12: 2987. https://doi.org/10.3390/microorganisms11122987
APA StyleKulyashov, M. A., Kolmykov, S. K., Khlebodarova, T. M., & Akberdin, I. R. (2023). State-of the-Art Constraint-Based Modeling of Microbial Metabolism: From Basics to Context-Specific Models with a Focus on Methanotrophs. Microorganisms, 11(12), 2987. https://doi.org/10.3390/microorganisms11122987