

Analysis of the Frequency of Mutations at Diagnostic Oligonucleotide Sites and Their Impact on the Efficiency of PCR for HIV-1

Abstract
:1. Introduction
2. Materials and Methods
2.1. Nucleotide Sequence Databases and Alignments
2.2. DNA Constructs and Site-Directed Mutagenesis
2.3. Determination of Concentrations of DNA Constructs
2.4. Real-Time PCR
2.5. Statistics
3. Results
3.1. Oligonucleotide Selection for Real-Time PCR
3.2. HIV-1 Nucleotide Sequence Analysis
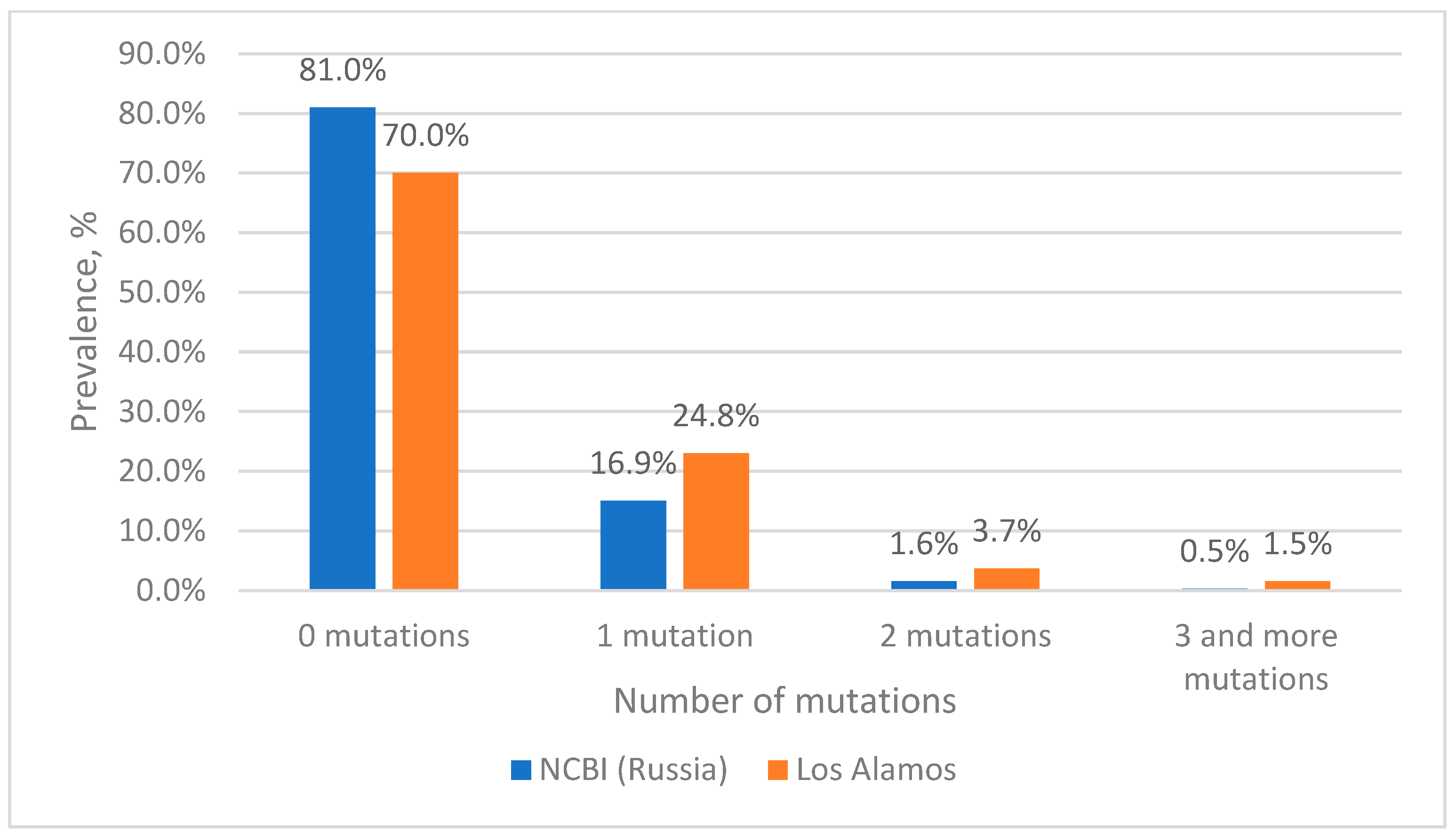
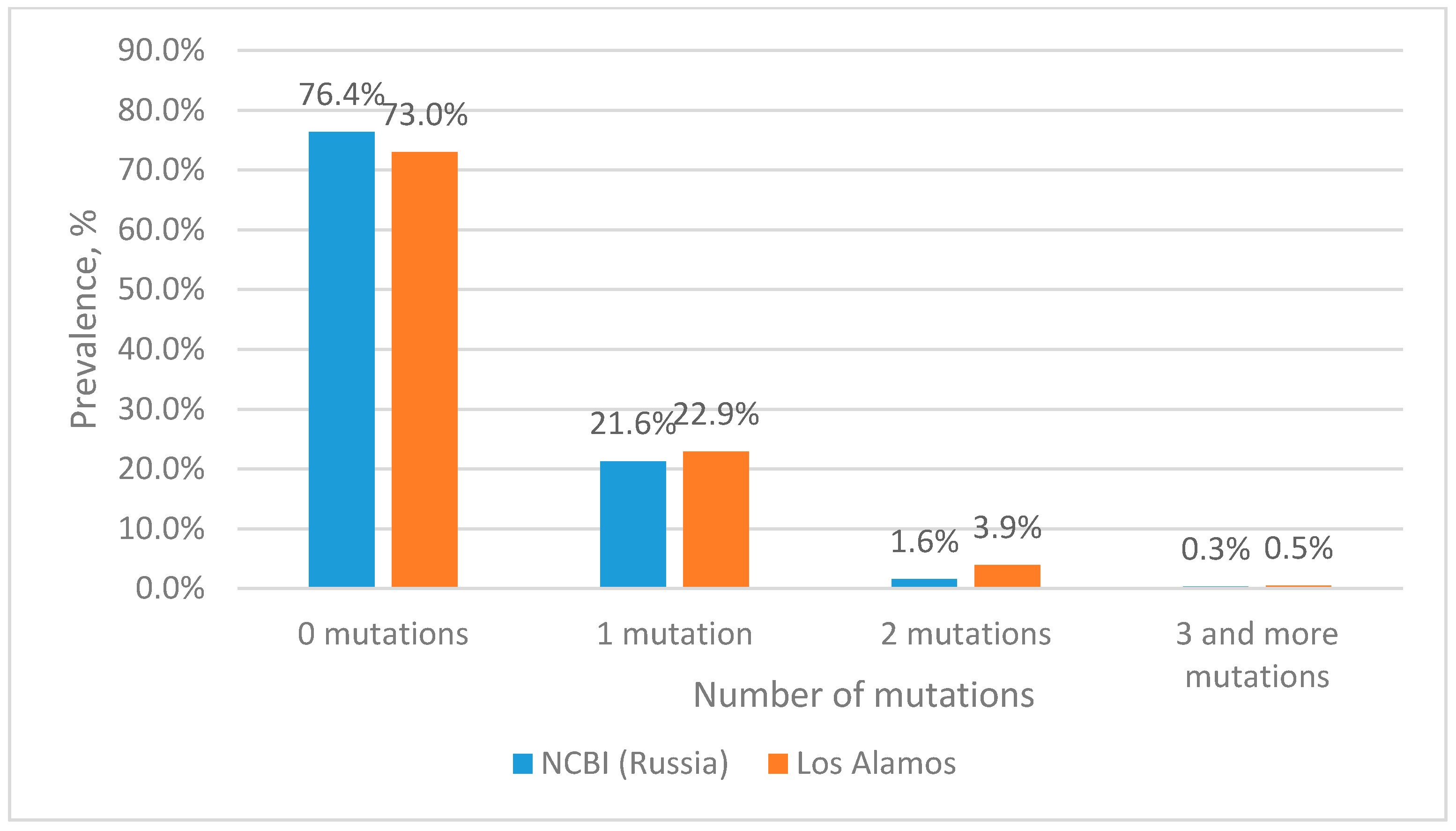
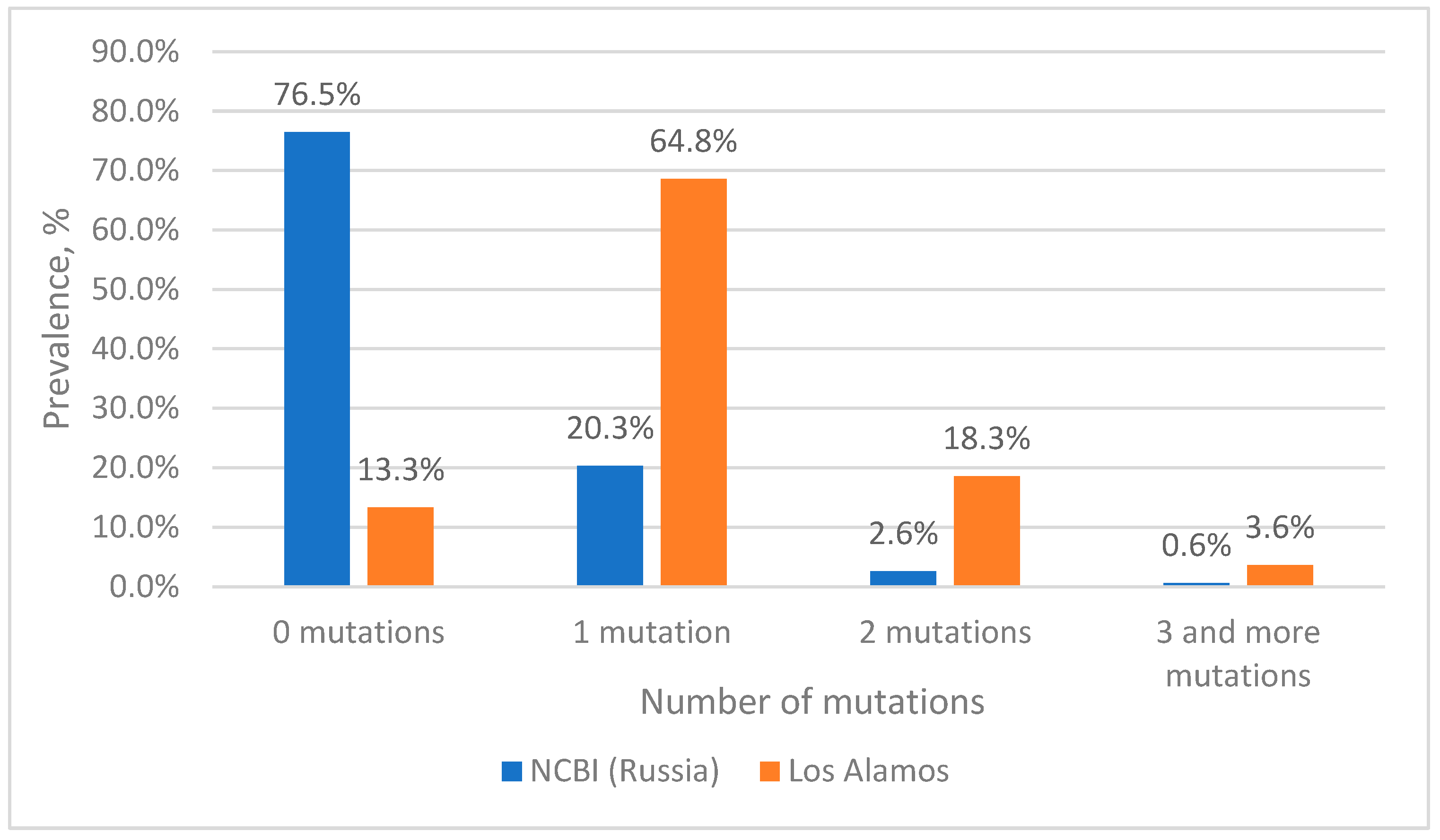
Estimation of Mutation Frequency at Primer and Probe Binding Sites
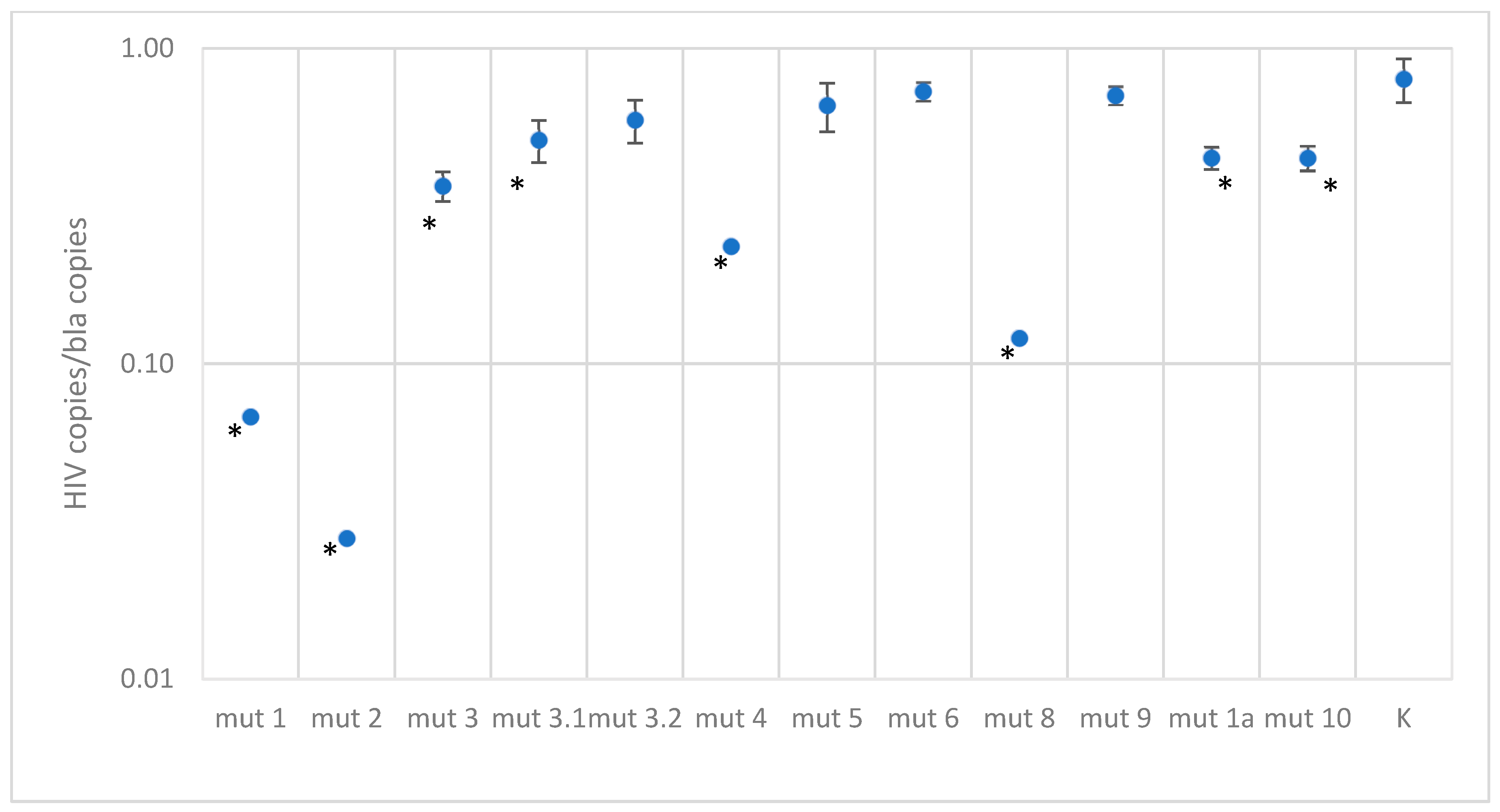
3.3. Efficiency of Primers and Probe in Real-Time PCR in Case of Mismatches at Binding Sites
3.4. Selection of a Conserved Target for Discordant Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Maksimenko, L.V.; Totmenin, A.V.; Gashnikova, M.P.; Astakhova, E.M.; Skudarnov, S.E.; Ostapova, T.S.; Yaschenko, S.V.; Meshkov, I.O.; Bocharov, E.F.; Maksyutov, R.; et al. Genetic Diversity of HIV-1 in Krasnoyarsk Krai: Area with High Levels of HIV-1 Recombination in Russia. BioMed Res. Int. 2020, 2020, 9057541. [Google Scholar] [CrossRef] [PubMed]
- Beloukas, A.; Psarris, A.; Giannelou, P.; Kostaki, E.; Hatzakis, A.; Paraskevis, D. Molecular epidemiology of HIV-1 infection in Europe: An overview. Infect. Genet. Evol. 2016, 46, 180–189. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xing, H.; Ruan, Y.; Hong, K.; Cheng, C.; Hu, Y.; Xin, R.; Wei, J.; Feng, Y.; Hsi, J.H.; et al. A comprehensive mapping of HIV-1 genotypes in various risk groups and regions across China based on a nationwide molecular epidemiologic survey. PLoS ONE 2012, 7, e47289. [Google Scholar] [CrossRef] [PubMed]
- GenBank. Available online: https://www.ncbi.nlm.nih.gov/genbank (accessed on 22 December 2021).
- HIV Sequence Database. Available online: https://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html (accessed on 9 March 2022).
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Piampongsant, S.; Faria, N.R.; Voet, A.; Pineda-Peña, A.-C.; Khouri, R.; Lemey, P.; Vandamme, A.-M.; Theys, K. An integrated map of HIV genome-wide variation from a population perspective. Retrovirology 2015, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- HIV Sequence Compendium 2021. Available online: https://www.hiv.lanl.gov/content/sequence/HIV/COMPENDIUM/2021compendium.html (accessed on 21 March 2018).
- Korn, K.; Weissbrich, B.; Henke-Gendo, C.; Heim, A.; Jauer, C.M.; Taylor, N.; Eberle, J. Single-point mutations causing more than 100-fold underestimation of human immunodeficiency virus type 1 (HIV-1) load with the Cobas TaqMan HIV-1 real-time PCR assay. J. Clin. Microbiol. 2009, 47, 1238–1240. [Google Scholar] [CrossRef] [PubMed]
- Damond, F.; Avettand-Fenoel, V.; Collin, G.; Roquebert, B.; Plantier, J.C.; Ganon, A.; Sizmann, D.; Babiel, R.; Glaubitz, J.; Chaix, M.L.; et al. Evaluation of an upgraded version of the Roche Cobas AmpliPrep/Cobas TaqMan HIV-1 test for HIV-1 load quantification. J. Clin. Microbiol. 2010, 48, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Karasi, J.; Dziezuk, F.; Quennery, L.; Förster, S.; Reischl, U.; Colucci, G.; Schoener, D.; Seguin-Devaux, C.; Schmit, J. High correlation between the Roche COBAS® AmpliPrep/COBAS® TaqMan® HIV-1, v2.0 and the Abbott m2000 RealTime HIV-1 assays for quantification of viral load in HIV-1 B and non-B subtypes. J. Clin. Virol. 2011, 52, 181–186. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct Name | Binding Sites | Russian Strains (NCBI) | International Strains (Los Alamos) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| % | Claster | Predominant Genotype | % | Claster | Predominant Genotype | ||||
| DNA constructs with mutations in the TaqMan probe binding site | |||||||||
| Mut 5 | gggattgggggatacagtgcaggg | 2.41% | 2 | B | 48% | 9.8% | 1 | B | 47% |
| Mut 3-2 | gggattggggggtacagtgcagga | 2.16% | 3 | B | 50% | 4.41% | 2 | B | 37.9% |
| Mut 3-1 | gggattggggggtacactgcaggg | 1.25% | 4 | A6/B | 40%/40% | 0.87% | 6 | B | 43.4% |
| Mut 6 | gggattggggggtactgtgcaggg | 0.58% | 6 | B | 100% | 0.22% | 19 | B | 75% |
| Mut 9 | gggattgggggatacagtgcagga | 0.42% | 11 | B | 80% | 0.63% | 8 | B | 60.5% |
| Mut 3 | gggattggggggtacactgcagga | 0 | ND | ND | ND | 0.68% | 7 | O | 90% |
| Mut 1a | gggattggggactacagtgcaggg | 0.17% | 17 | A6/CRF63_02A1 | 50%/50% | 0.33% | 10 | B | 47.4% |
| Mut 10 | gggattgggggatatagtgcaggg | 0 | ND | ND | ND | 0.24% | 16 | B | 46% |
| Mut 8 | gggattggggaatacagtgcagga | 0.25% | 14 | B | 100% | 0.03% | 49 | B | 100% |
| Mut 1 | gggattggggactactgtgcaggg | 0 | ND | ND | ND | 0.28% | 12 | B/01_AE | 37.5%/37.5% |
| Mut 2 | gggattggggactatagtgcaggg | 0.08% * | 40 | A1 | 100% | 0.27% | 14 | B | 33% |
| Mut 4 | gggattggggaccacagtgcaggg | 0 | ND | ND | ND | 0.02% * | 133 | 75_BF1 | 100% |
| DNA constructs with mutations in the primers binding sites | |||||||||
| Primer Int for | cagcagtacaaatggcagtattcatycaca | ||||||||
| Mut 1 | cagcagtacaaatggcagtatttgtgcaca | 0 | ND | ND | ND | 0.02% * | 209 | O | 100% |
| Mut 2 | cagcagtacaaatggcagtctttgtccaca | 0 | ND | ND | ND | 0.06% | 43 | O | 100% |
| Mut 3 | cagcagtactaatggcagtatacatccata | 0 | ND | ND | ND | 0.02% * | 104 | B | 100% |
| Mut 4 | caacagtccaaataacaatattcatccaca | 0 | ND | ND | ND | 0 | ND | ND | ND |
| Mut 5 | cagcagtgcaaatggcggttttcatccaca | 0 | ND | ND | ND | 0.08% | 33 | N | 100% |
| Primer Int rev | cctgtattacyactgccccttcacctttcca | ||||||||
| Mut 6 | cttggattaccactgccccttctcctttcca | 0 | ND | ND | ND | 0.16% | 20 | N | 100% |
| Mut 7 | cttgtatgactactgctccctcacctttcca | 0 | ND | ND | ND | 0.65% | 8 | O | 89.7% |
| Mut 8 | cttgtatgactactgcccctttaccttttca | 0 | ND | ND | ND | 0 | ND | ND | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogoslovskaya, E.V.; Tsyganova, G.M.; Nosova, A.O.; Shipulin, G.A. Analysis of the Frequency of Mutations at Diagnostic Oligonucleotide Sites and Their Impact on the Efficiency of PCR for HIV-1. Microorganisms 2023, 11, 2838. https://doi.org/10.3390/microorganisms11122838
Bogoslovskaya EV, Tsyganova GM, Nosova AO, Shipulin GA. Analysis of the Frequency of Mutations at Diagnostic Oligonucleotide Sites and Their Impact on the Efficiency of PCR for HIV-1. Microorganisms. 2023; 11(12):2838. https://doi.org/10.3390/microorganisms11122838
Chicago/Turabian StyleBogoslovskaya, Elena V., Galina M. Tsyganova, Anastasiia O. Nosova, and German A. Shipulin. 2023. "Analysis of the Frequency of Mutations at Diagnostic Oligonucleotide Sites and Their Impact on the Efficiency of PCR for HIV-1" Microorganisms 11, no. 12: 2838. https://doi.org/10.3390/microorganisms11122838
APA StyleBogoslovskaya, E. V., Tsyganova, G. M., Nosova, A. O., & Shipulin, G. A. (2023). Analysis of the Frequency of Mutations at Diagnostic Oligonucleotide Sites and Their Impact on the Efficiency of PCR for HIV-1. Microorganisms, 11(12), 2838. https://doi.org/10.3390/microorganisms11122838