

Clinical Whole-Genome Sequencing Assay for Rapid Mycobacterium tuberculosis Complex First-Line Drug Susceptibility Testing and Phylogenetic Relatedness Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. Clinical Samples
2.3. Isolate Preparation, DNA Extraction and WGS
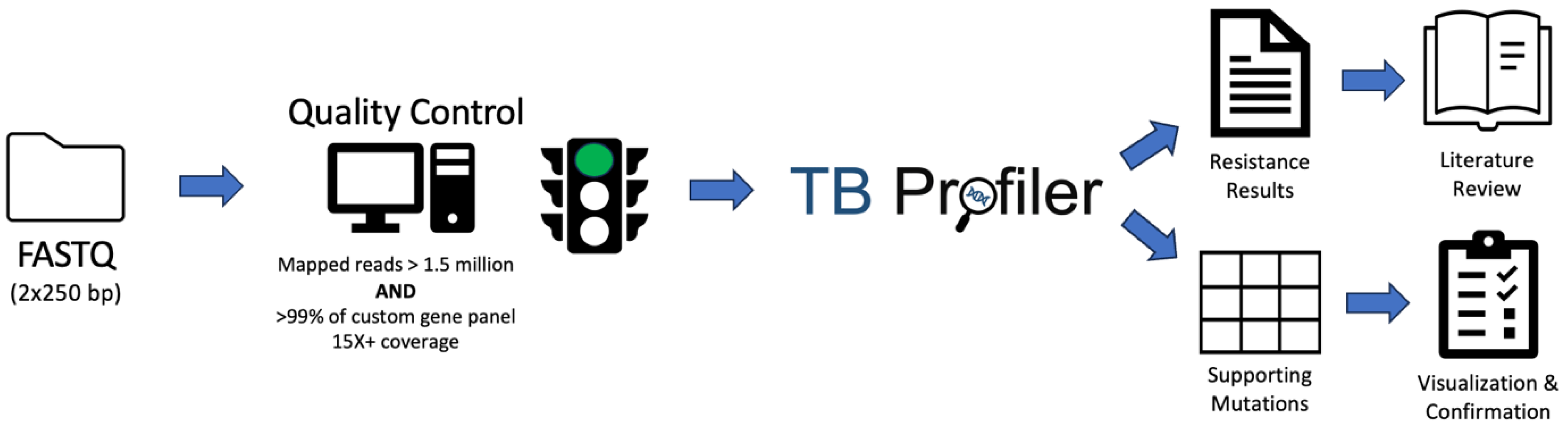
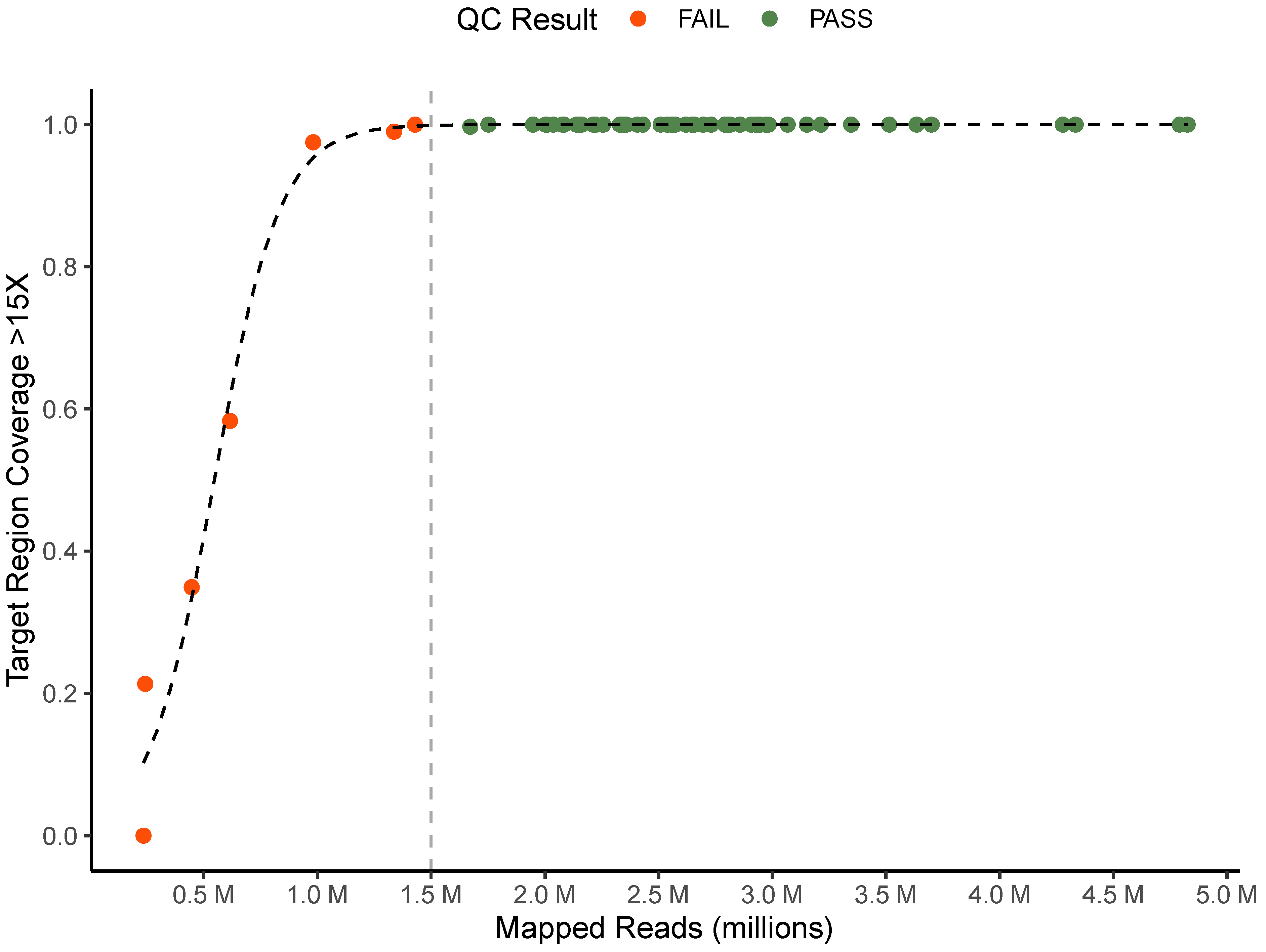
2.4. Sequence Data Quality Control Criteria
2.5. Mutation Profiling and Resistance Calling Using TB-Profiler
2.6. Reproducibility and Repeatability Studies
2.7. Cross-Validation of Culture Media
2.8. Clinical Metadata and Phenotypic DST Results
2.9. Prospective Evaluation
2.10. Genomic Epidemiology
2.11. Statistical Analysis
3. Results
3.1. Development and Testing of Quality Control Criteria
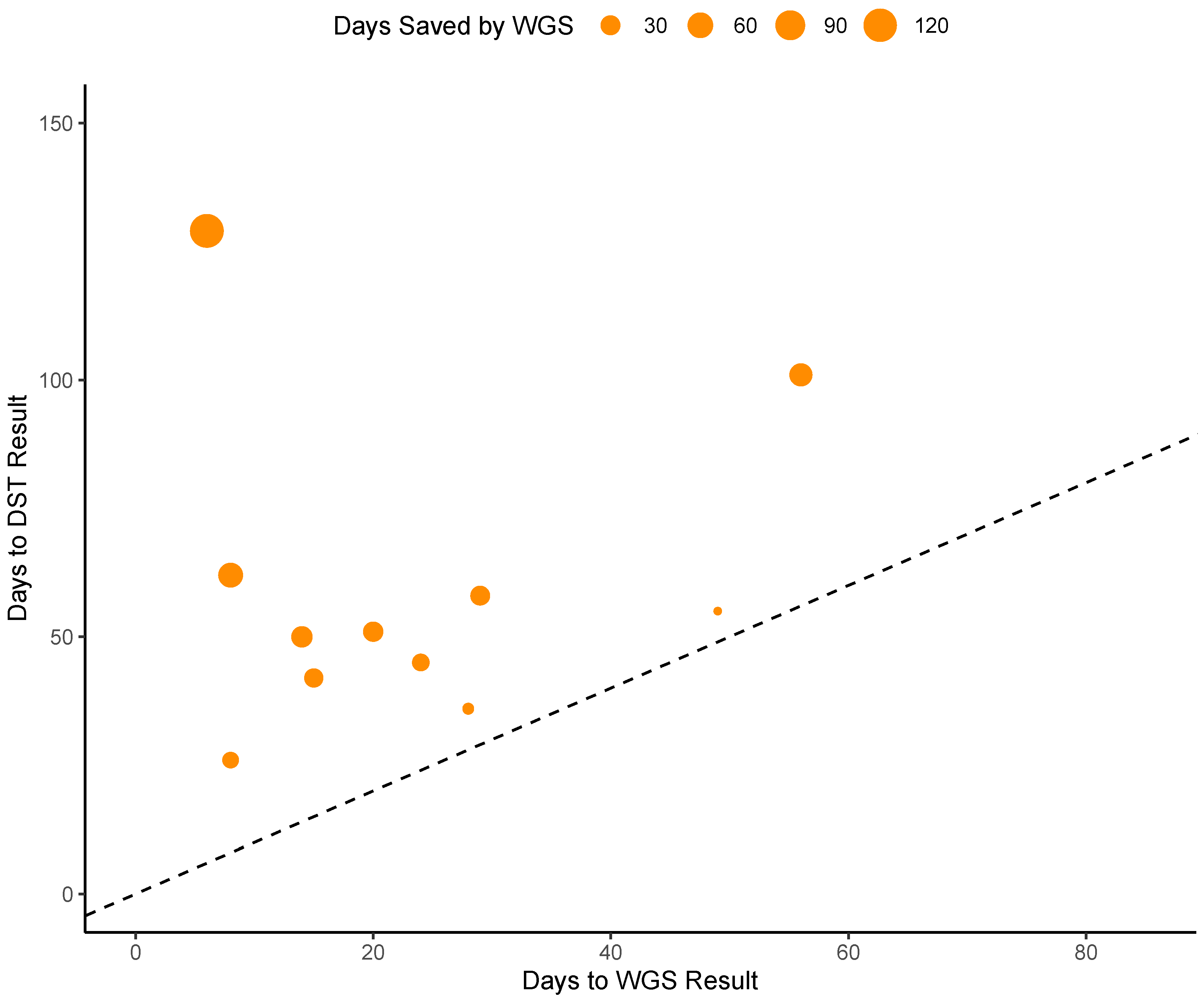
3.2. Turnaround Time
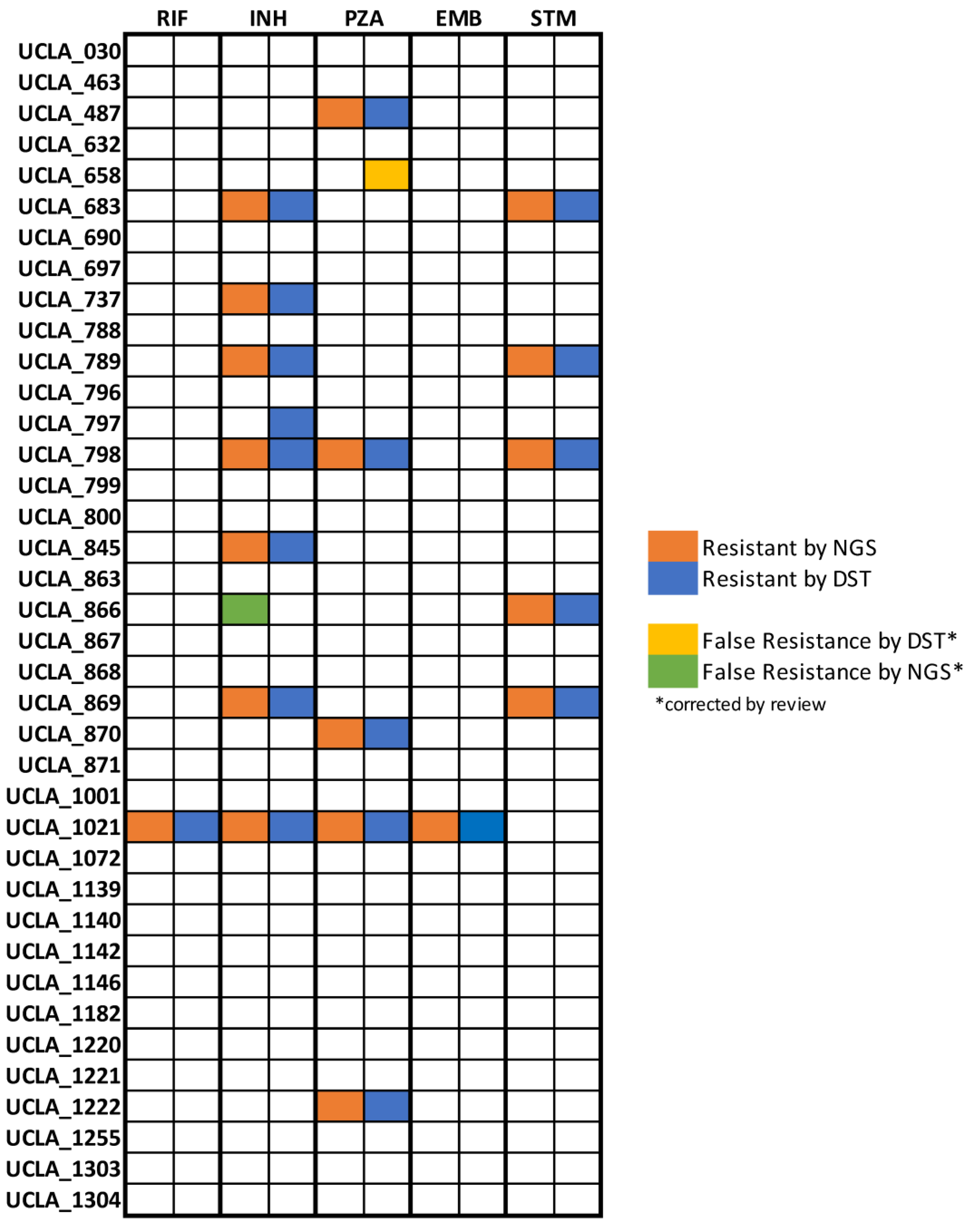
3.3. Accuracy
3.4. Precision and MGIT vs. Solid Agar Isolates Cross Validation
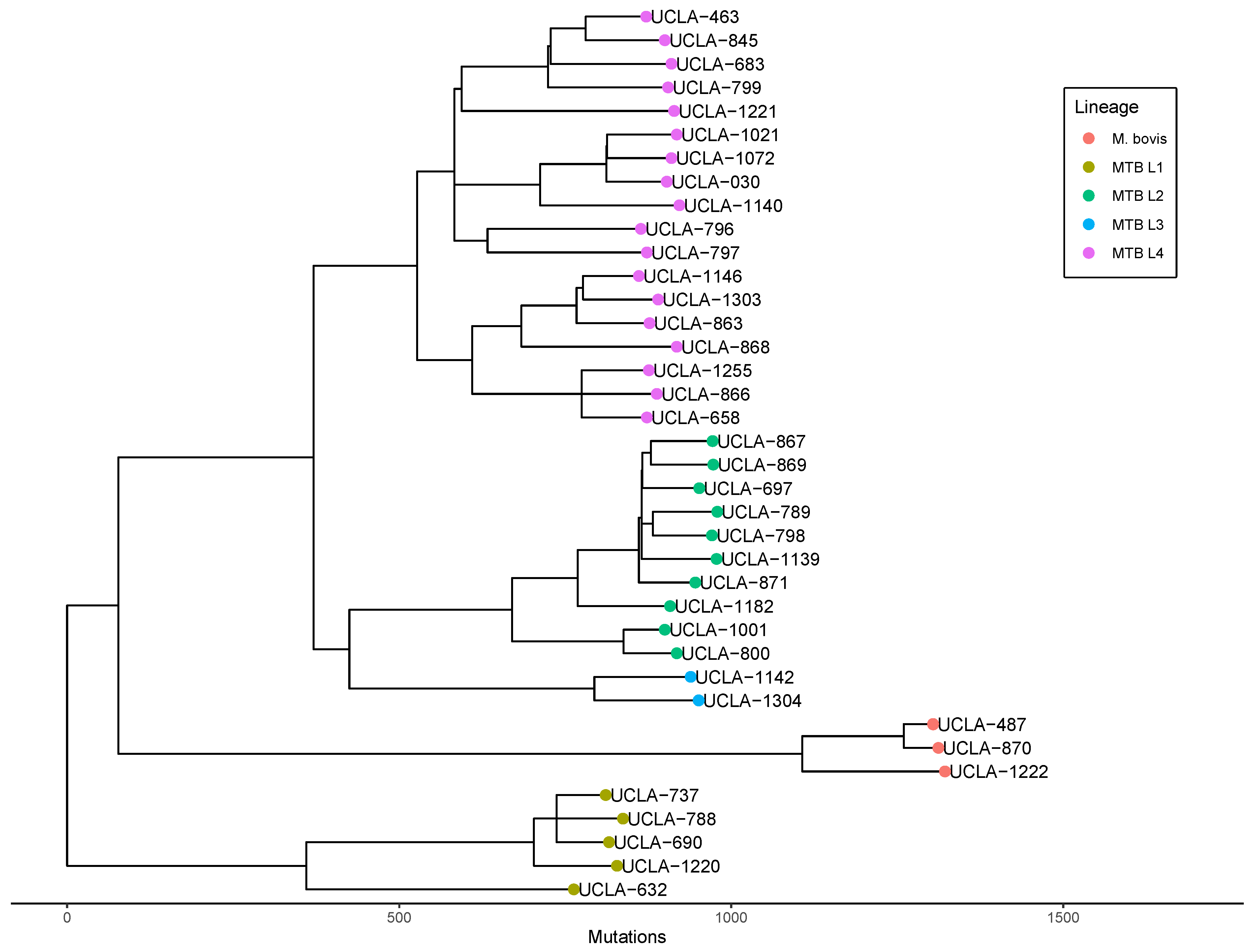
3.5. Genomic Epidemiology
3.6. Phylogenetic Analysis for Laboratory Contamination Investigation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tuberculosis | Newsroom | Global Health | CDC. Available online: https://www.cdc.gov/globalhealth/newsroom/topics/tb/index.html (accessed on 1 May 2023).
- Tuberculosis. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis. (accessed on 1 May 2023).
- Pai, M.; Kasaeva, T.; Swaminathan, S. COVID-19’s Devastating Effect on Tuberculosis Care—A Path to Recovery. N. Engl. J. Med. 2022, 386, 1490–1493. [Google Scholar] [CrossRef] [PubMed]
- Global Tuberculosis Report 2021. Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 1 May 2023).
- Khawbung, J.L.; Nath, D.; Chakraborty, S. Drug resistant Tuberculosis: A review. Comp. Immunol. Microbiol. Infect. Dis. 2021, 74, 101574. [Google Scholar] [CrossRef]
- Chung-Delgado, K.; Guillen-Bravo, S.; Revilla-Montag, A.; Bernabe-Ortiz, A. Mortality among MDR-TB cases: Comparison with drug-susceptible tuberculosis and associated factors. PLoS ONE 2015, 10, e0119332. [Google Scholar] [CrossRef] [PubMed]
- Tiberi, S.; Carvalho, A.C.C.; Sulis, G.; Vaghela, D.; Rendon, A.; Mello, F.C.d.Q.; Rahman, A.; Matin, N.; Zumla, A.; Pontali, E. The cursed duet today: Tuberculosis and HIV-coinfection. Presse Méd. 2017, 46, e23–e39. [Google Scholar] [CrossRef]
- O’donnell, M.R.; Padayatchi, N.; Kvasnovsky, C.; Werner, L.; Master, I.; Horsburgh, C.R. Treatment Outcomes for Extensively Drug-Resistant Tuberculosis and HIV Co-infection. Emerg. Infect. Dis. 2013, 19, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Global Tuberculosis Report 2022. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (accessed on 1 May 2023).
- Böttger, E.C. The ins and outs of Mycobacterium tuberculosis drug susceptibility testing. Clin. Microbiol. Infect. 2011, 17, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Miotto, P.; Zhang, Y.; Cirillo, D.M.; Yam, W.C. Drug resistance mechanisms and drug susceptibility testing for tuberculo-sis. Respirology 2018, 23, 1098–1113. [Google Scholar] [CrossRef] [PubMed]
- Satoshi, M.; Kato, S.; Ogata, H.; Aono, A.; Chikamatsu, K.; Mizuno, K.; Toyota, E.; Sejimo, A.; Suzuki, K.; Yoshida, S.; et al. Comprehensive Multicenter Evaluation of a New Line Probe Assay Kit for Identification of Mycobacterium Species and Detection of Drug-Resistant Mycobacterium tuberculosis. J. Clin. Microbiol. 2020, 50, 884–890. [Google Scholar]
- Wu, X.; Tan, G.; Sha, W.; Liu, H.; Yang, J.; Guo, Y.; Shen, X.; Wu, Z.; Shen, H.; Yu, F. Use of Whole-Genome Sequencing to Predict Mycobacterium tuberculosis Complex Drug Resistance from Early Positive Liquid Cultures. Microbiol. Spectr. 2022, 10, e0251621. [Google Scholar] [CrossRef]
- Shea, J.; Halse, T.A.; Lapierre, P.; Shudt, M.; Kohlerschmidt, D.; Van Roey, P.; Limberger, R.; Taylor, J.; Escuyer, V.; Musser, K.A. Comprehensive Whole-Genome Sequencing and Reporting of Drug Resistance Profiles on Clinical Cases of Mycobacterium tuberculosis in New York State. J. Clin. Microbiol. 2017, 55, 1871–1882. [Google Scholar] [CrossRef]
- Coll, F.; McNerney, R.; Preston, M.D.; Guerra-Assunção, J.A.; Warry, A.; Hill-Cawthorne, G.; Mallard, K.; Nair, M.; Miranda, A.; Alves, A.; et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.; Pallen, M. XDR-TB genome sequencing: A glimpse of the microbiology of the future. Future Microbiol. 2008, 3, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Witney, A.A.; Cosgrove, C.A.; Arnold, A.; Hinds, J.; Stoker, N.G.; Butcher, P.D. Clinical use of whole genome sequencing for Mycobacterium tuberculosis. BMC Med. 2016, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Katale, B.Z.; Mbelele, P.M.; Lema, N.A.; Campino, S.; Mshana, S.E.; Rweyemamu, M.M.; Phelan, J.E.; Keyyu, J.D.; Majigo, M.; Mbugi, E.V.; et al. Whole genome sequencing of Mycobacterium tuberculosis isolates and clinical outcomes of patients treated for multidrug-resistant tuberculosis in Tanzania. BMC Genom. 2020, 21, 174. [Google Scholar] [CrossRef]
- Genestet, C.; Hodille, E.; Berland, J.; Ginevra, C.; Bryant, J.E.; Ader, F.; Lina, G.; Dumitrescu, O.; Lyon TB Study Group. Whole-genome sequencing in drug susceptibility testing of Mycobacterium tuber-culosis in routine practice in Lyon, France. Int. J. Antimicrob. Agents 2020, 55, 105912. [Google Scholar] [CrossRef]
- Maladan, Y.; Krismawati, H.; Wahyuni, T.; Tanjung, R.; Awaludin, K.; Audah, K.A.; Parikesit, A.A. The whole-genome sequencing in predicting Mycobacterium tuberculosis drug susceptibility and resistance in Papua, Indonesia. BMC Genom. 2021, 22, 844. [Google Scholar] [CrossRef]
- Walker, T.M.; Kohl, T.A.; Omar, S.V.; Hedge, J.; Elías, C.D.O.; Bradley, P.; Iqbal, Z.; Feuerriegel, S.; Niehaus, K.E.; Wilson, D.J.; et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: A retrospective cohort study. Lancet Infect. Dis. 2015, 15, 1193–1202. [Google Scholar] [CrossRef]
- Kamolwat, P.; Nonghanphithak, D.; Smithtikarn, S.; Pugrassami, P.; Faksri, K. Diagnostic performance of whole-genome se-quencing for identifying drug-resistant TB in Thailand. Int. J. Tuberc. Lung Dis. 2021, 25, 754–760. [Google Scholar] [CrossRef]
- The CRyPTIC Consortium A data compendium associating the genomes of 12,289 Mycobacterium tuberculosis isolates with quantitative resistance phenotypes to 13 antibiotics. PLoS Biol. 2022, 20, e3001721. [CrossRef]
- Gordon, A.K.; Marais, B.; Walker, T.M.; Sintchenko, V. Clinical and public health utility of Mycobacterium tuberculosis whole genome sequencing. Int. J. Infect. Dis. 2021, 113, S40–S42. [Google Scholar] [CrossRef]
- Kato-Maeda, M.; Ho, C.; Passarelli, B.; Banaei, N.; Grinsdale, J.; Flores, L.; Anderson, J.; Murray, M.; Rose, G.; Kawamura, L.M.; et al. Use of Whole Genome Sequencing to Determine the Microevolution of Mycobacterium tuberculosis during an Outbreak. PLoS ONE 2013, 8, e58235. [Google Scholar] [CrossRef]
- Bryant, J.M.; Schürch, A.C.; van Deutekom, H.; Harris, S.R.; de Beer, J.L.; de Jager, V.; Kremer, K.; van Hijum, S.A.; Siezen, R.J.; Borgdorff, M.; et al. Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data. BMC Infect. Dis. 2013, 13, 110. [Google Scholar] [CrossRef]
- Huddleston, J.; Hadfield, J.; Sibley, T.R.; Lee, J.; Fay, K.; Ilcisin, M.; Harkins, E.; Bedford, T.; Neher, R.A.; Hodcroft, E.B. Augur: A bioinformatics toolkit for phylogenetic analyses of human pathogens. J. Open Source Softw. 2021, 6, 2906. [Google Scholar] [CrossRef]
- Phelan, J.; O’Sullivan, D.M.; Machado, D.; Ramos, J.; Oppong, Y.E.A.; Campino, S.; O’Grady, J.; McNerney, R.; Hibberd, M.L.; Viveiros, M.; et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to an-ti-tuberculous drugs. Genome. Med. 2019, 11, 41. [Google Scholar] [CrossRef]
- Price, T.K.; Realegeno, S.; Mirasol, R.; Tsan, A.; Chandrasekaran, S.; Garner, O.B.; Yang, S. Validation, Implementation, and Clinical Utility of Whole Genome Sequence-Based Bacterial Identification in the Clinical Microbiology Laboratory. J. Mol. Diagn. 2021, 23, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Vasimuddin; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. GGTREE: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Campbell, E.M.; Boyles, A.; Shankar, A.; Kim, J.; Knyazev, S.; Cintron, R.; Switzer, W.M. MicrobeTrace: Retooling molecular epidemiology for rapid public health response. PLoS Comput. Biol. 2021, 17, e1009300. [Google Scholar] [CrossRef]
- Shi, W. Activity of Pyrazinamide against Mycobacterium tuberculosis at Neutral pH in PZA-S1 Minimal Medium. Antibiotics 2021, 10, 909. [Google Scholar] [CrossRef] [PubMed]
- Gagneux, S. Strain Variation in the Mycobacterium tuberculosis Complex: Its Role in Biology, Epidemiology and Control; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; Volume 1019. [Google Scholar]
- World Health Organization. The Use of Next-Generation Sequencing Technologies for the Detection of Mutations Associated with Drug Resistance in Mycobacterium tuberculosis Complex: Technical Guide. 2018. Available online: https://apps.who.int/iris/handle/10665/274443 (accessed on 1 May 2023).
- Laurent, S.; Zakham, F.; Bertelli, C.; Merz, L.; Nicod, L.; Mazza-Stalder, J.; Greub, G.; Jaton, K.; Opota, O. Genome sequencing of Mycobacterium tuberculosis clinical isolates revealed isoniazid resistance mechanisms undetected by conventional molecular methods. Int. J. Antimicrob. Agents 2020, 56, 106068. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.N.; Talarico, S.; Poonja, S.; McDaniel, C.J.; Cilnis, M.; Chang, A.H.; Raz, K.; Noboa, W.S.; Cowan, L.; Shaw, T.; et al. Mutation of Mycobacterium tuberculosis and Implications for Using Whole-Genome Sequencing for In-vestigating Recent Tuberculosis Transmission. Front. Public Health 2022, 9, 790544. [Google Scholar] [CrossRef]
- Pfyffer, G.E.; Welscher, H.M.; Kissling, P.; Cieslak, C.; Casal, M.J.; Gutierrez, J.; Rüsch-Gerdes, S. Comparison of the Mycobacteria Growth Indicator Tube (MGIT) with radiometric and solid culture for recovery of acid-fast bacilli. J. Clin. Microbiol. 1997, 35, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.; Wang, F.; Hernandez, K.M.; Zhang, Z.; Grossman, R.L. Accurate and rapid prediction of tuberculosis drug re-sistance from genome sequence data using traditional machine learning algorithms and CNN. Sci. Rep. 2022, 12, 2427. [Google Scholar] [CrossRef]
- Gröschel, M.I.; Owens, M.; Freschi, L.; Vargas, R., Jr.; Marin, M.G.; Phelan, J.; Iqbal, Z.; Dixit, A.; Farhat, M.R. GenTB: A user-friendly genome-based predictor for tuberculosis resistance powered by machine learning. Genome Med. 2021, 13, 138. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaw, B.; von Bredow, B.; Tsan, A.; Garner, O.; Yang, S. Clinical Whole-Genome Sequencing Assay for Rapid Mycobacterium tuberculosis Complex First-Line Drug Susceptibility Testing and Phylogenetic Relatedness Analysis. Microorganisms 2023, 11, 2538. https://doi.org/10.3390/microorganisms11102538
Shaw B, von Bredow B, Tsan A, Garner O, Yang S. Clinical Whole-Genome Sequencing Assay for Rapid Mycobacterium tuberculosis Complex First-Line Drug Susceptibility Testing and Phylogenetic Relatedness Analysis. Microorganisms. 2023; 11(10):2538. https://doi.org/10.3390/microorganisms11102538
Chicago/Turabian StyleShaw, Bennett, Benjamin von Bredow, Allison Tsan, Omai Garner, and Shangxin Yang. 2023. "Clinical Whole-Genome Sequencing Assay for Rapid Mycobacterium tuberculosis Complex First-Line Drug Susceptibility Testing and Phylogenetic Relatedness Analysis" Microorganisms 11, no. 10: 2538. https://doi.org/10.3390/microorganisms11102538
APA StyleShaw, B., von Bredow, B., Tsan, A., Garner, O., & Yang, S. (2023). Clinical Whole-Genome Sequencing Assay for Rapid Mycobacterium tuberculosis Complex First-Line Drug Susceptibility Testing and Phylogenetic Relatedness Analysis. Microorganisms, 11(10), 2538. https://doi.org/10.3390/microorganisms11102538