
Illuminating the Genomic Landscape of Lactiplantibacillus plantarum PU3—A Novel Probiotic Strain Isolated from Human Breast Milk, Explored through Nanopore Sequencing

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Sample Collection
2.2. Bacterial Isolation and Identification
2.3. DNA Extraction, Sequencing, Assembly
2.4. Genome-Based Identification and MultiLocus Sequence Typing (MLST)
2.5. Genome Annotation
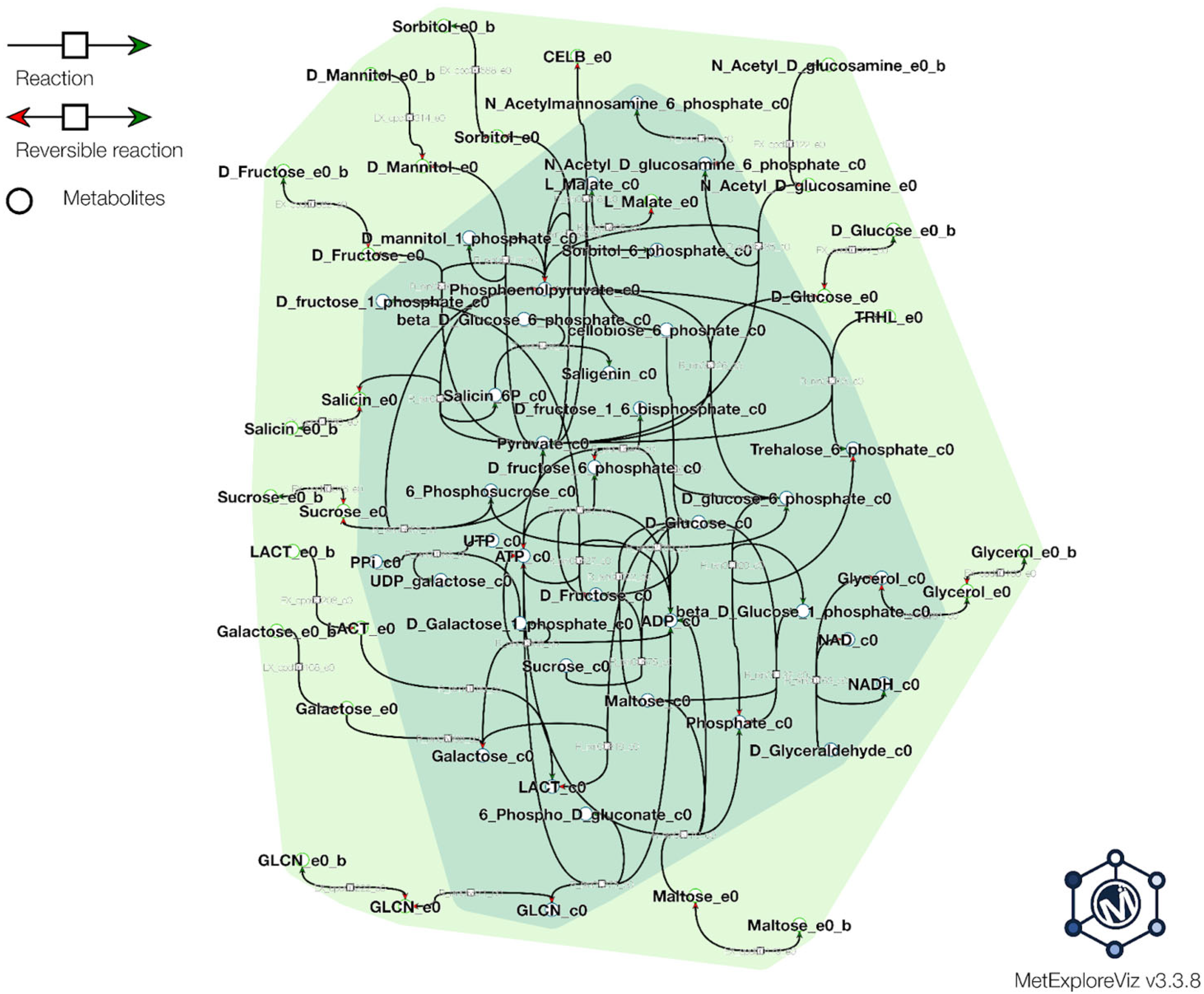
2.6. Metabolic Modeling
2.7. Assimilation of Different Types of Carbon Sources and Osmotic Sensitivity
2.8. Bile Salt Tolerance
2.9. Acid Tolerance
2.10. Determination of Antibiotic Sensitivity of Strain
3. Results and Discussion
3.1. In Silico Genomic Insights of the Strain Lactiplantibacillus plantarum PU3
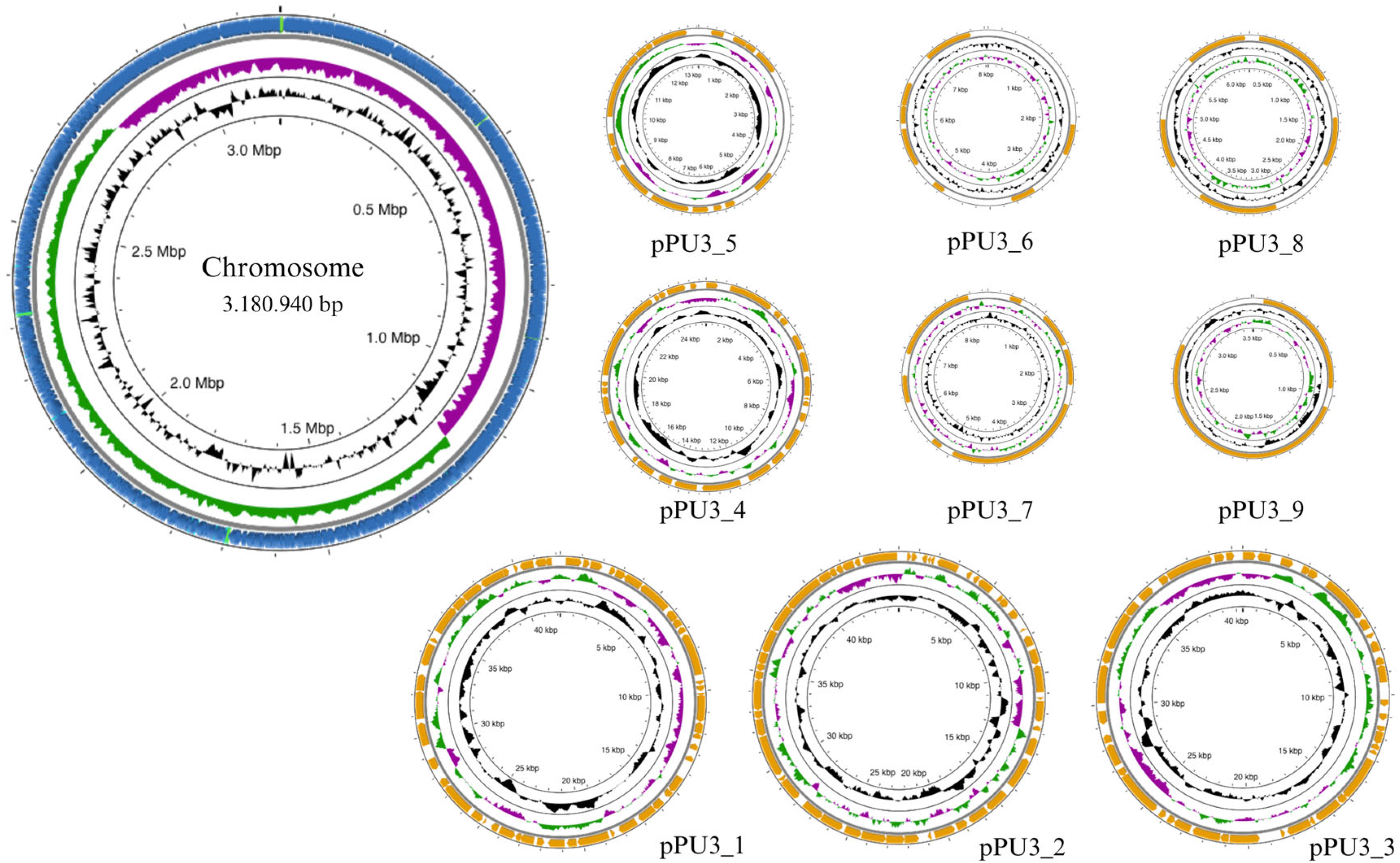
3.1.1. Genome Overview
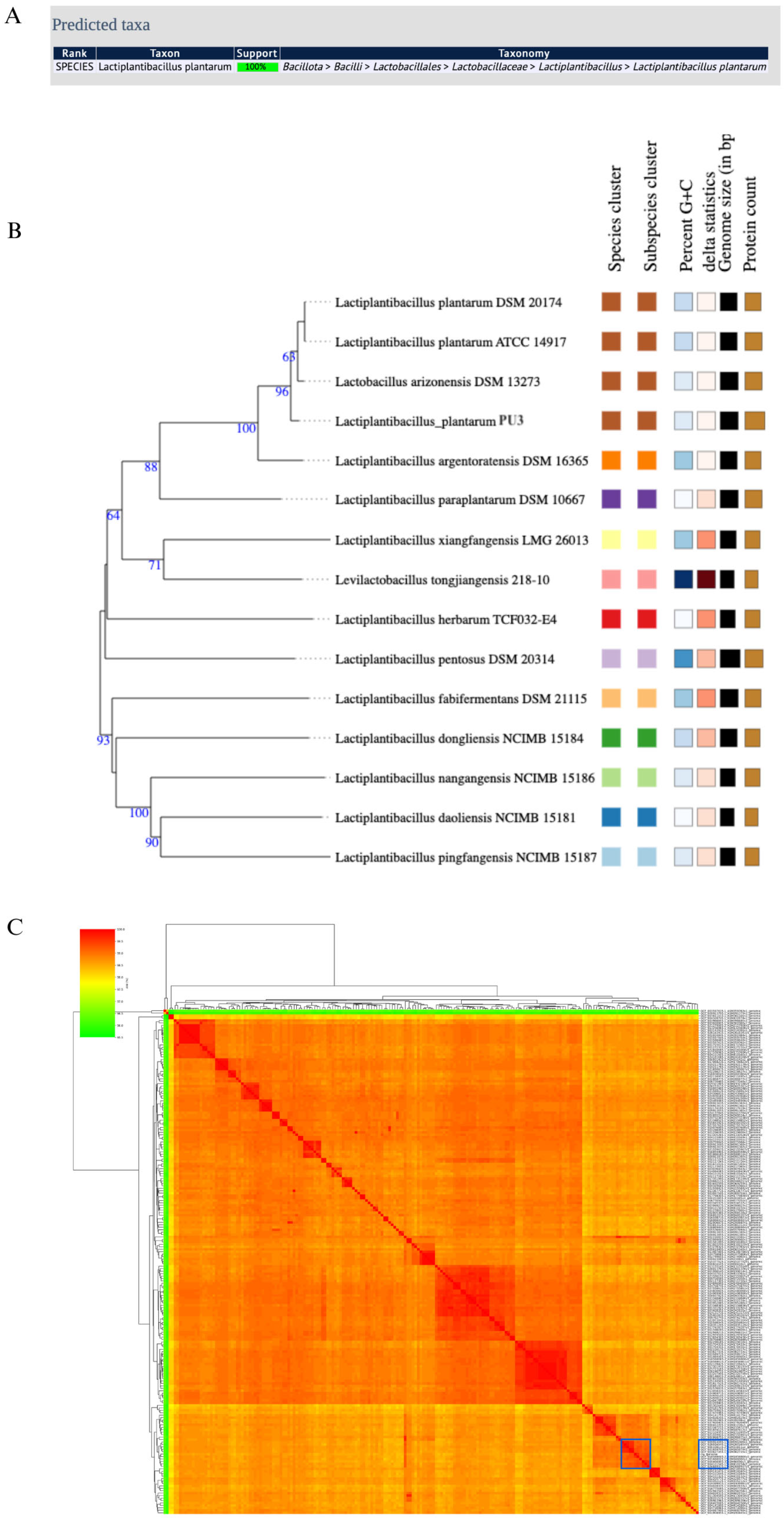
3.1.2. Species Confirmation and Average Nucleotide Identity (ANI) of the Strain
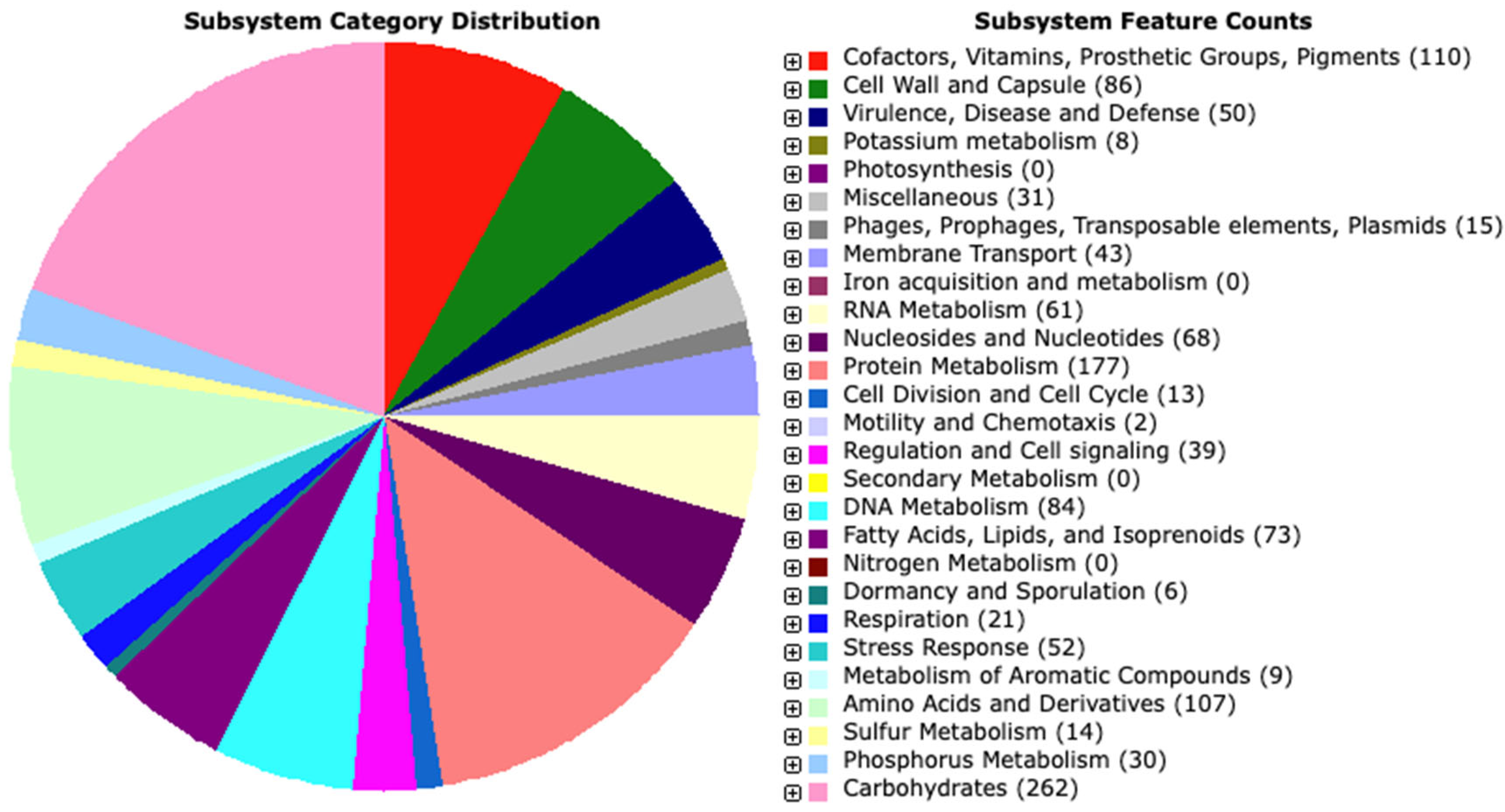
3.1.3. Genome Annotation
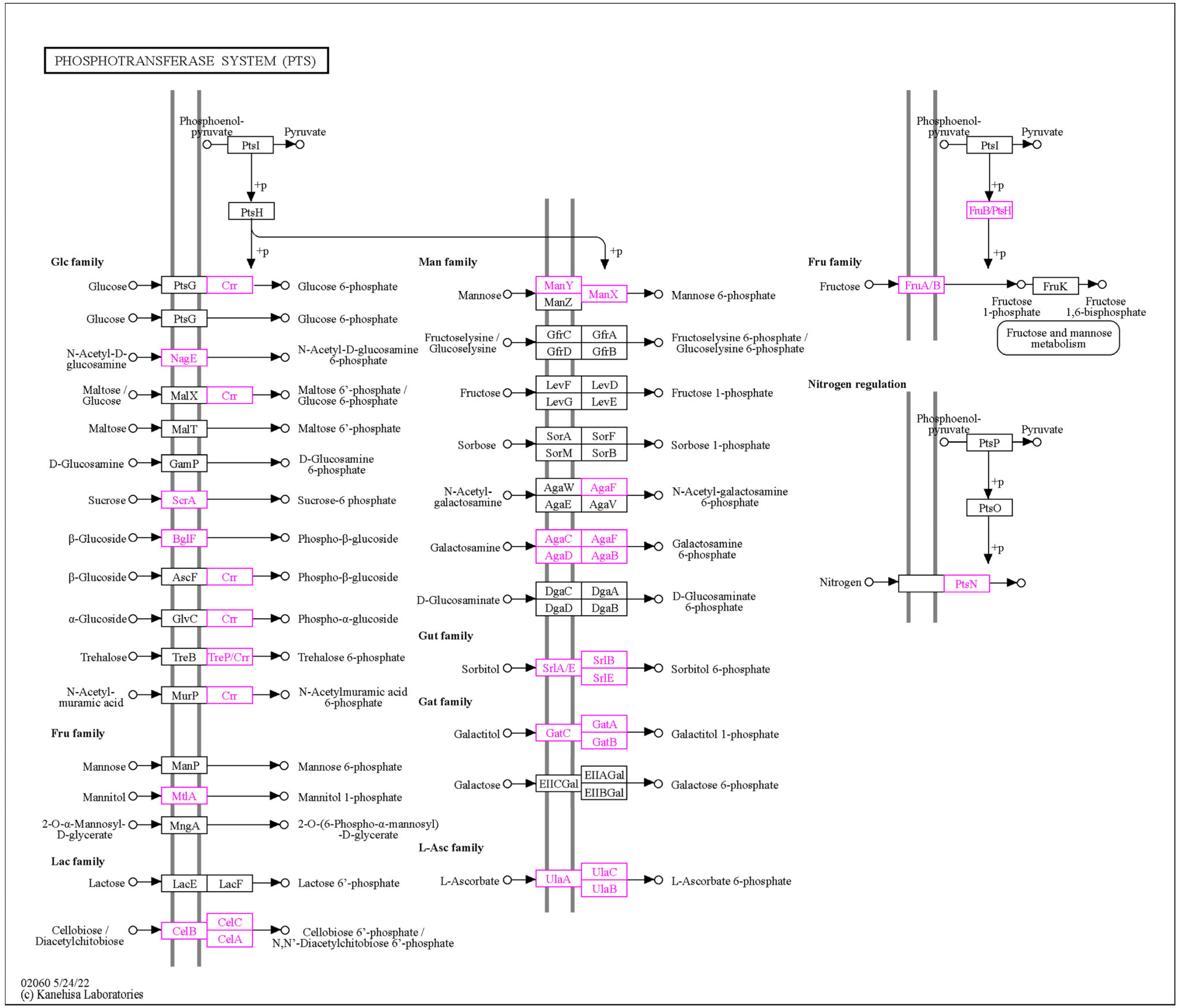
3.1.4. Carbohydrate-Active Enzyme (CAZyme) Annotation
3.1.5. Identification of Probiotic Genes
3.1.6. Antibiotic Resistance Gene (AMR) Prediction
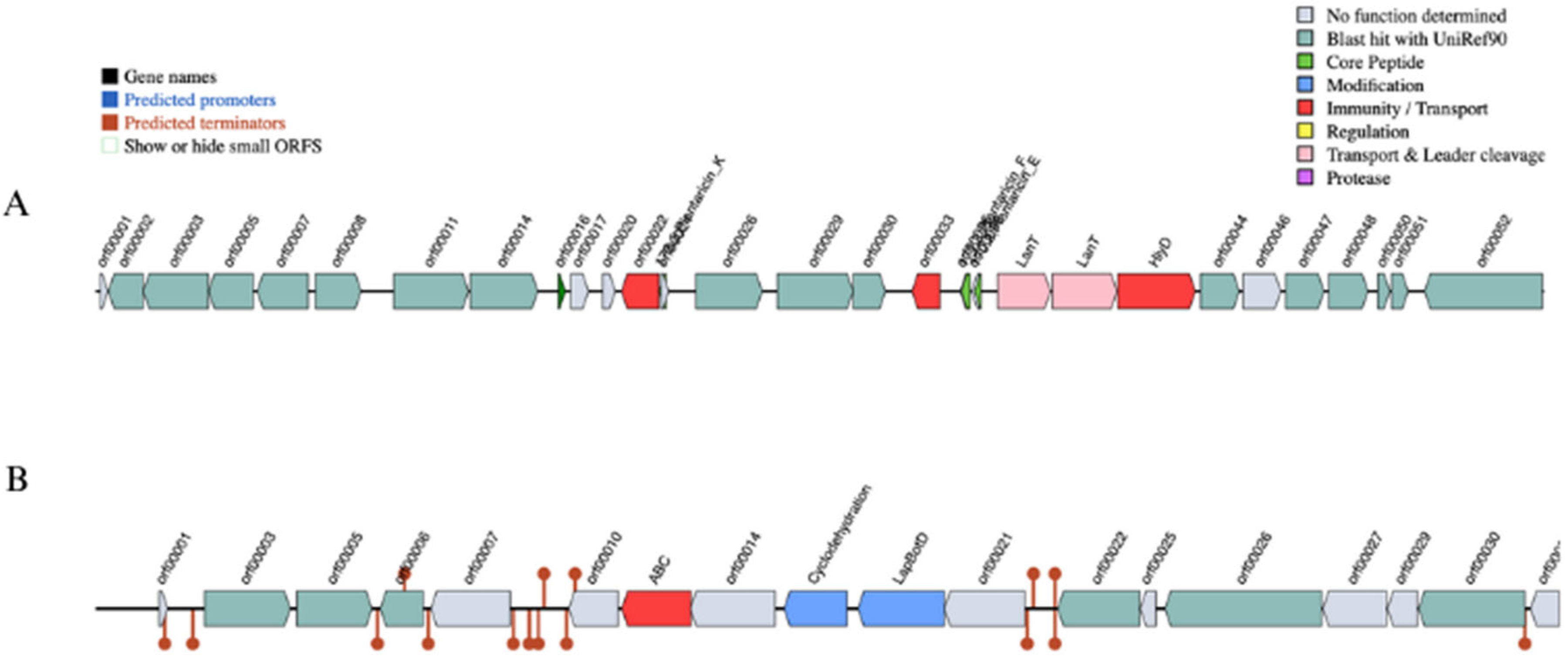
3.1.7. Virulence Factors and Bacteriocin-Encoding Genes
3.1.8. CRISPR–Cas
3.2. Characterization and Functional Analysis of the Strain L. plantarum PU3
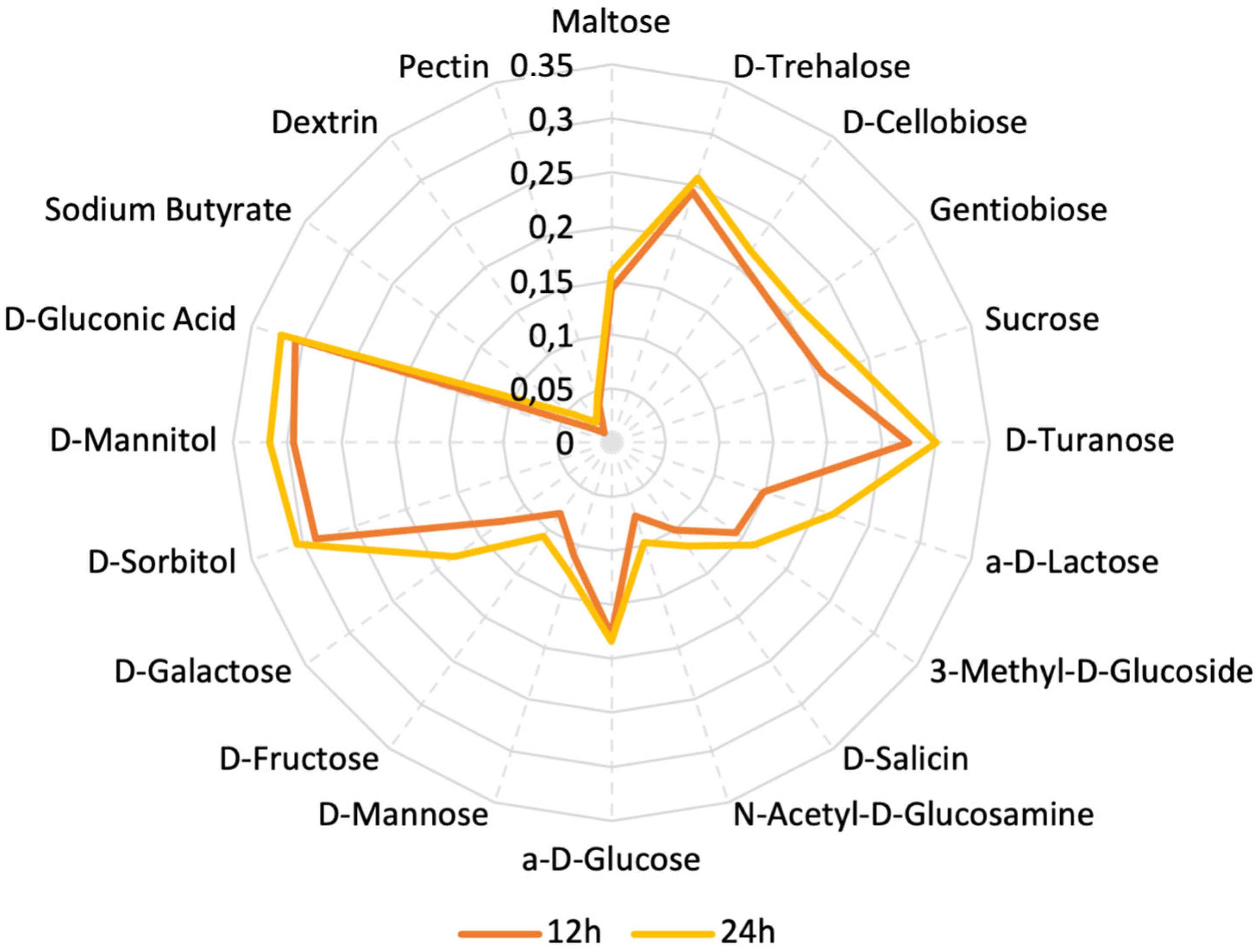
3.2.1. Assimilation of Various Carbon Sources by L. plantarum PU3
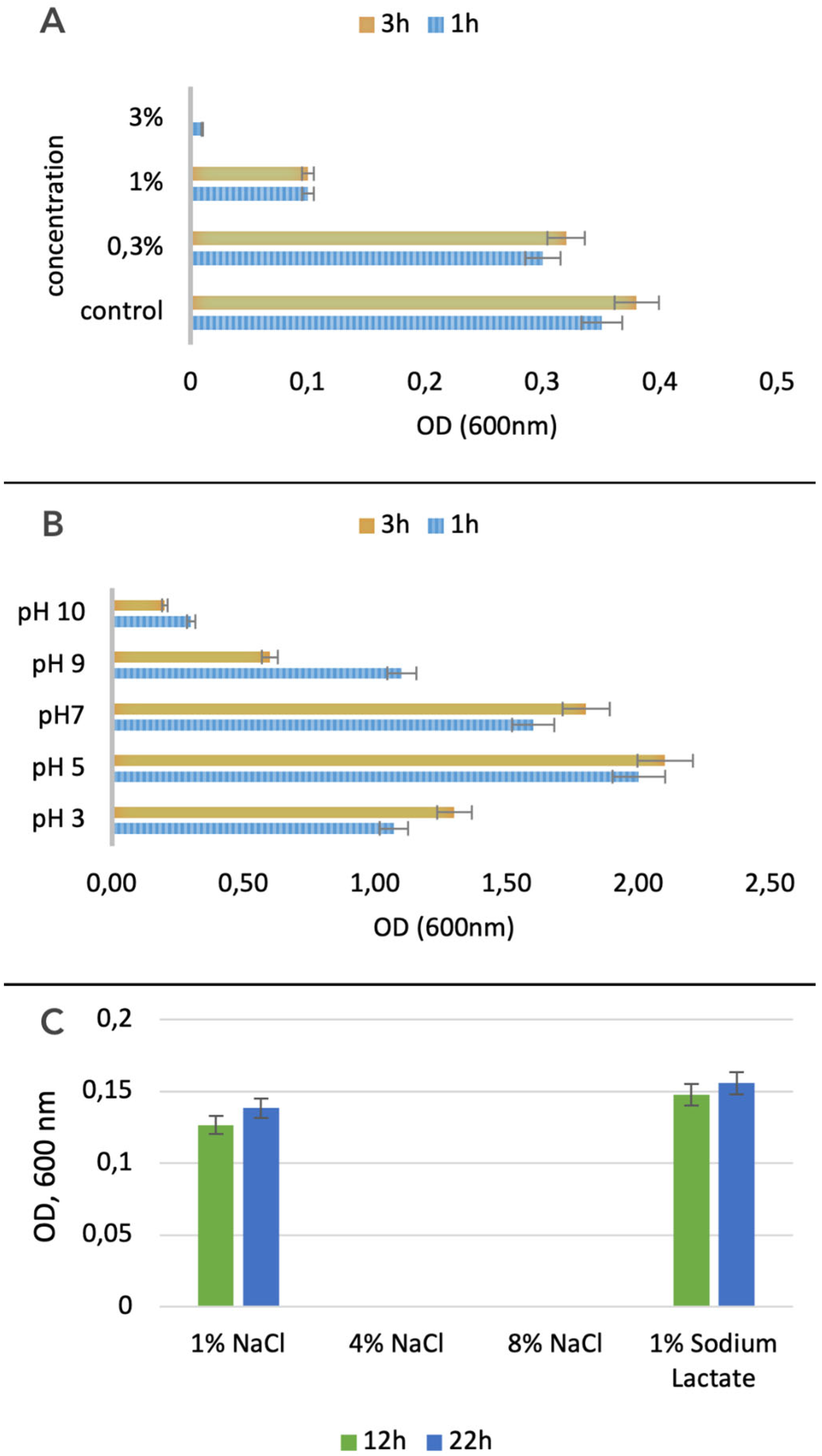
3.2.2. Bile Salts and Acid Tolerance
3.2.3. Osmotic Sensitivity of L. plantarum PU3
3.2.4. Determination of Antibiotic Sensitivity of Strain L. plantarum PU3
- (1)
- Inhibitors of cell wall synthesis: Penicillin (10 µg), Amoxicillin (30 µg), III generation cephalosporins—Ceftriaxone (10 µg);
- (2)
- Protein synthesis inhibitors: aminoglycosides—Amikacin (10 µg), Kanamycin (30 µg), Chloramphenicol (10 µg), macrolides-Erythromycin (15 µg);
- (3)
- Inhibitors of nucleic acid synthesis: quinolones-Ciproflaoxacin (10 µg).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rautava, S. Early Microbial Contact, the Breast Milk Microbiome and Child Health. J. Dev. Orig. Health Dis. 2016, 7, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, H.; Rodríguez, J.M.; Salminen, S.; Szajewska, H. Probiotics in Human Milk and Probiotic Supplementation in Infant Nutrition: A Workshop Report. Br. J. Nutr. 2014, 112, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Cacho, N.T.; Lawrence, R.M. Innate Immunity and Breast Milk. Front. Immunol. 2017, 8, 584. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, S.; Gras-Leguen, C.; Le Vacon, F.; Potel, G.; de La Cochetiere, M.-F. Development of Intestinal Microbiota in Infants and Its Impact on Health. Trends Microbiol. 2013, 21, 167–173. [Google Scholar] [CrossRef]
- McGuire, M.K.; McGuire, M.A. Got Bacteria? The Astounding, Yet Not-so-Surprising, Microbiome of Human Milk. Curr. Opin. Biotechnol. 2017, 44, 63–68. [Google Scholar] [CrossRef]
- Osmanagaoglu, O.; Kiran, F.; Ataoglu, H. Evaluation of in Vitro Probiotic Potential of Pediococcus Pentosaceus OZF Isolated from Human Breast Milk. Probiot. Antimicrob. Proteins 2010, 2, 162–174. [Google Scholar] [CrossRef]
- Filannino, P.; Di Cagno, R.; Gobbetti, M. Metabolic and Functional Paths of Lactic Acid Bacteria in Plant Foods: Get out of the Labyrinth. Curr. Opin. Biotechnol. 2018, 49, 64–72. [Google Scholar] [CrossRef]
- Filannino, P.; De Angelis, M.; Di Cagno, R.; Gozzi, G.; Riciputi, Y.; Gobbetti, M. How Lactobacillus plantarum Shapes Its Transcriptome in Response to Contrasting Habitats. Environ. Microbiol. 2018, 20, 3700–3716. [Google Scholar] [CrossRef]
- Carpi, F.M.; Coman, M.M.; Silvi, S.; Picciolini, M.; Verdenelli, M.C.; Napolioni, V. Comprehensive Pan-genome Analysis of Lactiplantibacillus plantarum Complete Genomes. J. Appl. Microbiol. 2022, 132, 592–604. [Google Scholar] [CrossRef]
- Siezen, R.J.; Tzeneva, V.A.; Castioni, A.; Wels, M.; Phan, H.T.K.; Rademaker, J.L.W.; Starrenburg, M.J.C.; Kleerebezem, M.; Molenaar, D.; van Hylckama Vlieg, J.E.T. Phenotypic and Genomic Diversity of Lactobacillus plantarum Strains Isolated from Various Environmental Niches. Environ. Microbiol. 2010, 12, 758–773. [Google Scholar] [CrossRef]
- de Vries, M.C.; Vaughan, E.E.; Kleerebezem, M.; de Vos, W.M. Lactobacillus plantarum—Survival, Functional and Potential Probiotic Properties in the Human Intestinal Tract. Int. Dairy J. 2006, 16, 1018–1028. [Google Scholar] [CrossRef]
- Villena, J.; Li, C.; Vizoso-Pinto, M.G.; Sacur, J.; Ren, L.; Kitazawa, H. Lactiplantibacillus plantarum as a Potential Adjuvant and Delivery System for the Development of SARS-CoV-2 Oral Vaccines. Microorganisms 2021, 9, 683. [Google Scholar] [CrossRef] [PubMed]
- Behera, S.S.; Ray, R.C.; Zdolec, N. Lactobacillus plantarum with Functional Properties: An Approach to Increase Safety and Shelf-Life of Fermented Foods. Biomed. Res. Int. 2018, 2018, 9361614. [Google Scholar] [CrossRef]
- Fidanza, M.; Panigrahi, P.; Kollmann, T.R. Lactiplantibacillus plantarum–Nomad and Ideal Probiotic. Front. Microbiol. 2021, 12, 712236. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, L.; Rud, I.; Naterstad, K.; Blom, H.; Renckens, B.; Boekhorst, J.; Kleerebezem, M.; van Hijum, S.; Siezen, R.J. Genome Sequence of the Naturally Plasmid-Free Lactobacillus plantarum Strain NC8 (CCUG 61730). J. Bacteriol. 2012, 194, 2391–2392. [Google Scholar] [CrossRef]
- Liu, C.-J.; Wang, R.; Gong, F.-M.; Liu, X.-F.; Zheng, H.-J.; Luo, Y.-Y.; Li, X.-R. Complete Genome Sequences and Comparative Genome Analysis of Lactobacillus plantarum Strain 5-2 Isolated from Fermented Soybean. Genomics 2015, 106, 404–411. [Google Scholar] [CrossRef]
- Leggett, R.M.; Clark, M.D. A World of Opportunities with Nanopore Sequencing. J. Exp. Bot. 2017, 68, 5419–5429. [Google Scholar] [CrossRef]
- Leggett, R.M.; Alcon-Giner, C.; Heavens, D.; Caim, S.; Brook, T.C.; Kujawska, M.; Martin, S.; Peel, N.; Acford-Palmer, H.; Hoyles, L.; et al. Rapid MinION Profiling of Preterm Microbiota and Antimicrobial-Resistant Pathogens. Nat. Microbiol. 2019, 5, 430–442. [Google Scholar] [CrossRef]
- Berbers, B.; Saltykova, A.; Garcia-Graells, C.; Philipp, P.; Arella, F.; Marchal, K.; Winand, R.; Vanneste, K.; Roosens, N.H.C.; De Keersmaecker, S.C.J. Combining Short and Long Read Sequencing to Characterize Antimicrobial Resistance Genes on Plasmids Applied to an Unauthorized Genetically Modified Bacillus. Sci. Rep. 2020, 10, 4310. [Google Scholar] [CrossRef]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef]
- Sanders, C.I.; Ne Ville, C.J.; Orwin, P.M. Complete Genome Sequences of Four Isolated Bacteria from an Undergraduate Microbiology Course Using a Hybrid Assembly Approach. Microbiol. Resour. Announc. 2022, 11, e01022-21. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.L.; Maghini, D.G.; Bhatt, A.S. Complete, Closed Bacterial Genomes from Microbiomes Using Nanopore Sequencing. Nat. Biotechnol. 2020, 38, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Murigneux, V.; Roberts, L.W.; Forde, B.M.; Phan, M.-D.; Nhu, N.T.K.; Irwin, A.D.; Harris, P.N.A.; Paterson, D.L.; Schembri, M.A.; Whiley, D.M.; et al. MicroPIPE: Validating an End-to-End Workflow for High-Quality Complete Bacterial Genome Construction. BMC Genom. 2021, 22, 474. [Google Scholar] [CrossRef]
- Buntin, N.; Hongpattarakere, T.; Ritari, J.; Douillard, F.P.; Paulin, L.; Boeren, S.; Shetty, S.A.; de Vos, W.M. An Inducible Operon Is Involved in Inulin Utilization in Lactobacillus plantarum Strains, as Revealed by Comparative Proteogenomics and Metabolic Profiling. Appl. Environ. Microbiol. 2017, 83, e02402-16. [Google Scholar] [CrossRef]
- Fuhren, J.; Rösch, C.; ten Napel, M.; Schols, H.A.; Kleerebezem, M. Synbiotic Matchmaking in Lactobacillus plantarum: Substrate Screening and Gene-Trait Matching to Characterize Strain-Specific Carbohydrate Utilization. Appl. Environ. Microbiol. 2020, 86, e01081-20. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Yin, R.; Li, X.; Cui, S.; Zhang, H.; Zhao, J.; Chen, W. Comparative Genomic Analysis of Lactiplantibacillus plantarum Isolated from Different Niches. Genes 2021, 12, 241. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Parrello, B.; Butler, R.; Chlenski, P.; Olson, R.; Overbeek, J.; Pusch, G.D.; Vonstein, V.; Overbeek, R. A Machine Learning-Based Service for Estimating Quality of Genomes Using PATRIC. BMC Bioinform. 2019, 20, 486. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST.Org Website and Their Applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. PGAP: Pan-Genomes Analysis Pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A Modular and Extensible Implementation of the RAST Algorithm for Building Custom Annotation Pipelines and Annotating Batches of Genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of Microbial Genomes Using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2019, 48, D517–D525. [Google Scholar] [CrossRef]
- Bonin, N.; Doster, E.; Worley, H.; Pinnell, L.J.; Bravo, J.E.; Ferm, P.; Marini, S.; Prosperi, M.; Noyes, N.; Morley, P.S.; et al. MEGARes and AMR++, v3.0: An Updated Comprehensive Database of Antimicrobial Resistance Determinants and an Improved Software Pipeline for Classification Using High-Throughput Sequencing. Nucleic Acids Res. 2023, 51, D744–D752. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A General Classification Scheme for Bacterial Virulence Factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef]
- Van Heel, A.J.; de Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A User-Friendly Web Server to Thoroughly Mine RiPPs and Bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an Update of CRISRFinder, Includes a Portable Version, Enhanced Performance and Integrates Search for Cas Proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the Integration of Metabolic Annotations and the Reconstruction, Comparison and Analysis of Metabolic Models for Plants, Fungi and Microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef] [PubMed]
- Cottret, L.; Wildridge, D.; Vinson, F.; Barrett, M.P.; Charles, H.; Sagot, M.-F.; Jourdan, F. MetExplore: A Web Server to Link Metabolomic Experiments and Genome-Scale Metabolic Networks. Nucleic Acids Res. 2010, 38, W132–W137. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Zhang, Y.; Zhang, Y.; Liu, Y.; Wang, S.; Dong, X.; Wang, Y.; Zhang, H. Screening of Potential Probiotic Properties of Lactobacillus Fermentum Isolated from Traditional Dairy Products. Food Control 2010, 21, 695–701. [Google Scholar] [CrossRef]
- Ramos, C.L.; Thorsen, L.; Schwan, R.F.; Jespersen, L. Strain-Specific Probiotics Properties of Lactobacillus fermentum, Lactobacillus plantarum and Lactobacillus brevis Isolates from Brazilian Food Products. Food Microbiol. 2013, 36, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Tomar, S.K.; Sangwan, V.; Goswami, P.; Singh, R. Antibiotic Resistance of Lactobacillus sp. Isolated from Commercial Probiotic Preparations. J. Food Saf. 2016, 36, 38–51. [Google Scholar] [CrossRef]
- Umanets, A.; Surono, I.S.; Venema, K. I Am Better than I Look: Genome Based Safety Assessment of the Probiotic Lactiplantibacillus plantarum IS-10506. BMC Genom. 2023, 24, 518. [Google Scholar] [CrossRef]
- Prete, R.; Long, S.L.; Joyce, S.A.; Corsetti, A. Genotypic and Phenotypic Characterization of Food-Associated Lactobacillus plantarum Isolates for Potential Probiotic Activities. FEMS Microbiol. Lett. 2020, 367, fnaa076. [Google Scholar] [CrossRef]
- Xu, H.; Liu, W.; Zhang, W.; Yu, J.; Song, Y.; Menhe, B.; Zhang, H.; Sun, Z. Use of Multilocus Sequence Typing to Infer Genetic Diversity and Population Structure of Lactobacillus plantarum Isolates from Different Sources. BMC Microbiol. 2015, 15, 241. [Google Scholar] [CrossRef]
- Abriouel, H.; Pérez Montoro, B.; Casimiro-Soriguer, C.S.; Pérez Pulido, A.J.; Knapp, C.W.; Caballero Gómez, N.; Castillo-Gutiérrez, S.; Estudillo-Martínez, M.D.; Gálvez, A.; Benomar, N. Insight into Potential Probiotic Markers Predicted in Lactobacillus Pentosus MP-10 Genome Sequence. Front. Microbiol. 2017, 8, 891. [Google Scholar] [CrossRef]
- Albarracin, L.; Raya Tonetti, F.; Fukuyama, K.; Suda, Y.; Zhou, B.; Baillo, A.A.; Fadda, S.; Saavedra, L.; Kurata, S.; Hebert, E.M.; et al. Genomic Characterization of Lactiplantibacillus plantarum Strains Possessing Differential Antiviral Immunomodulatory Activities. Bacteria 2022, 1, 136–160. [Google Scholar] [CrossRef]
- Goel, A.; Halami, P.M.; Tamang, J.P. Genome Analysis of Lactobacillus plantarum Isolated from Some Indian Fermented Foods for Bacteriocin Production and Probiotic Marker Genes. Front. Microbiol. 2020, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, L.C.L.; Aburjaile, F.F.; Sousa, T.D.J.; Felice, A.G.; Soares, S.D.C.; Alcantara, L.C.J.; Azevedo, V.A.D.C. Genomic Characterization of Lactobacillus delbrueckii Strains with Probiotics Properties. Front. Bioinform. 2022, 2, 912795. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Yang, S.-M.; Kim, D.; Kim, H.-Y. Complete Genome Sequencing and Comparative Genomics of Three Potential Probiotic Strains, Lacticaseibacillus casei FBL6, Lacticaseibacillus chiayiensis FBL7, and Lacticaseibacillus zeae FBL8. Front. Microbiol. 2022, 12, 794315. [Google Scholar] [CrossRef]
- Kiousi, D.E.; Efstathiou, C.; Tegopoulos, K.; Mantzourani, I.; Alexopoulos, A.; Plessas, S.; Kolovos, P.; Koffa, M.; Galanis, A. Genomic Insight into Lacticaseibacillus paracasei SP5, Reveals Genes and Gene Clusters of Probiotic Interest and Biotechnological Potential. Front. Microbiol. 2022, 13, 922689. [Google Scholar] [CrossRef]
- Tenea, G.N. Decoding the Gene Variants of Two Native Probiotic Lactiplantibacillus plantarum Strains through Whole-Genome Resequencing: Insights into Bacterial Adaptability to Stressors and Antimicrobial Strength. Genes 2022, 13, 443. [Google Scholar] [CrossRef]
- Deckers-Hebestreit, G.; Altendorf, K. THE F0 F1 -TYPE ATP synthases of bacteria: Structure and Function of the F0 Complex. Annu. Rev. Microbiol. 1996, 50, 791–824. [Google Scholar] [CrossRef]
- Kwak, W.; Kim, K.; Lee, C.; Lee, C.; Kang, J.; Cho, K.; Yoon, S.H.; Kang, D.-K.; Kim, H.; Heo, J.; et al. Comparative Analysis of the Complete Genome of Lactobacillus plantarum GB-LP2 and Potential Candidate Genes for Host Immune System Enhancement. J. Microbiol. Biotechnol. 2016, 26, 684–692. [Google Scholar] [CrossRef]
- Liu, N.; Qin, L.; Miao, S. Regulatory Mechanisms of L-Lactic Acid and Taste Substances in Chinese Acid Rice Soup (Rice-Acid) Fermented with a Lacticaseibacillus paracasei and Kluyveromyces marxianus. Front. Microbiol. 2021, 12, 594631. [Google Scholar] [CrossRef]
- Van Pijkeren, J.-P.; Canchaya, C.; Ryan, K.A.; Li, Y.; Claesson, M.J.; Sheil, B.; Steidler, L.; O’Mahony, L.; Fitzgerald, G.F.; van Sinderen, D.; et al. Comparative and Functional Analysis of Sortase-Dependent Proteins in the Predicted Secretome of Lactobacillus salivarius UCC118. Appl. Environ. Microbiol. 2006, 72, 4143–4153. [Google Scholar] [CrossRef]
- Granato, D.; Bergonzelli, G.E.; Pridmore, R.D.; Marvin, L.; Rouvet, M.; Corthésy-Theulaz, I.E. Cell Surface-Associated Elongation Factor Tu Mediates the Attachment of Lactobacillus johnsonii NCC533 (La1) to Human Intestinal Cells and Mucins. Infect. Immun. 2004, 72, 2160–2169. [Google Scholar] [CrossRef] [PubMed]
- Bergonzelli, G.E.; Granato, D.; Pridmore, R.D.; Marvin-Guy, L.F.; Donnicola, D.; Corthésy-Theulaz, I.E. GroEL of Lactobacillus johnsonii La1 (NCC 533) Is Cell Surface Associated: Potential Role in Interactions with the Host and the Gastric Pathogen Helicobacter Pylori. Infect. Immun. 2006, 74, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Antikainen, J.; Kupannen, V.; Lähteenmäki, K.; Korhonen, T.K. PH-Dependent Association of Enolase and Glyceraldehyde-3-Phosphate Dehydrogenase of Lactobacillus crispatus with the Cell Wall and Lipoteichoic Acids. J. Bacteriol. 2007, 189, 4539–4543. [Google Scholar] [CrossRef] [PubMed]
- Mack, D.R. Extracellular MUC3 Mucin Secretion Follows Adherence of Lactobacillus strains to Intestinal Epithelial Cells in Vitro. Gut 2003, 52, 827–833. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, W.; Dotson, D.L.; Beckstein, O.; Shen, J. Mechanism of PH-Dependent Activation of the Sodium-Proton Antiporter NhaA. Nat. Commun. 2016, 7, 12940. [Google Scholar] [CrossRef]
- Angelin, J.; Kavitha, M. Exopolysaccharides from Probiotic Bacteria and Their Health Potential. Int. J. Biol. Macromol. 2020, 162, 853–865. [Google Scholar] [CrossRef]
- Riaz Rajoka, M.S.; Wu, Y.; Mehwish, H.M.; Bansal, M.; Zhao, L. Lactobacillus Exopolysaccharides: New Perspectives on Engineering Strategies, Physiochemical Functions, and Immunomodulatory Effects on Host Health. Trends Food Sci. Technol. 2020, 103, 36–48. [Google Scholar] [CrossRef]
- Deo, D.; Davray, D.; Kulkarni, R. A Diverse Repertoire of Exopolysaccharide Biosynthesis Gene Clusters in Lactobacillus Revealed by Comparative Analysis in 106 Sequenced Genomes. Microorganisms 2019, 7, 444. [Google Scholar] [CrossRef]
- Madeira, J.-P.; Alpha-Bazin, B.M.; Armengaud, J.; Duport, C. Methionine Residues in Exoproteins and Their Recycling by Methionine Sulfoxide Reductase AB Serve as an Antioxidant Strategy in Bacillus Cereus. Front. Microbiol. 2017, 8, 1342. [Google Scholar] [CrossRef]
- De Clerck, E.; Rodriguez-Diaz, M.; Forsyth, G.; Lebbe, L.; Logan, N.A.; DeVos, P. Polyphasic Characterization of Bacillus coagulans Strains, Illustrating Heterogeneity within This Species, and Emended Description of the Species. Syst. Appl. Microbiol. 2004, 27, 50–60. [Google Scholar] [CrossRef]
- Jiménez, N.; Curiel, J.A.; Reverón, I.; de las Rivas, B.; Muñoz, R. Uncovering the Lactobacillus plantarum WCFS1 Gallate Decarboxylase Involved in Tannin Degradation. Appl. Environ. Microbiol. 2013, 79, 4253–4263. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, Y.; Sasaki, E.; Fujisawa, T.; Osawa, R.o. Genotypic Analyses of Lactobacilli with a Range of Tannase Activities Isolated from Human Feces and Fermented Foods. Syst. Appl. Microbiol. 2004, 27, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, I.; Marcobal, Á.; Muñoz, R. Tannase Activity by Lactic Acid Bacteria Isolated from Grape Must and Wine. Int. J. Food Microbiol. 2004, 96, 199–204. [Google Scholar] [CrossRef]
- Crawley, A.B.; Henriksen, E.D.; Stout, E.; Brandt, K.; Barrangou, R. Characterizing the Activity of Abundant, Diverse and Active CRISPR-Cas Systems in Lactobacilli. Sci. Rep. 2018, 8, 11544. [Google Scholar] [CrossRef]
- Yang, X.; Teng, K.; Su, R.; Li, L.; Zhang, T.; Fan, K.; Zhang, J.; Zhong, J. AcrR and Rex Control Mannitol and Sorbitol Utilization through Their Cross-Regulation of Aldehyde-Alcohol Dehydrogenase (AdhE) in Lactobacillus plantarum. Appl. Environ. Microbiol. 2019, 85, e02035-18. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, B.-C.; Zou, L.-K.; Cheng, J.-G.; Cai, Y.-H.; Kang, J.-P.; Li, B.; Gao, X.-H.; Wang, P.; Xiao, J.-J. Identification and Characterization of Lactic Acid Bacteria from Forest Musk Deer Feces. Afr. J. Microbiol. Res. 2012, 6, 5871–5881. [Google Scholar] [CrossRef]
- Wauters, L.; Van Oudenhove, L.; Accarie, A.; Geboers, K.; Geysen, H.; Toth, J.; Luypaerts, A.; Verbeke, K.; Smokvina, T.; Raes, J.; et al. Lactobacillus rhamnosus CNCM I-3690 Decreases Subjective Academic Stress in Healthy Adults: A Randomized Placebo-Controlled Trial. Gut Microbes 2022, 14, 2031695. [Google Scholar] [CrossRef] [PubMed]
- Van Zyl, W.F.; Deane, S.M.; Dicks, L.M.T. Molecular Insights into Probiotic Mechanisms of Action Employed against Intestinal Pathogenic Bacteria. Gut Microbes 2020, 12, 1831339. [Google Scholar] [CrossRef]
- Kenfack, C.; Kaktcham, P.; Ngoufack, F.; Wang, Y.; Yin, L.; Zhu, T. Screening and Characterization of Putative Probiotic Lactobacillus Strains from Honey Bee Gut (Apis mellifera). J. Adv. Microbiol. 2018, 10, 1–18. [Google Scholar] [CrossRef]
- Ruiz, L.; Margolles, A.; Sánchez, B. Bile Resistance Mechanisms in Lactobacillus and Bifidobacterium. Front. Microbiol. 2013, 4, 396. [Google Scholar] [CrossRef]
- Reale, A.; Di Renzo, T.; Rossi, F.; Zotta, T.; Iacumin, L.; Preziuso, M.; Parente, E.; Sorrentino, E.; Coppola, R. Tolerance of Lactobacillus Casei, Lactobacillus paracasei and Lactobacillus rhamnosus Strains to Stress Factors Encountered in Food Processing and in the Gastro-Intestinal Tract. LWT—Food Sci. Technol. 2015, 60, 721–728. [Google Scholar] [CrossRef]
- Barbosa, J.; Borges, S.; Teixeira, P. Pediococcus acidilactici as a Potential Probiotic to Be Used in Food Industry. Int. J. Food Sci. Technol. 2015, 50, 1151–1157. [Google Scholar] [CrossRef]
- Lovmar, M.; Nilsson, K.; Vimberg, V.; Tenson, T.; Nervall, M.; Ehrenberg, M. The Molecular Mechanism of Peptide-Mediated Erythromycin Resistance. J. Biol. Chem. 2006, 281, 6742–6750. [Google Scholar] [CrossRef]
- ur Rahman, S.; Ali, T.; Ali, I.; Khan, N.A.; Han, B.; Gao, J. The Growing Genetic and Functional Diversity of Extended Spectrum Beta-Lactamases. Biomed. Res. Int. 2018, 2018, 9519718. [Google Scholar] [CrossRef] [PubMed]
- Dicks, L.; Botes, M. Probiotic Lactic Acid Bacteria in the Gastro-Intestinal Tract: Health Benefits, Safety and Mode of Action. Benef. Microbes 2010, 1, 11–29. [Google Scholar] [CrossRef]
- Radovanovic, M.; Kekic, D.; Gajic, I.; Kabic, J.; Jovicevic, M.; Kekic, N.; Opavski, N.; Ranin, L. Potential Influence of Antimicrobial Resistance Gene Content in Probiotic Bacteria on the Gut Resistome Ecosystems. Front. Nutr. 2023, 10, 1054555. [Google Scholar] [CrossRef]
- Montassier, E.; Valdés-Mas, R.; Batard, E.; Zmora, N.; Dori-Bachash, M.; Suez, J.; Elinav, E. Probiotics Impact the Antibiotic Resistance Gene Reservoir along the Human GI Tract in a Person-Specific and Antibiotic-Dependent Manner. Nat. Microbiol. 2021, 6, 1043–1054. [Google Scholar] [CrossRef]
- Courvalin, P. Antibiotic Resistance: The Pros and Cons of Probiotics. Dig. Liver Dis. 2006, 38, S261–S265. [Google Scholar] [CrossRef]
- Lu, N.; Hu, Y.; Zhu, L.; Yang, X.; Yin, Y.; Lei, F.; Zhu, Y.; Du, Q.; Wang, X.; Meng, Z.; et al. DNA Microarray Analysis Reveals That Antibiotic Resistance-Gene Diversity in Human Gut Microbiota Is Age Related. Sci. Rep. 2014, 4, 4302. [Google Scholar] [CrossRef]
- Sharma, P.; Tomar, S.K.; Goswami, P.; Sangwan, V.; Singh, R. Antibiotic Resistance among Commercially Available Probiotics. Food Res. Int. 2014, 57, 176–195. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Size | GC% | Relaxase_Type(S) | Predicted_Mobility | Nearest_Neighbor | Neighbor_Identification |
|---|---|---|---|---|---|---|
| PPU3_1 | 44900 | 39.00 | MOBQ, MOBQ | conjugative | CP015967 | Lactobacillus plantarum |
| PPU3_2 | 42197 | 39.94 | MOBQ, MOBQ | mobilizable | CP035023 | Lactobacillus plantarum |
| PPU3_3 | 40483 | 39.77 | MOBQ | mobilizable | CP025284 | Lactobacillus plantarum subsp. plantarum |
| PPU3_4 | 25867 | 41.09 | MOBP, MOBQ | mobilizable | CP026509 | Lactobacillus plantarum |
| PPU3_5 | 13241 | 39.27 | - | non-mobilizable | CP010527 | Lactobacillus plantarum subsp. plantarum P-8 |
| PPU3_6 | 8689 | 35.93 | - | non-mobilizable | CP005948 | Lactobacillus plantarum subsp. plantarum P-8 |
| PPU3_7 | 8053 | 35.23 | - | non-mobilizable | KT149389 | Lactobacillus plantarum |
| PPU3_8 | 6492 | 35.32 | - | non-mobilizable | CP015127 | Lactobacillus plantarum |
| PPU3_9 | 3512 | 37.30 | MOBV | mobilizable | JX174167 | Lactobacillus plantarum |
| Probiotic Activity | Genes |
|---|---|
| Acid and bile tolerance | rpsS, ppk, ackA_1, ackA_2, fabH_1, fabH_2, pgm, pepF, arcB, copA, dnaK, eno, pgk, dnaJ, groS/groES, grpE, htrA |
| Acid stress | atpA, atpB, atpC, atpD, atpE, atpF, atpG, atpH, clpB, clpP, dltC, gap, groL/groEL, ldh_1, ldh_2, ldh_3, ldh_4, pgi, plsC, pyk, recA, relA, tpiA, yjbM, ywaC, nhaC, lepA_1, lepA_2, |
| Bile resistance | argS, bsh_1, bsh_2, bsh_3, dps, glf_1, glf_2, glnA, nagB/gnp, oppA_1, oppA_2, oppA_3, oppA_4, pyrG, rplE, rplF, rpsC, rpsE, ppaC, cfa_1, cfa_2, cbh, celB |
| Biofilm formation | luxS, comC_1, comC_2, comD_1, comD_2, comE |
| Adhesion | exoA, lspA, PrtF, tuf, gpr, gapA, bgaB, epsA, epsB, pgaC_1, pgaC_2, pgaC_3, EpsC_1, EpsC_2, glnH, hsp33/hslO, srtA, dacC_1, dacC_2, cpb_1, cpb_2, EpsF_1, EpsF_2 |
| Oxidative stress | fnr, nrdH, gpx, gsr_1, gsr_2, gsr_3, mntH_1, mntH_2, mntB, mntC, ndh_1, ndh_2, npr_1, npr_2, poxL_1, poxL_2, poxL_3, poxL_4, trxA, trxB, msrA_1, msrA_2, msrA_3, msrB, msrC, tpx, gor |
| Temperature stress | cspA_1, cspA_2, cspA_3, cspA_4, rnr, clpC, clpL, htpX, hrcA, hslV, HSP20, ctsR |
| Immunomodulation | dltD, dltC_1, dltC_2, dltB |
| Ionic and heavy metal stress | zntA, czcD, corA_1, corA_2 |
| Osmotic stress | glpF_1, glpF_2, glpF_3, opuC, opuCA, opuBD_1, opuBD_2, opuA_1, opuA_2 |
| Antibiotic Discs | L. plantarum PU3 | |
|---|---|---|
| CLSI a | Mean ± Standard Deviation | |
| AK (10 µg) | R | b |
| AMC (30 µg) | S | 29.300 ± 0.500 |
| CIP (10 µg) | R | b |
| C (10 µg) | S | 25.364 ± 0.350 |
| E (15 µg) | R | b |
| CRO (10 µg) | R | b |
| K (30 µg) | S | 24.300 ± 0.150 |
| P (10 µg) | S | 20.435 ± 0.210 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollova, D.; Gozmanova, M.; Apostolova, E.; Yahubyan, G.; Iliev, I.; Baev, V. Illuminating the Genomic Landscape of Lactiplantibacillus plantarum PU3—A Novel Probiotic Strain Isolated from Human Breast Milk, Explored through Nanopore Sequencing. Microorganisms 2023, 11, 2440. https://doi.org/10.3390/microorganisms11102440
Mollova D, Gozmanova M, Apostolova E, Yahubyan G, Iliev I, Baev V. Illuminating the Genomic Landscape of Lactiplantibacillus plantarum PU3—A Novel Probiotic Strain Isolated from Human Breast Milk, Explored through Nanopore Sequencing. Microorganisms. 2023; 11(10):2440. https://doi.org/10.3390/microorganisms11102440
Chicago/Turabian StyleMollova, Daniela, Mariyana Gozmanova, Elena Apostolova, Galina Yahubyan, Ilia Iliev, and Vesselin Baev. 2023. "Illuminating the Genomic Landscape of Lactiplantibacillus plantarum PU3—A Novel Probiotic Strain Isolated from Human Breast Milk, Explored through Nanopore Sequencing" Microorganisms 11, no. 10: 2440. https://doi.org/10.3390/microorganisms11102440
APA StyleMollova, D., Gozmanova, M., Apostolova, E., Yahubyan, G., Iliev, I., & Baev, V. (2023). Illuminating the Genomic Landscape of Lactiplantibacillus plantarum PU3—A Novel Probiotic Strain Isolated from Human Breast Milk, Explored through Nanopore Sequencing. Microorganisms, 11(10), 2440. https://doi.org/10.3390/microorganisms11102440