



New Insights into the Effect of Fipronil on the Soil Bacterial Community

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
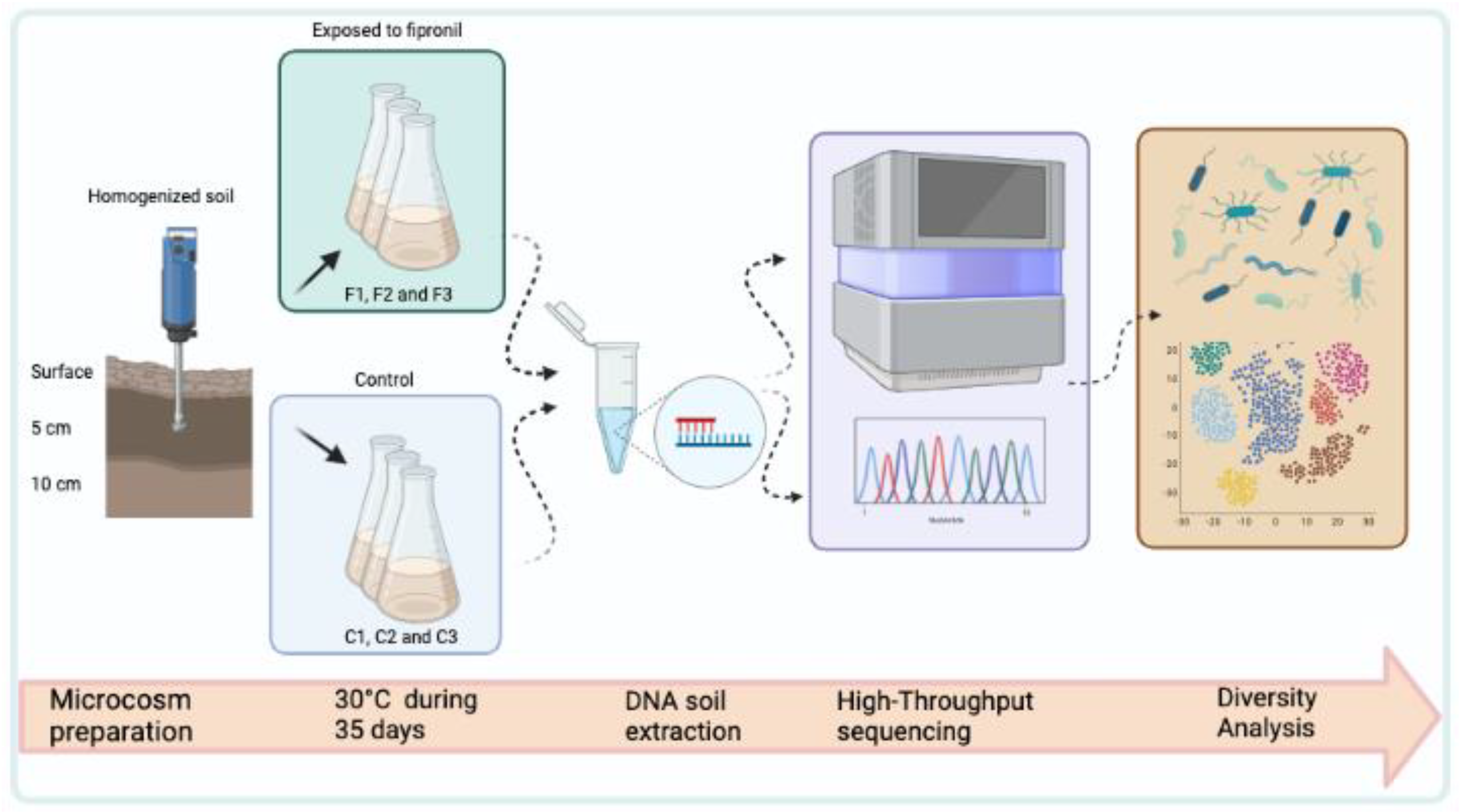
2.1. Soil Sampling and Microcosm
2.2. DNA Extraction and Metagenomic Library Construction
2.3. High-Throughput Sequencing
2.4. Diversity Analysis
2.5. Statistical Analysis
3. Results
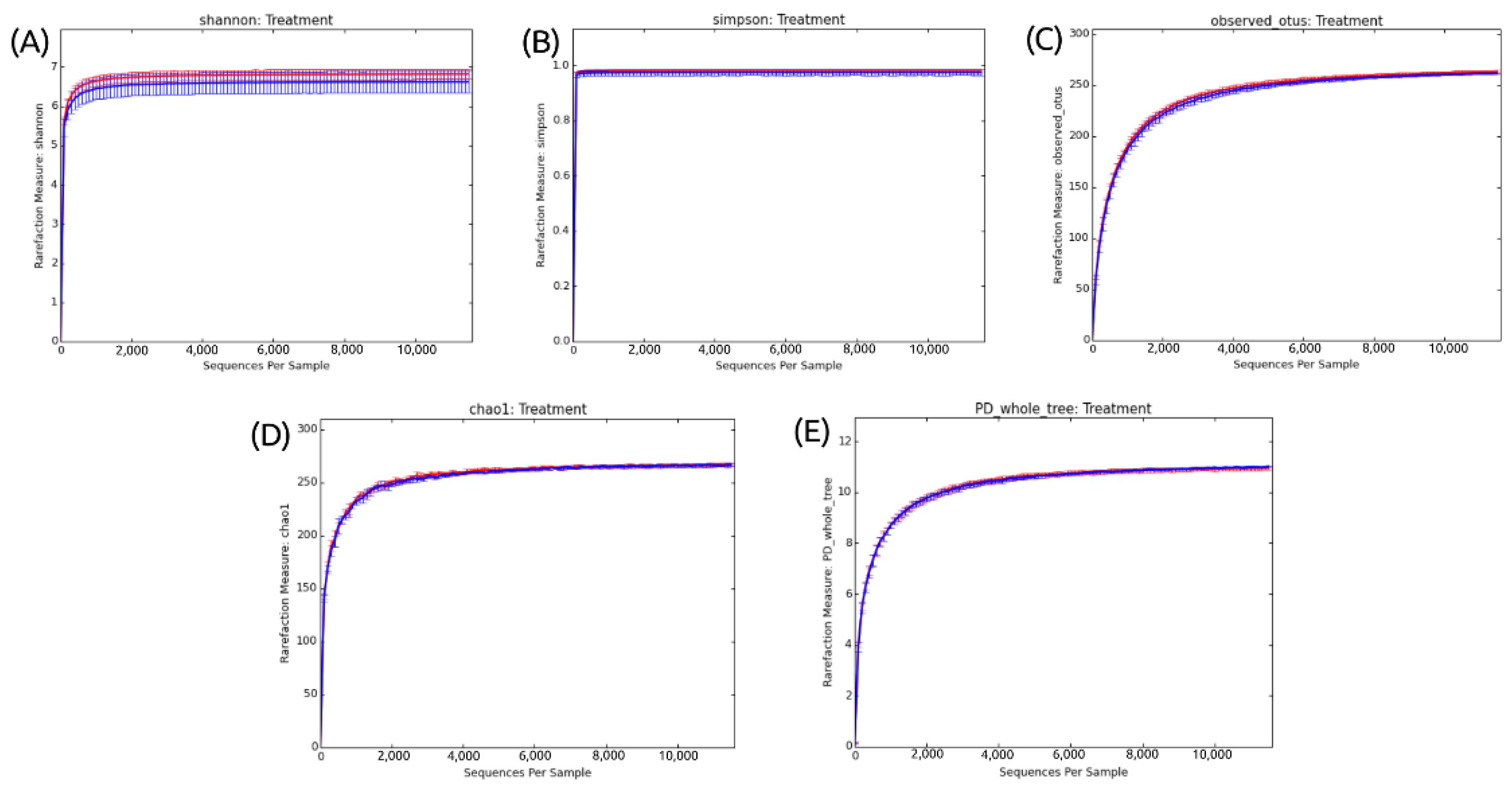
3.1. General Information about Sampling and Sequencing
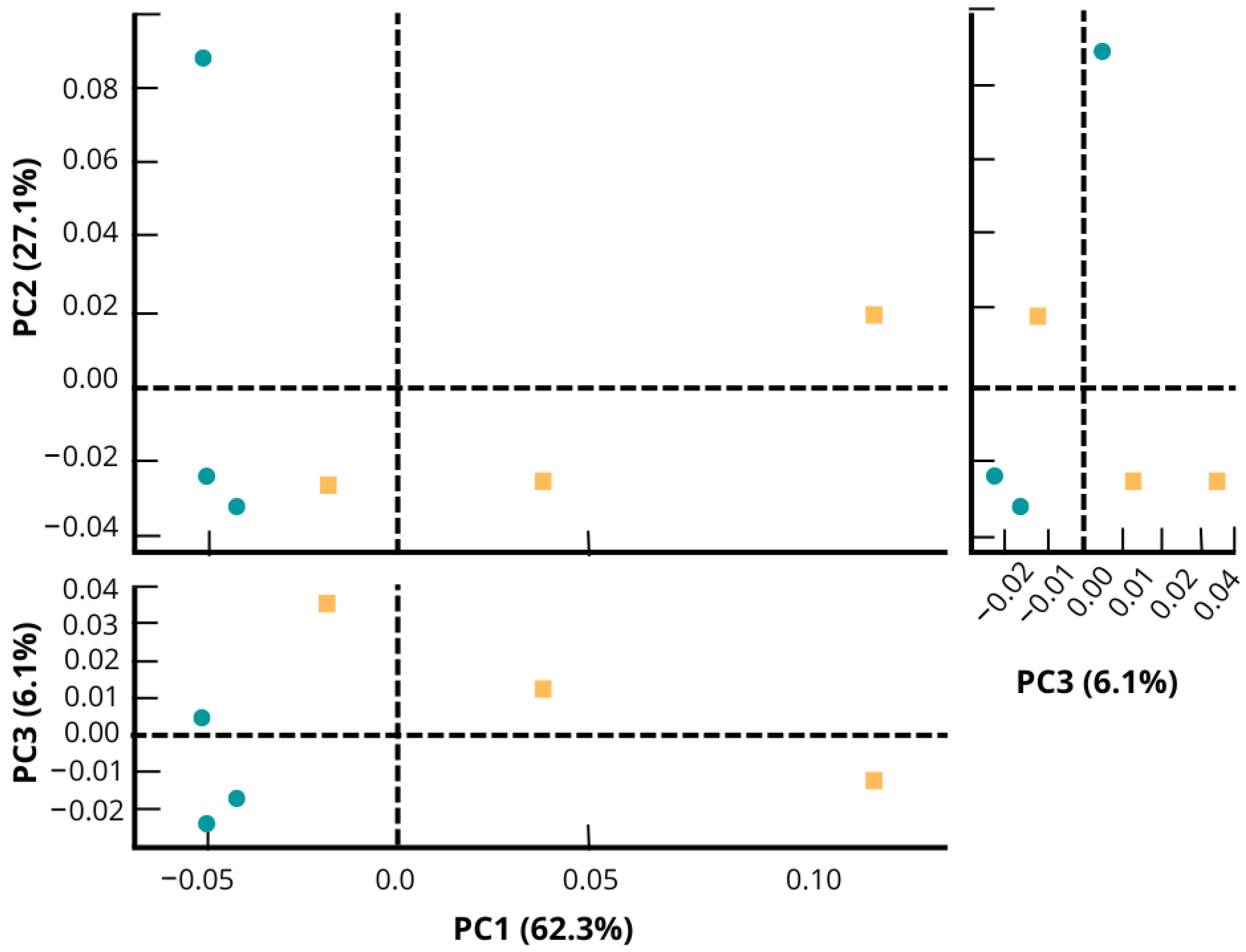
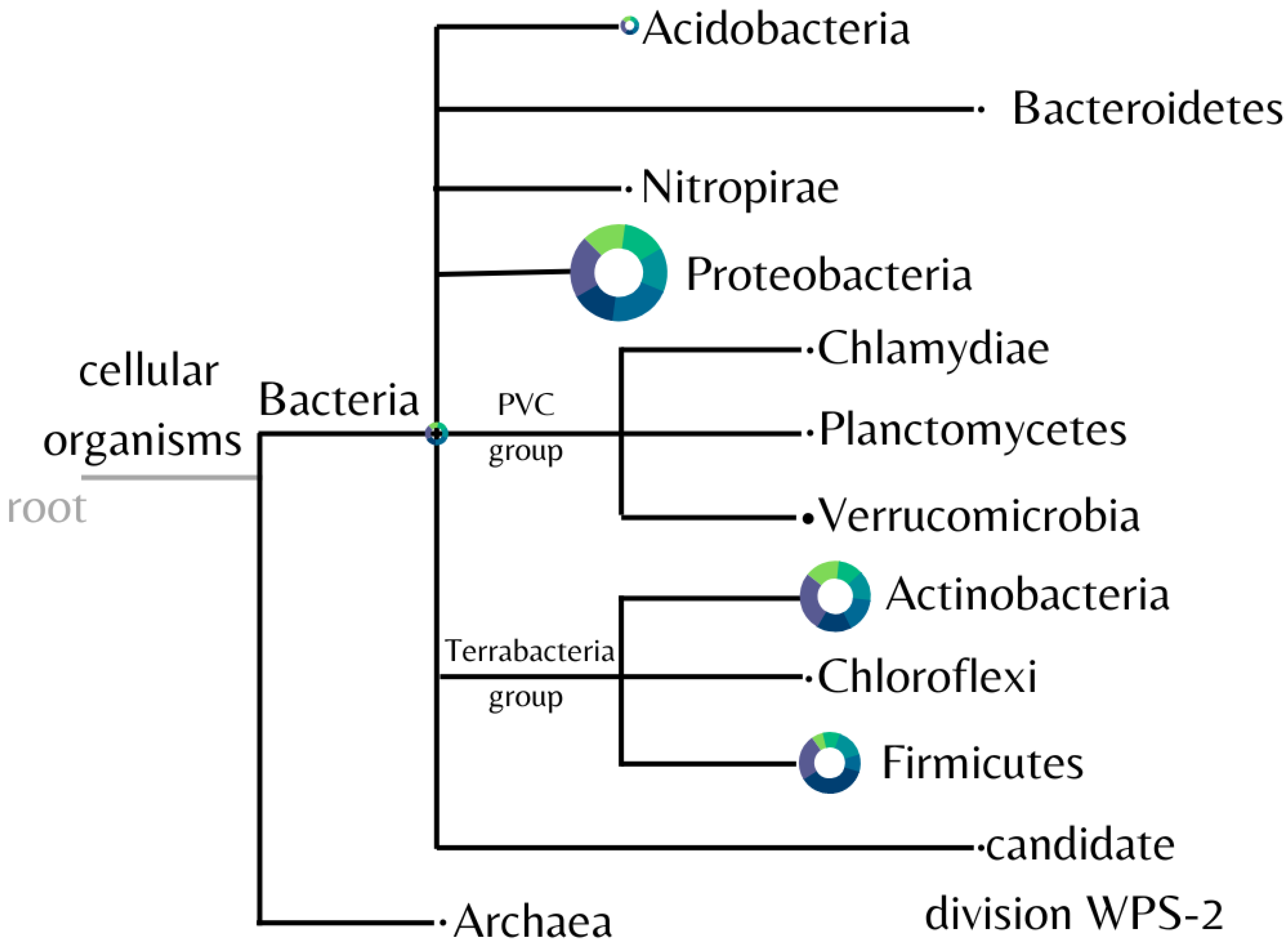
3.2. Composition of the Microbial Community
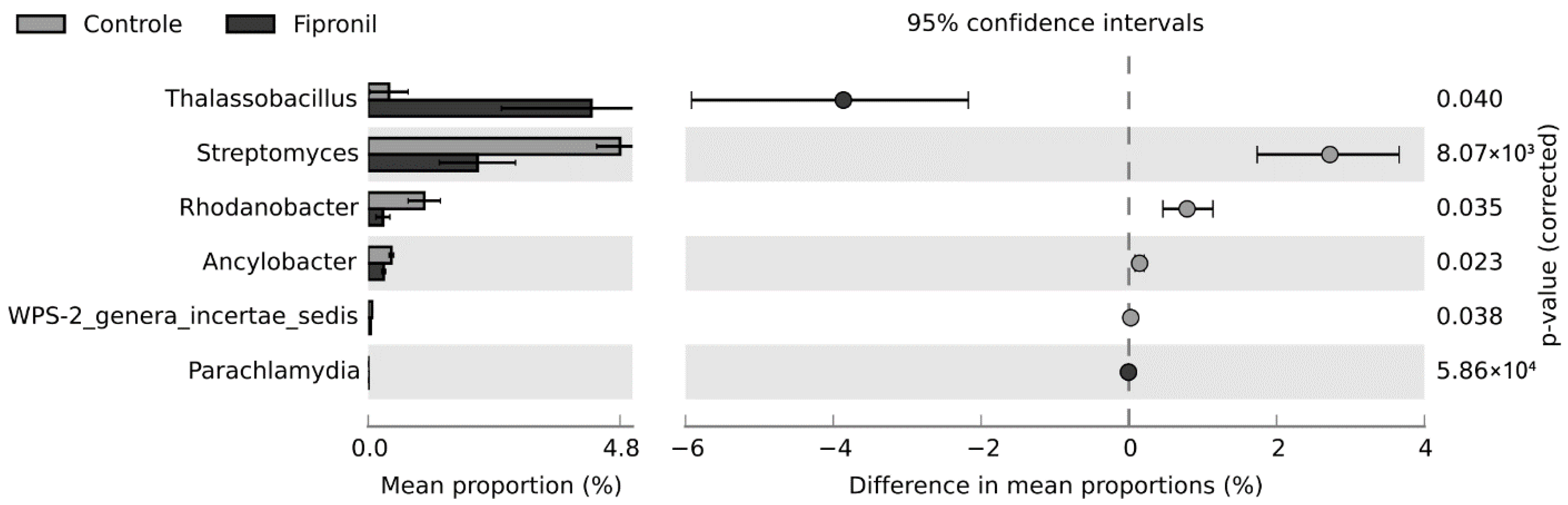
3.3. Statistically Significant Changes
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gonçalves, S.; Vasconcelos, M.W.; Mota, T.F.M.; Lopes, J.M.H.; Guimaraes, L.J.; Miglioranza, K.S.B.; Ghisi, N.d.C. Identifying Global Trends and Gaps in Research on Pesticide Fipronil: A Scientometric Review. Environ. Sci. Pollut. Res. 2022, 29, 79111–79125. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Ferreira, M.; Maleski, A.L.A.; Balan-Lima, L.; Bernardo, J.T.G.; Hipolito, L.M.; Seni-Silva, A.C.; Batista-Filho, J.; Falcao, M.A.P.; Lima, C. Impact of Pesticides on Human Health in the Last Six Years in Brazil. Int. J. Environ. Res. Public Health 2022, 19, 3198. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.H.; Koshlukova, S. Comparative Toxicity of Endosulfan and Fipronil Insecticides: Utilizing in Vivo and in Vitro Data. J. Med. Toxicol. Clin. Forensic Med. 2016, 1, 1. [Google Scholar] [CrossRef][Green Version]
- Gibbons, D.; Morrissey, C.; Mineau, P. A Review of the Direct and Indirect Effects of Neonicotinoids and Fipronil on Vertebrate Wildlife. Environ. Sci. Pollut. Res. 2015, 22, 103–118. [Google Scholar] [CrossRef]
- De Oliveira Silva, R.; Scorza Júnior, R.P.; Bonfá, M.R.L.; Campanari, M.F.Z.; Mendes, I.d.C. Degradation and Sorption of Fipronil and Atrazine in Latossols with Organic Residues from Sugarcane Crop. Ciência Rural 2016, 46, 1172–1177. [Google Scholar] [CrossRef][Green Version]
- Kanat, Ö.N.; Selmanoğlu, G. Neurotoxic Effect of Fipronil in Neuroblastoma SH-SY5Y Cell Line. Neurotox. Res. 2020, 37, 30–40. [Google Scholar] [CrossRef]
- Romero, A.; Ramos, E.; Ares, I.; Castellano, V.; Martínez, M.; Martínez-Larrañaga, M.R.; Anadón, A.; Martínez, M.A. Fipronil Sulfone Induced Higher Cytotoxicity than Fipronil in SH-SY5Y Cells: Protection by Antioxidants. Toxicol. Lett. 2016, 252, 42–49. [Google Scholar] [CrossRef]
- Vidau, C.; González-Polo, R.A.; Niso-Santano, M.; Gómez-Sánchez, R.; Bravo-San Pedro, J.M.; Pizarro-Estrella, E.; Blasco, R.; Brunet, J.-L.; Belzunces, L.P.; Fuentes, J.M. Fipronil Is a Powerful Uncoupler of Oxidative Phosphorylation That Triggers Apoptosis in Human Neuronal Cell Line SHSY5Y. Neurotoxicology 2011, 32, 935–943. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, S.; Yan, Y.; Lin, F.; Zhang, L.; Zhao, W.; Zhao, C.; Xu, H. Design, Synthesis, and Insecticidal Activity of 5, 5-Disubstituted 4, 5-Dihydropyrazolo [1, 5-a] Quinazolines as Novel Antagonists of GABA Receptors. J. Agric. Food Chem. 2020, 68, 15005–15014. [Google Scholar] [CrossRef]
- Tian, Y.; Gao, Y.; Chen, Y.; Liu, G.; Ju, X. Identification of the Fipronil Resistance Associated Mutations in Nilaparvata lugens GABA Receptors by Molecular Modeling. Molecules 2019, 24, 4116. [Google Scholar] [CrossRef]
- Godinho, A.F.; de Oliveira Souza, A.C.; Carvalho, C.C.; Horta, D.F.; de Fraia, D.; Anselmo, F.; Chaguri, J.L.; Faria, C.A. Memory Impairment Due to Fipronil Pesticide Exposure Occurs at the GABAA Receptor Level, in Rats. Physiol. Behav. 2016, 165, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Conte, F.M.; Cestonaro, L.V.; Piton, Y.V.; Guimarães, N.; Garcia, S.C.; da Silva, D.D.; Arbo, M.D. Toxicity of Pesticides Widely Applied on Soybean Cultivation: Synergistic Effects of Fipronil, Glyphosate and Imidacloprid in HepG2 Cells. Toxicol. Vitr. 2022, 84, 105446. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, P.R.; Bechara, G.H.; Denardi, S.E.; Oliveira, R.J.; Mathias, M.I.C. Cytotoxicity of Fipronil on Mice Liver Cells. Microsc. Res. Tech. 2012, 75, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Mohafrash, M.M. Sub-Chronic Exposure to Fipronil Induced Oxidative Stress, Biochemical and Histotopathological Changes in the Liver and Kidney of Male Albino Rats. Toxicol. Rep. 2015, 2, 775–784. [Google Scholar]
- Mandal, K.; Singh, B.; Jariyal, M.; Gupta, V.K. Microbial Degradation of Fipronil by Bacillus Thuringiensis. Ecotoxicol. Environ. Saf. 2013, 93, 87–92. [Google Scholar] [CrossRef]
- Vogel, T.M.; Simonet, P.; Jansson, J.K.; Hirsch, P.R.; Tiedje, J.M.; van Elsas, J.D.; Bailey, M.J.; Nalin, R.; Philippot, L. TerraGenome: A Consortium for the Sequencing of a Soil Metagenome. Nat. Rev. Microbiol. 2009, 7, 252. [Google Scholar] [CrossRef]
- Chu, H.; Gao, G.-F.; Ma, Y.; Fan, K.; Delgado-Baquerizo, M. Soil Microbial Biogeography in a Changing World: Recent Advances and Future Perspectives. mSystems 2020, 5, e00803-19. [Google Scholar] [CrossRef]
- Coughlan, L.M.; Cotter, P.D.; Hill, C.; Alvarez-Ordóñez, A. Biotechnological Applications of Functional Metagenomics in the Food and Pharmaceutical Industries. Front. Microbiol. 2015, 6, 672. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, D.; Parales, R.E.; Jiang, J. Oxygenases as Powerful Weapons in the Microbial Degradation of Pesticides. Annu. Rev. Microbiol. 2022, 76, 91758. [Google Scholar] [CrossRef]
- Singh, N.S.; Sharma, R.; Singh, S.K.; Singh, D.K. A Comprehensive Review of Environmental Fate and Degradation of Fipronil and Its Toxic Metabolites. Environ. Res. 2021, 199, 111316. [Google Scholar] [CrossRef]
- Zhou, Z.; Wu, X.; Lin, Z.; Pang, S.; Mishra, S.; Chen, S. Biodegradation of Fipronil: Current State of Mechanisms of Biodegradation and Future Perspectives. Appl. Microbiol. Biotechnol. 2021, 105, 7695–7708. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, S.A.; Chen, W.Y.; Wong, T.; Kanakamedala, B.S.; Deveson, I.W.; Ongley, S.E.; Santini, N.S.; Marcellin, E.; Smith, M.A.; Nielsen, L.K. Synthetic Microbe Communities Provide Internal Reference Standards for Metagenome Sequencing and Analysis. Nat. Commun. 2018, 9, 3096. [Google Scholar] [CrossRef] [PubMed]
- Francioli, D.; Lentendu, G.; Lewin, S.; Kolb, S. DNA Metabarcoding for the Characterization of Terrestrial Microbiota—Pitfalls and Solutions. Microorganisms 2021, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kumar, M.; Kumar, J.; Ahmad, V.; Pandey, R.; Chauhan, N.S. Systemic Analysis of Soil Microbiome Deciphers Anthropogenic Influence on Soil Ecology and Ecosystem Functioning. Int. J. Environ. Sci. Technol. 2017, 14, 2229–2238. [Google Scholar] [CrossRef]
- Hemmat-Jou, M.H.; Safari-Sinegani, A.A.; Mirzaie-Asl, A.; Tahmourespour, A. Analysis of Microbial Communities in Heavy Metals-Contaminated Soils Using the Metagenomic Approach. Ecotoxicology 2018, 27, 1281–1291. [Google Scholar] [CrossRef]
- Almeida, O.G.G.; de Martinis, E.C.P. Bioinformatics Tools to Assess Metagenomic Data for Applied Microbiology. Appl. Microbiol. Biotechnol. 2019, 103, 69–82. [Google Scholar] [CrossRef]
- Mandal, K.; Singh, B.; Jariyal, M.; Gupta, V.K. Bioremediation of Fipronil by a Bacillus firmus Isolate from Soil. Chemosphere 2014, 101, 55–60. [Google Scholar] [CrossRef]
- Uniyal, S.; Paliwal, R.; Sharma, R.K.; Rai, J.P.N. Degradation of Fipronil by Stenotrophomonas acidaminiphila Isolated from Rhizospheric Soil of Zea Mays. 3 Biotech 2016, 6, 48. [Google Scholar] [CrossRef]
- Elizaquível, P.; Pérez-Cataluña, A.; Yépez, A.; Aristimuño, C.; Jiménez, E.; Cocconcelli, P.S.; Vignolo, G.; Aznar, R. Pyrosequencing vs. Culture-Dependent Approaches to Analyze Lactic Acid Bacteria Associated to Chicha, a Traditional Maize-Based Fermented Beverage from Northwestern Argentina. Int. J. Food Microbiol. 2015, 198, 9–18. [Google Scholar] [CrossRef]
- Bonfá, M.R.L.; Pereira, R.M.; Scorza Júnior, R.P.; Prado, C.C.A.D. Biodegradation of pesticides in Brazil and other tropical countries: Experimental and in silico studies. In Microbes and Enzymes in Soil Health and Bioremediation; Springer: Berlin/Heidelberg, Germany, 2019; pp. 277–304. [Google Scholar]
- Silva, C.M.M.S.; Roque, M.R.A.; Melo, I.S. Microbiologia Ambiental: Manual de Laboratório; Embrapa Meio Ambiente: Jaguariuna, Brazil, 2000. [Google Scholar]
- Quince, C.; Lanzen, A.; Davenport, R.J.; Turnbaugh, P.J. Removing Noise from Pyrosequenced Amplicons. BMC Bioinform. 2011, 12, 38. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality Control and Preprocessing of Metagenomic Datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Pylro, V.S.; Roesch, L.F.W.; Morais, D.K.; Clark, I.M.; Hirsch, P.R.; Tótola, M.R. Data Analysis for 16S Microbial Profiling from Different Benchtop Sequencing Platforms. J. Microbiol. Methods 2014, 107, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F. The Biological Observation Matrix (BIOM) Format or: How I Learned to Stop Worrying and Love the Ome-Ome. Gigascience 2012, 1, 2047X–2217X. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pẽa, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of RRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S RRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A Flexible Tool for Aligning Sequences to a Template Alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition-Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.B.; Forsythe, A.B. Robust Tests for the Equality of Variances. J. Am. Stat. Assoc. 1974, 69, 364–367. [Google Scholar] [CrossRef]
- Shapiro, S.S.; Wilk, M.B. An Analysis of Variance Test for Normality (Complete Samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G.I. Microbiome Helper: A Custom and Streamlined Workflow for Microbiome Research. mSystems 2017, 2, e00127-16. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical Methods for Detecting Differentially Abundant Features in Clinical Metagenomic Samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R. Predictive Functional Profiling of Microbial Communities Using 16S RRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Souza, R.C.; Mendes, I.C.; Reis-Junior, F.B.; Carvalho, F.M.; Nogueira, M.A.; Vasconcelos, A.T.R.; Vicente, V.A.; Hungria, M. Shifts in Taxonomic and Functional Microbial Diversity with Agriculture: How Fragile Is the Brazilian Cerrado? BMC Microbiol. 2016, 16, 42. [Google Scholar] [CrossRef]
- Procópio, L.; Barreto, C. The Soil Microbiomes of the Brazilian Cerrado. J. Soils Sediments 2021, 21, 2327–2342. [Google Scholar] [CrossRef]
- de Souza, L.C.; Procópio, L. The Profile of the Soil Microbiota in the Cerrado Is Influenced by Land Use. Appl. Microbiol. Biotechnol. 2021, 105, 4791–4803. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.S.F.; de Souza Oliveira, L.M.; Melo, V.M.M.; Antunes, J.E.L.; Araujo, F.F.; Mendes, L.W. Distinct Taxonomic Composition of Soil Bacterial Community across a Native Gradient of Cerrado-Ecotone-Caatinga. Appl. Soil Ecol. 2021, 161, 103874. [Google Scholar] [CrossRef]
- Walder, F.; Schmid, M.W.; Riedo, J.; Valzano-Held, A.Y.; Banerjee, S.; Büchi, L.; Bucheli, T.D.; van der Heijden, M.G.A. Soil Microbiome Signatures Are Associated with Pesticide Residues in Arable Landscapes. Soil Biol. Biochem. 2022, 174, 108830. [Google Scholar] [CrossRef]
- Rodríguez-Valdecantos, G.; Manzano, M.; Sánchez, R.; Urbina, F.; Hengst, M.B.; Lardies, M.A.; Ruz, G.A.; González, B. Early Successional Patterns of Bacterial Communities in Soil Microcosms Reveal Changes in Bacterial Community Composition and Network Architecture, Depending on the Successional Condition. Appl. Soil Ecol. 2017, 120, 44–54. [Google Scholar] [CrossRef]
- At, K.; Karthikeyan, S. Occurrence and Microbial Degradation of Fipronil Residues in Tropical Highland Rhizosphere Soils of Kerala, India. Soil Sediment Contam. Int. J. 2019, 28, 360–379. [Google Scholar] [CrossRef]
- Abraham, J.; Gajendiran, A. Biodegradation of Fipronil and Its Metabolite Fipronil Sulfone by Streptomyces Rochei Strain AJAG7 and Its Use in Bioremediation of Contaminated Soil. Pestic. Biochem. Physiol. 2019, 155, 90–100. [Google Scholar] [CrossRef]
- Cappelini, L.T.D.; Alberice, J.V.; Eugênio, P.F.M.; Pozzi, E.; Urbaczek, A.C.; Diniz, L.G.R.; Carrilho, E.N.V.M.; Carrilho, E.; Vieira, E.M. Burkholderia thailandensis: The Main Bacteria Biodegrading Fipronil in Fertilized Soil with Assessment by a QuEChERS/GC-MS Method. J. Braz. Chem. Soc. 2018, 29, 1934–1943. [Google Scholar] [CrossRef]
- Ahemad, M.; Khan, M.S. Insecticide-tolerant and Plant-growth-promoting Rhizobium Improves the Growth of Lentil (Lens esculentus) in Insecticide-stressed Soils. Pest. Manag. Sci. 2011, 67, 423–429. [Google Scholar] [CrossRef]
- Gangola, S.; Sharma, A.; Joshi, S.; Bhandari, G.; Prakash, O.; Govarthanan, M.; Kim, W.; Bhatt, P. Novel mechanism and degradation kinetics of pesticides mixture using Bacillus sp. strain 3C in contaminated sites. Pestic. Biochem. Physiol. 2022, 181, 104996. [Google Scholar] [CrossRef]
- Pacchioni, R.G.; Carvalho, F.M.; Thompson, C.E.; Faustino, A.L.F.; Nicolini, F.; Pereira, T.S.; Silva, R.C.B.; Cantão, M.E.; Gerber, A.; Vasconcelos, A.T.R. Taxonomic and Functional Profiles of Soil Samples from Atlantic Forest and Caatinga Biomes in Northeastern Brazil. Microbiologyopen 2014, 3, 299–315. [Google Scholar] [CrossRef]
- Tomazini, R.; Saia, F.T.; van der Zaan, B.; Grosseli, G.M.; Fadini, P.S.; de Oliveira, R.G.M.; Gregoracci, G.B.; Mozetto, A.; van Vugt-Lussenburg, B.; Brouwer, A. Biodegradation of Fipronil: Transformation Products, Microbial Characterisation and Toxicity Assessment. Water Air Soil Pollut. 2021, 232, 123. [Google Scholar] [CrossRef]
- Ahemad, M.; Khan, M.S. Effects of Pesticides on Plant Growth Promoting Traits of Mesorhizobium Strain MRC4. J. Saudi Soc. Agric. Sci. 2012, 11, 63–71. [Google Scholar] [CrossRef]
- Shamseldin, A.; Abdelkhalek, A.; Sadowsky, M.J. Recent Changes to the Classification of Symbiotic, Nitrogen-Fixing, Legume-Associating Bacteria: A Review. Symbiosis 2017, 71, 91–109. [Google Scholar]
- Bontemps, C.; Toussaint, M.; Revol, P.-V.; Hotel, L.; Jeanbille, M.; Uroz, S.; Turpault, M.-P.; Blaudez, D.; Leblond, P. Taxonomic and Functional Diversity of Streptomyces in a Forest Soil. FEMS Microbiol. Lett. 2013, 342, 157–167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Niu, G.; Chater, K.F.; Tian, Y.; Zhang, J.; Tan, H. Specialised Metabolites Regulating Antibiotic Biosynthesis in Streptomyces Spp. FEMS Microbiol. Rev. 2016, 40, 554–573. [Google Scholar] [CrossRef]
- Kashiwagi, N.; Ogino, C.; Kondo, A. Production of Chemicals and Proteins Using Biomass-Derived Substrates from a Streptomyces Host. Bioresour. Technol. 2017, 245, 1655–1663. [Google Scholar] [CrossRef]
- Bhatt, P.; Sharma, A.; Rene, E.R.; Kumar, A.J.; Zhang, W.; Chen, S. Bioremediation of Fipronil Using Bacillus sp. FA3: Mechanism, Kinetics and Resource Recovery Potential from Contaminated Environments. J. Water Process Eng. 2021, 39, 101712. [Google Scholar] [CrossRef]
- Bhatt, P.; Rene, E.R.; Kumar, A.J.; Gangola, S.; Kumar, G.; Sharma, A.; Zhang, W.; Chen, S. Fipronil Degradation Kinetics and Resource Recovery Potential of Bacillus sp. Strain FA4 Isolated from a Contaminated Agricultural Field in Uttarakhand, India. Chemosphere 2021, 276, 130156. [Google Scholar] [CrossRef]
- Tuesta-Popolizio, D.A.; Velázquez-Fernández, J.B.; Rodriguez-Campos, J.; Contreras-Ramos, S.M. Thalassobacillus, a Genus of Extreme to Moderate Environmental Halophiles with Biotechnological Potential. World J. Microbiol. Biotechnol. 2021, 37, 147. [Google Scholar] [CrossRef]
- Sánchez-Porro, C.; Amoozegar, M.A.; Rohban, R.; Hajighasemi, M.; Ventosa, A. Thalassobacillus cyri sp. nov., a Moderately Halophilic Gram-Positive Bacterium from a Hypersaline Lake. Int. J. Syst. Evol. Microbiol. 2009, 59, 2565–2570. [Google Scholar] [CrossRef]
- Sanchez-Porro, C.; Yilmaz, P.; Rafael, R.; Birbir, M.; Ventosa, A. Thalassobacillus pellis sp. nov., a Moderately Halophilic, Gram-Positive Bacterium Isolated from Salted Hides. Int. J. Syst. Evol. Microbiol. 2011, 61, 1206–1210. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Oh, T.-K.; Yoon, J.-H. Thalassobacillus hwangdonensis sp. nov., Isolated from a Tidal Flat Sediment. Int. J. Syst. Evol. Microbiol. 2010, 60, 2108–2112. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Analysis | Value |
|---|---|
| P (mg/dm3) | 3.46 |
| Ca (cmolc/dm3) | 4.04 |
| Mg (cmolc/dm3) | 1.93 |
| K (cmolc/dm3) | 0.27 |
| Al (cmolc/dm3) | 0.00 |
| H + Al (cmolc/dm3) | 2.30 |
| pH CaCl2 | 6.19 |
| pH H2O | 6.74 |
| pH SMP | 6.83 |
| T (cmolc/dm3) | 8.52 |
| SB (cmolc/dm3) | 6.23 |
| V% | 73.06 |
| Genera | C1 | C2 | C3 | Sum | Norm. | F1 | F2 | F3 | Sum | Norm. |
|---|---|---|---|---|---|---|---|---|---|---|
| Gaiella | 359 | 356 | 514 | 1229 | 0.057219 | 919 | 621 | 604 | 2144 | 0.099818 |
| Thalassobacillus | 0 | 102 | 38 | 140 | 0.006518 | 839 | 1632 | 480 | 2951 | 0.13739 |
| Mesorhizobium | 360 | 301 | 273 | 934 | 0.043484 | 693 | 485 | 623 | 1801 | 0.083849 |
| Bacillus | 1211 | 4 | 10 | 1225 | 0.057032 | 15 | 1214 | 15 | 1244 | 0.057917 |
| Streptomyces | 502 | 377 | 490 | 1369 | 0.063737 | 315 | 244 | 417 | 976 | 0.04544 |
| Mycobacterium | 252 | 191 | 255 | 698 | 0.032497 | 423 | 345 | 308 | 1076 | 0.050095 |
| Actinobacterium Gp-6 | 236 | 201 | 247 | 684 | 0.031845 | 347 | 377 | 276 | 1000 | 0.046557 |
| Bradyrhizobium | 199 | 185 | 186 | 570 | 0.026538 | 379 | 228 | 329 | 936 | 0.043577 |
| Pedomicrobium | 184 | 161 | 164 | 509 | 0.023698 | 303 | 271 | 283 | 857 | 0.039899 |
| Rhodoplanes | 138 | 134 | 153 | 425 | 0.019787 | 237 | 217 | 257 | 711 | 0.033102 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guima, S.E.S.; Piubeli, F.; Bonfá, M.R.L.; Pereira, R.M. New Insights into the Effect of Fipronil on the Soil Bacterial Community. Microorganisms 2023, 11, 52. https://doi.org/10.3390/microorganisms11010052
Guima SES, Piubeli F, Bonfá MRL, Pereira RM. New Insights into the Effect of Fipronil on the Soil Bacterial Community. Microorganisms. 2023; 11(1):52. https://doi.org/10.3390/microorganisms11010052
Chicago/Turabian StyleGuima, Suzana Eiko Sato, Francine Piubeli, Maricy Raquel Lindenbah Bonfá, and Rodrigo Matheus Pereira. 2023. "New Insights into the Effect of Fipronil on the Soil Bacterial Community" Microorganisms 11, no. 1: 52. https://doi.org/10.3390/microorganisms11010052
APA StyleGuima, S. E. S., Piubeli, F., Bonfá, M. R. L., & Pereira, R. M. (2023). New Insights into the Effect of Fipronil on the Soil Bacterial Community. Microorganisms, 11(1), 52. https://doi.org/10.3390/microorganisms11010052