

Genome Sequence of Eubacterium limosum B2 and Evolution for Growth on a Mineral Medium with Methanol and CO2 as Sole Carbon Sources




Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Growth Conditions
2.2. Adaptive Laboratory Evolution
2.3. Clone Isolation on Methanol Mineral Medium without Cysteine
2.4. Analytical Methods
2.5. Genomic DNA Extraction
2.6. Whole Genome Sequencing
2.7. Functional Annotation and Genome Analysis
2.8. Methylome and Type I Restriction-Modification System Analysis
2.9. Protein Isolation and Total Proteome Analysis
3. Results
3.1. De Novo Sequencing, Annotation and Analysis of the E. limosum B2 Genome
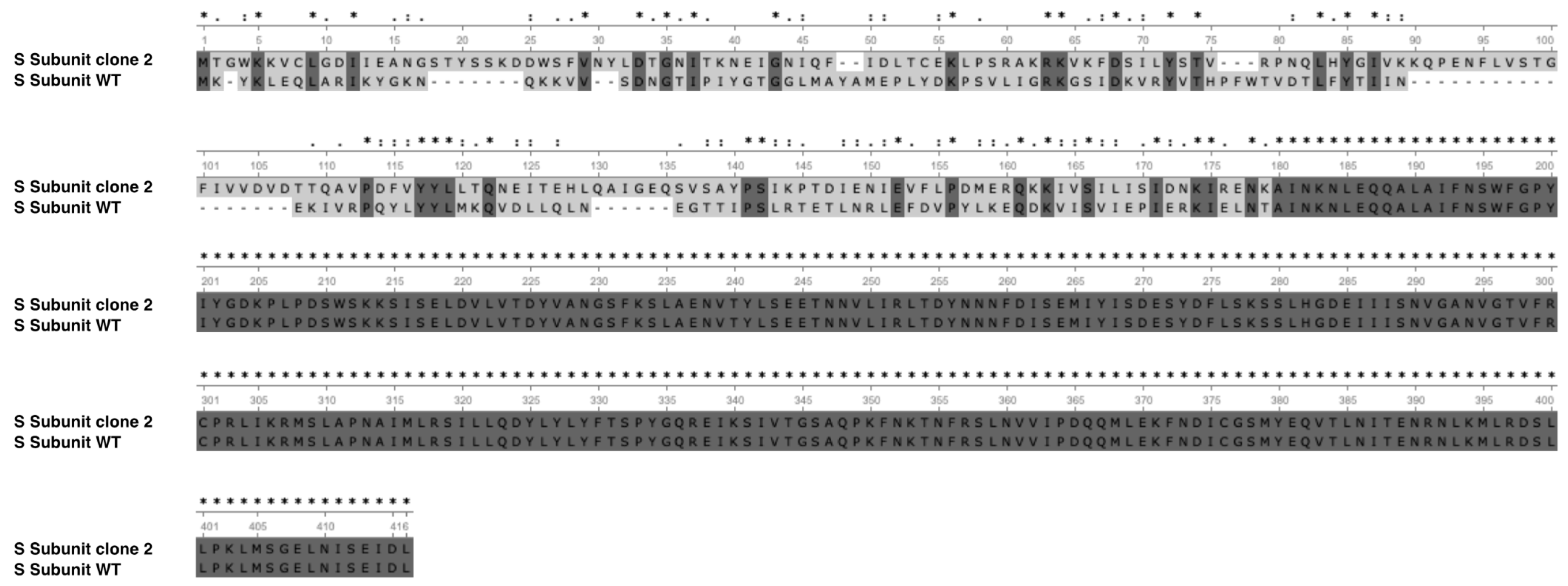
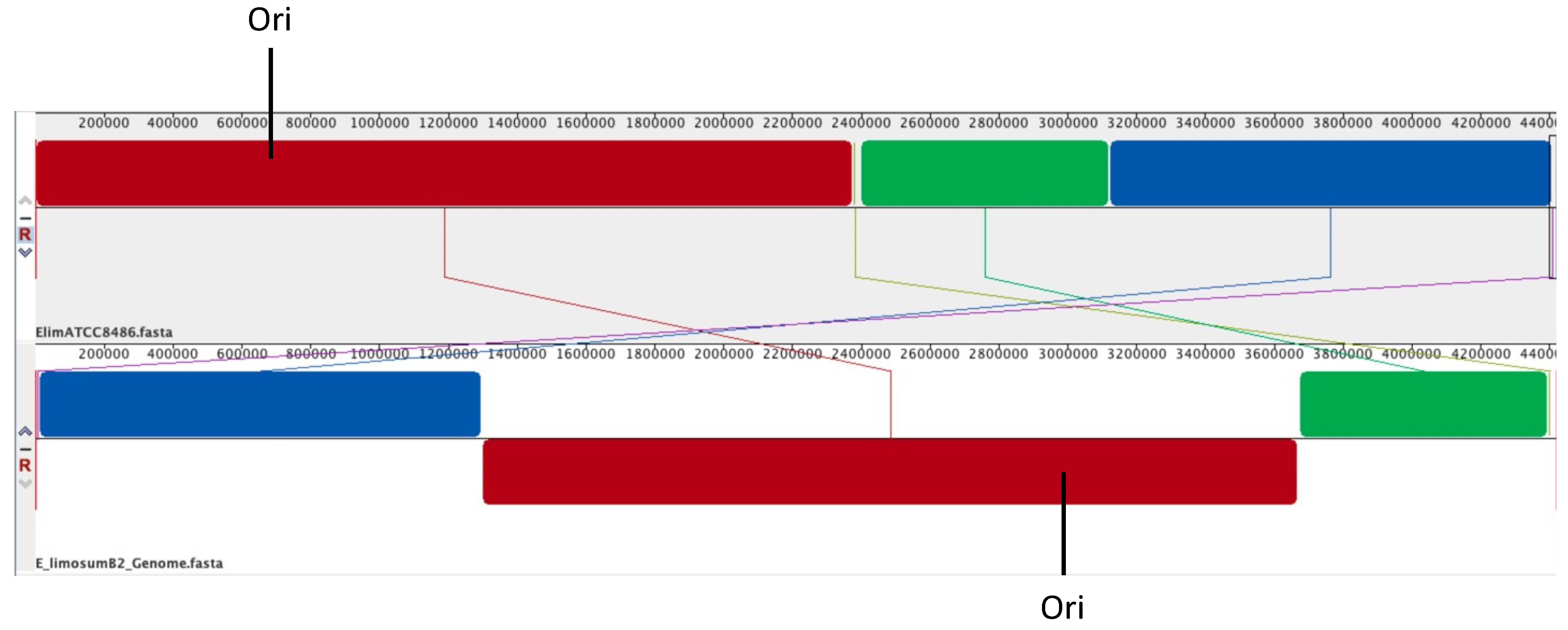
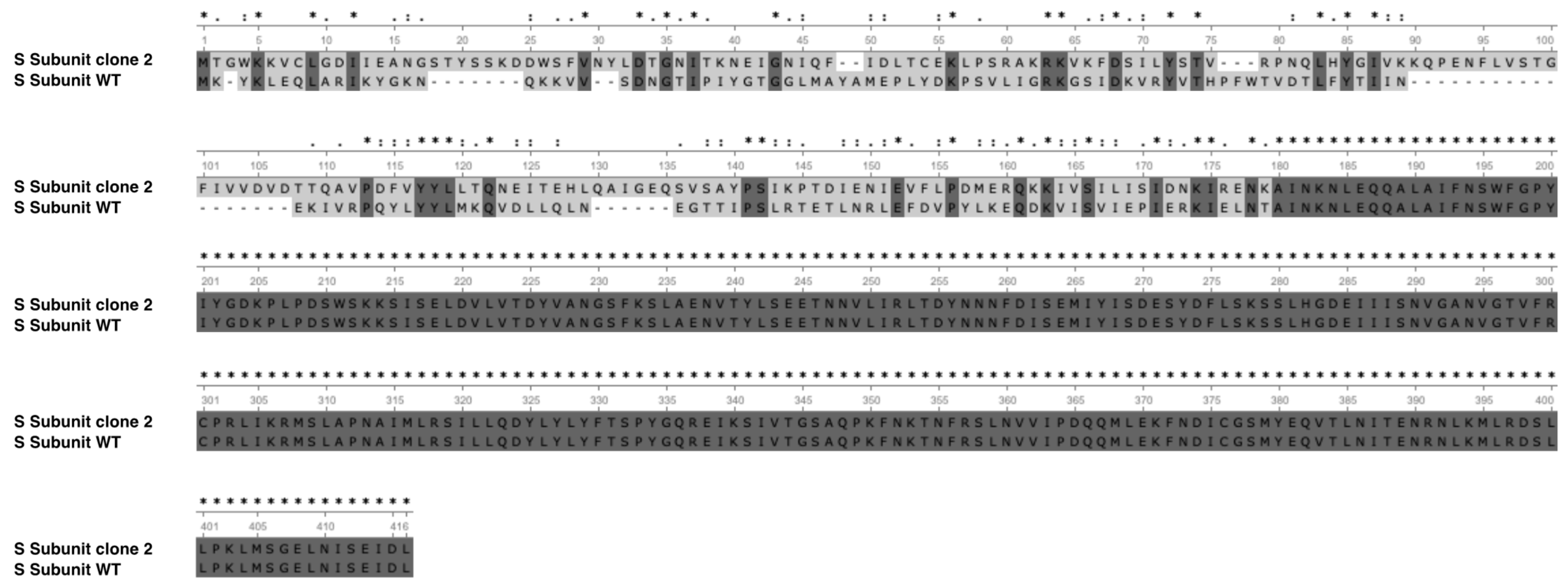
3.2. Genomic Comparison of E. limosum B2 vs. E. limosum ATCC 8486
3.3. Whole DNA Methylation Level
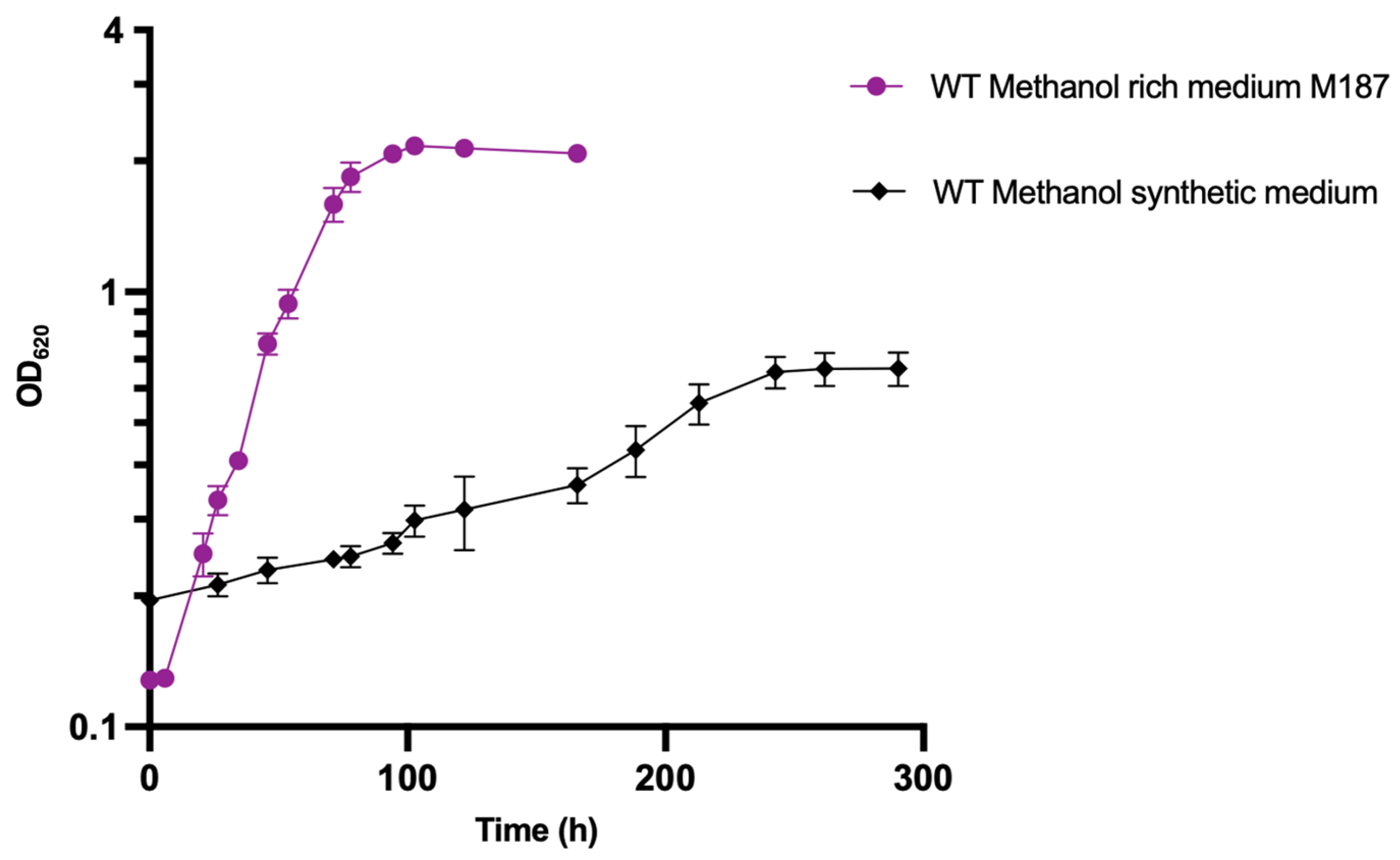
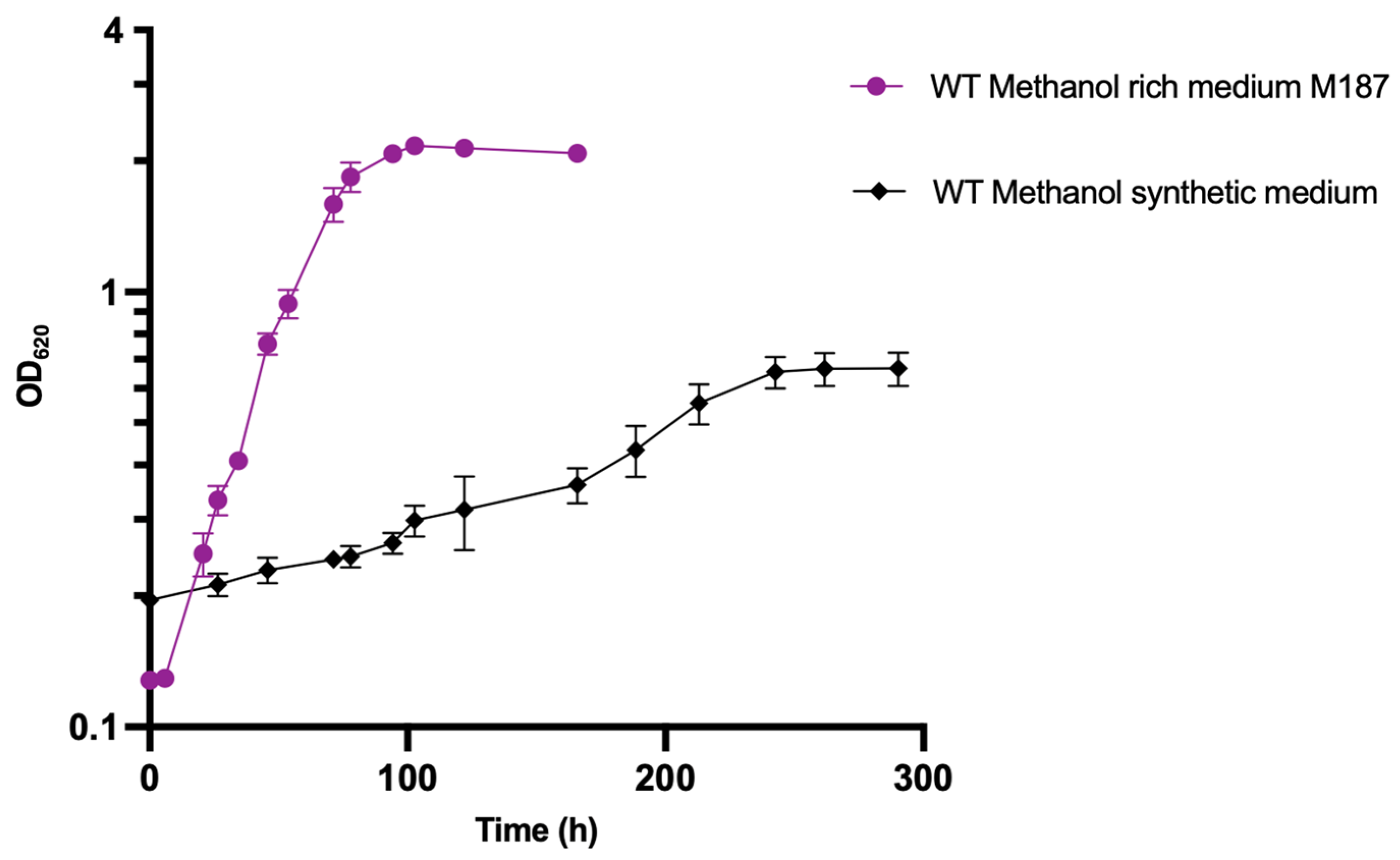
3.4. Strain Adaptation on Methanol-Defined Medium without Yeast Extract
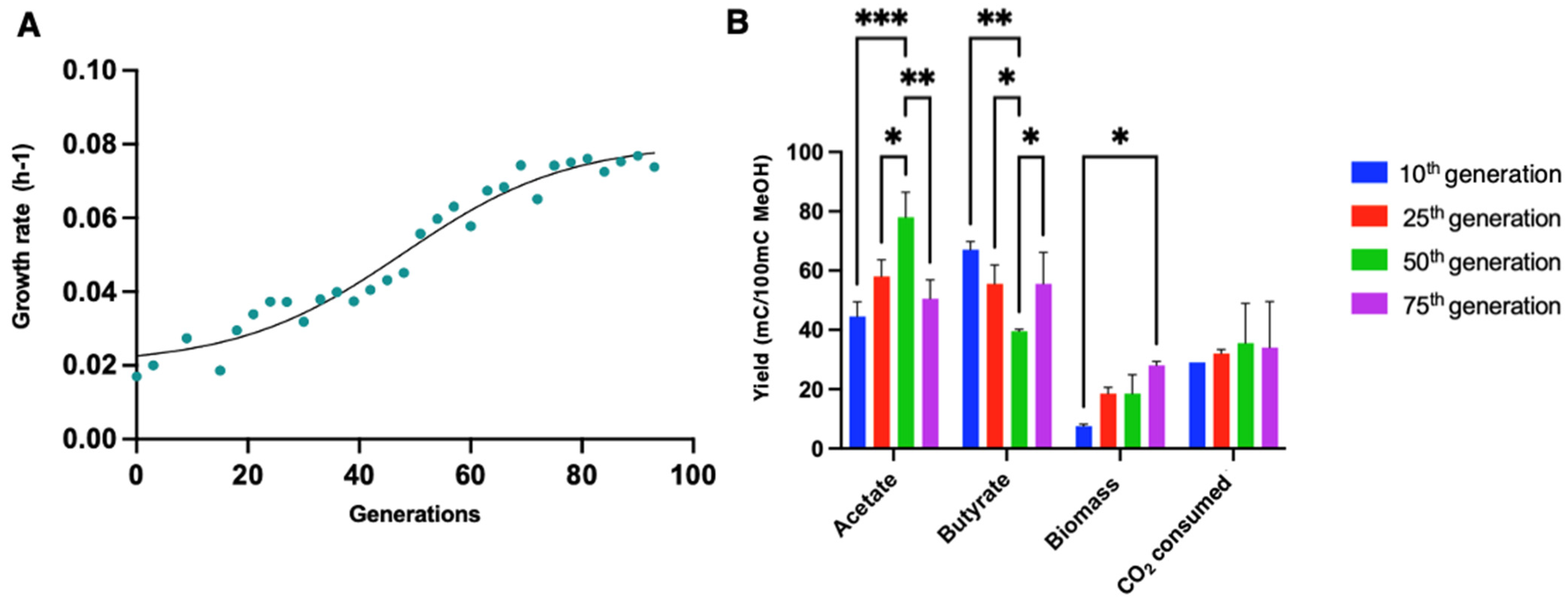
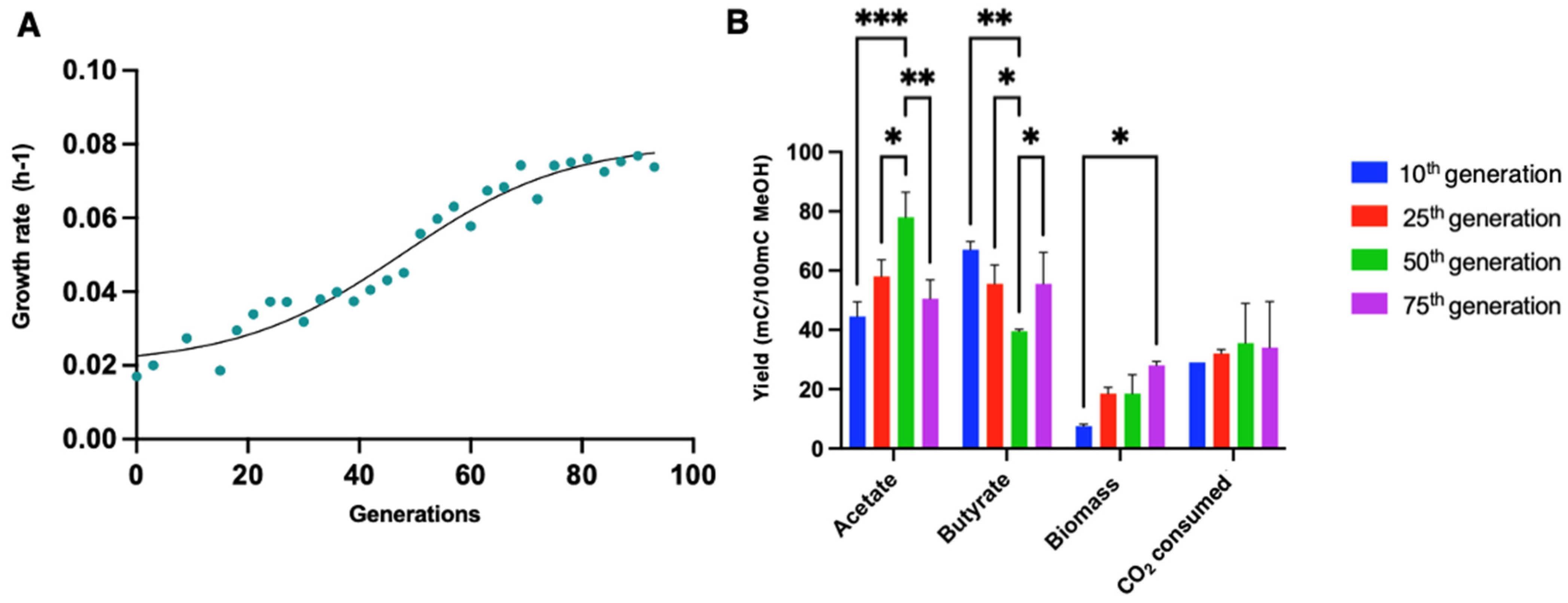
3.5. Adaptative Laboratory Evolution of E. limosum B2 in a Methanol-Defined Medium
3.6. Isolation of Individual Clones Growing on Methanol Mineral Medium
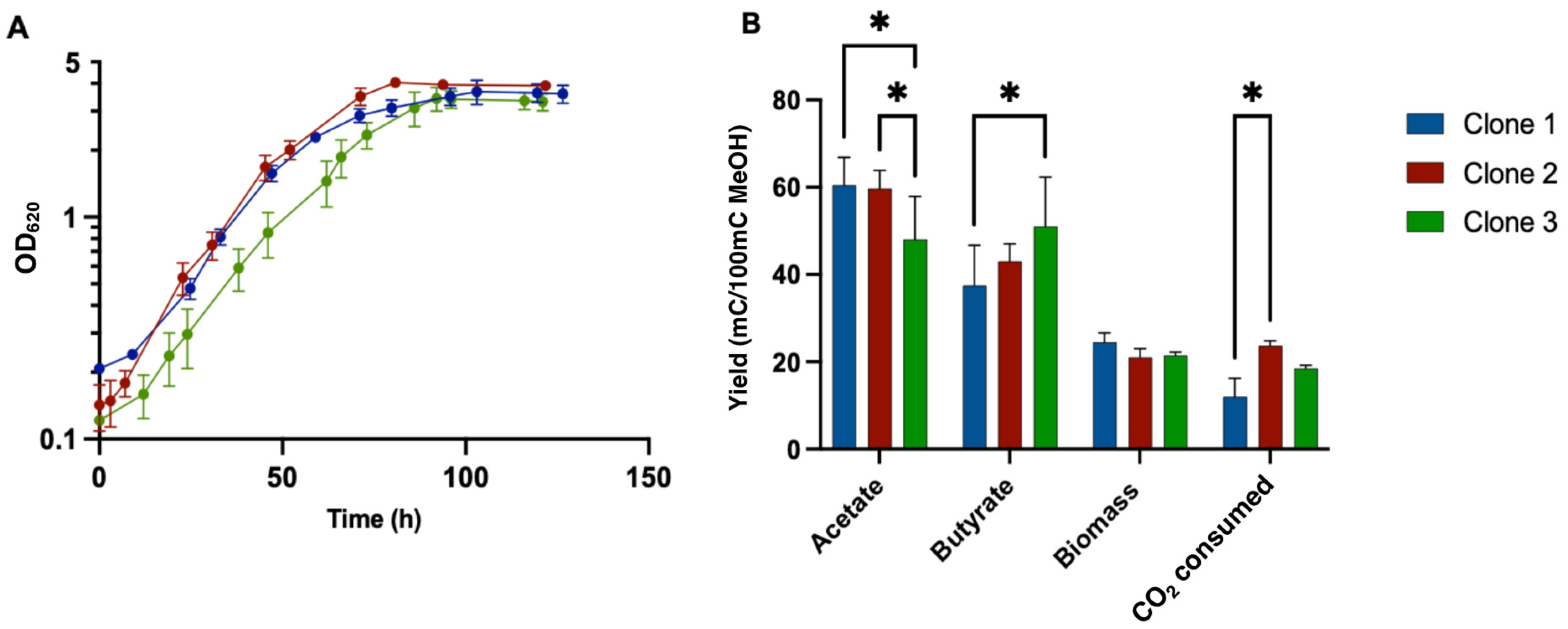
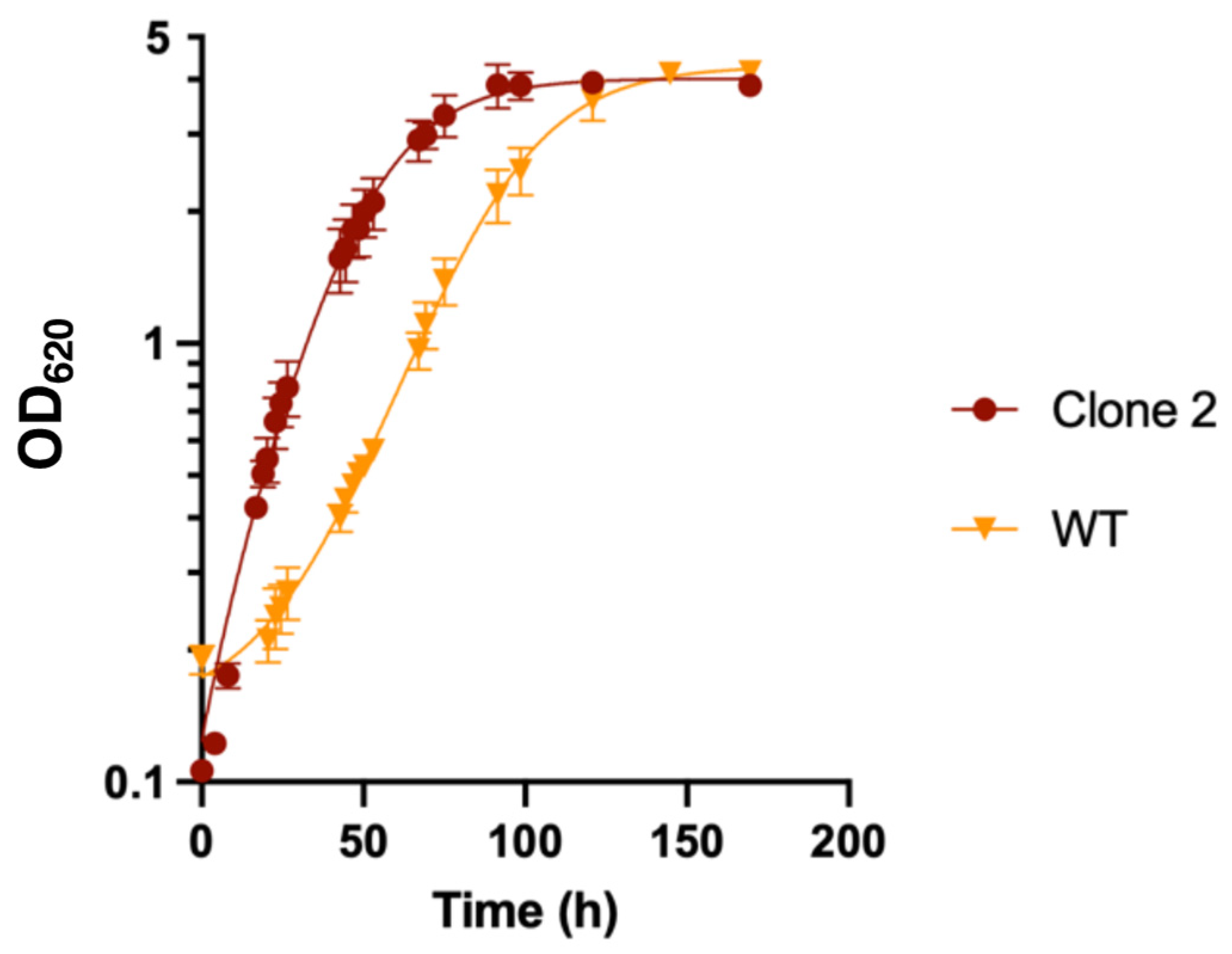
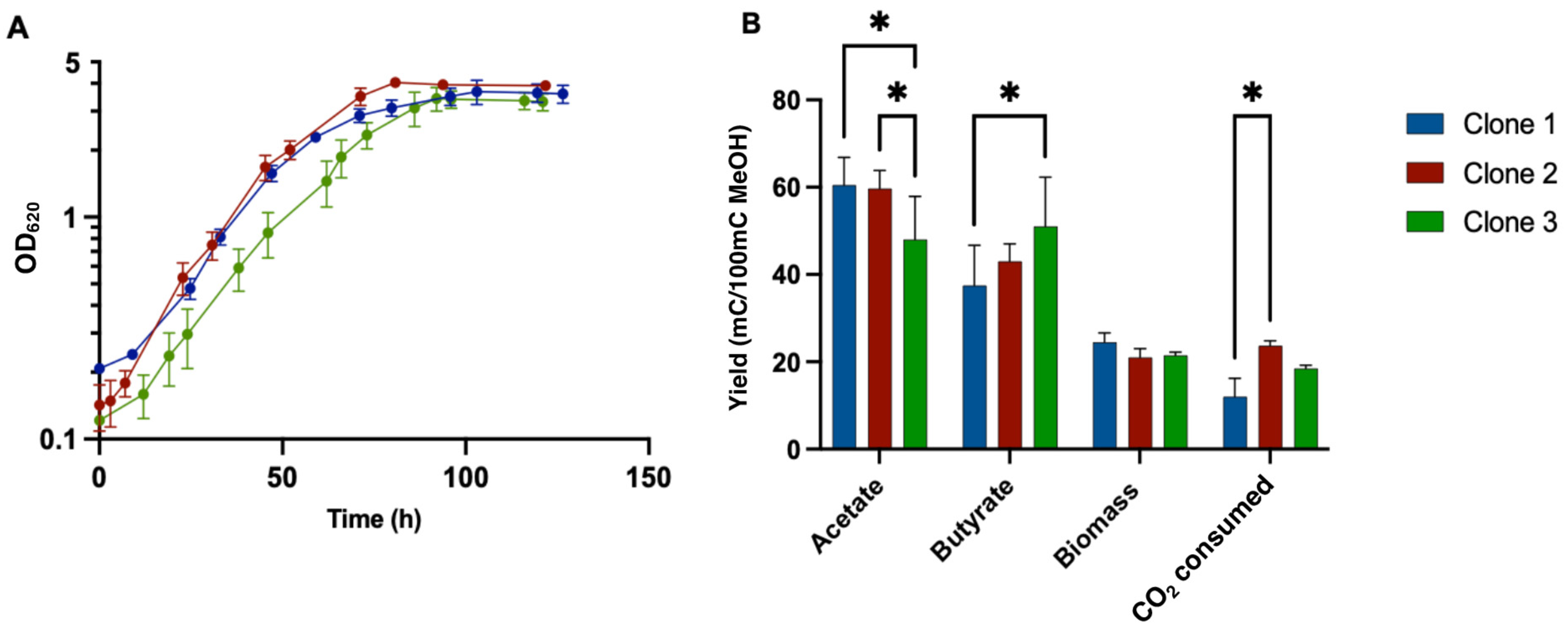
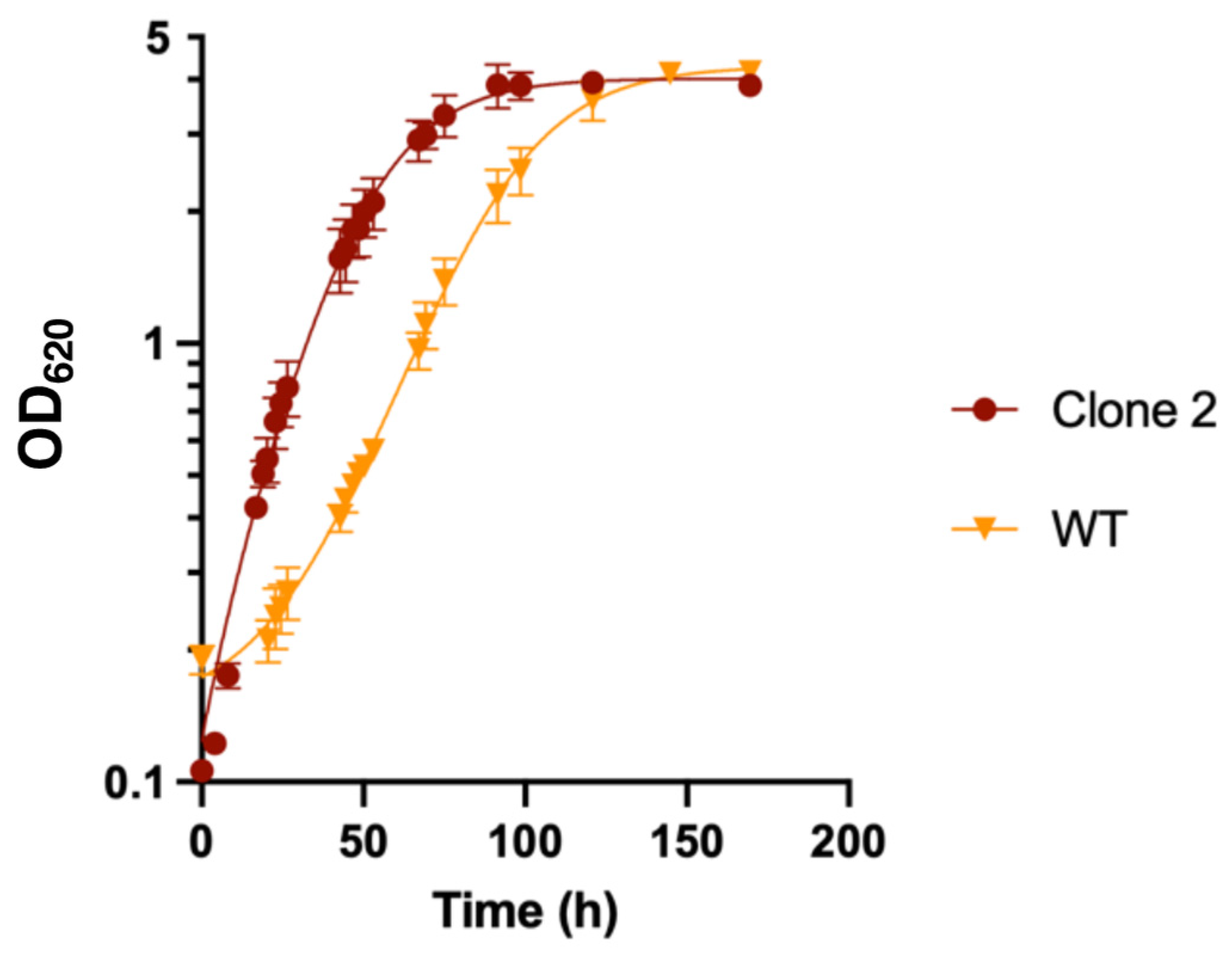
3.7. Growth Profile of Adapted Clones in Liquid MMM
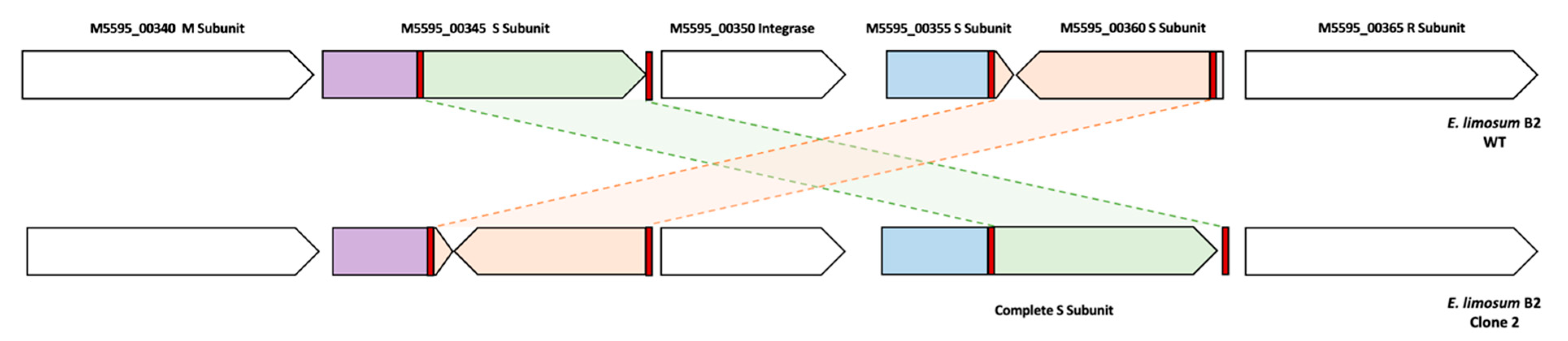
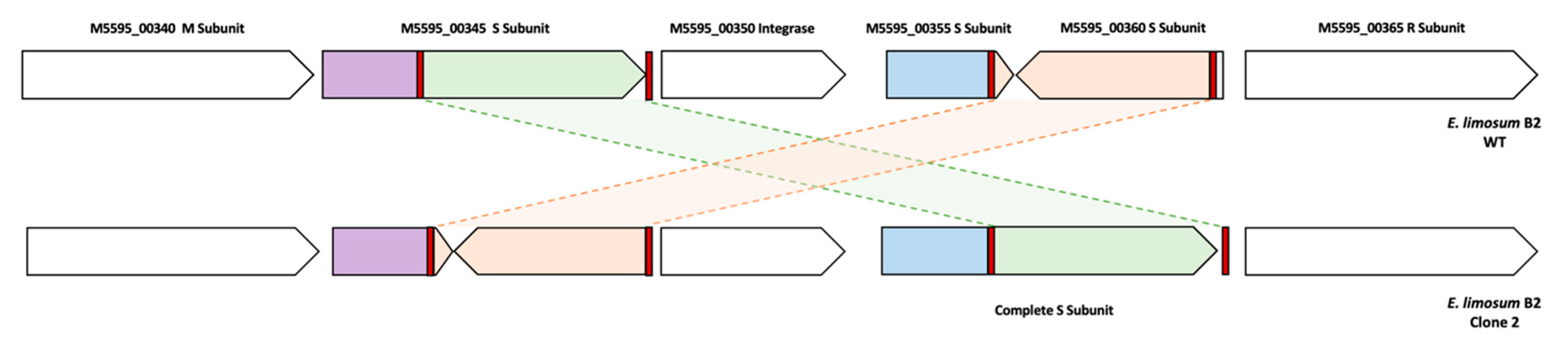
3.8. Mutation Profiles of the ALE Lineage by Whole Genome Resequencing
3.9. Proteomic Analysis of the WT and Evolved Clone 2
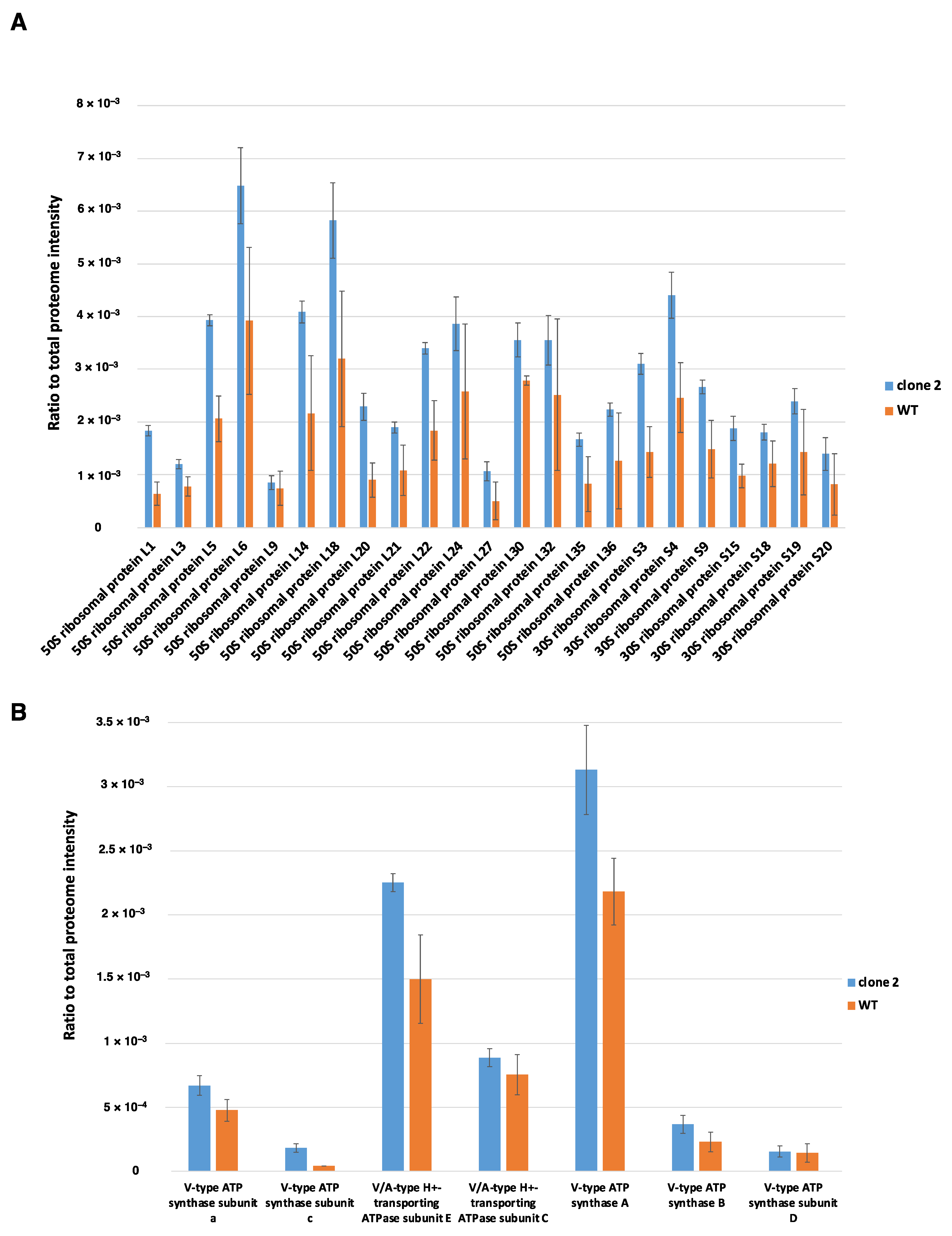
3.9.1. Global Analysis of the Proteomes
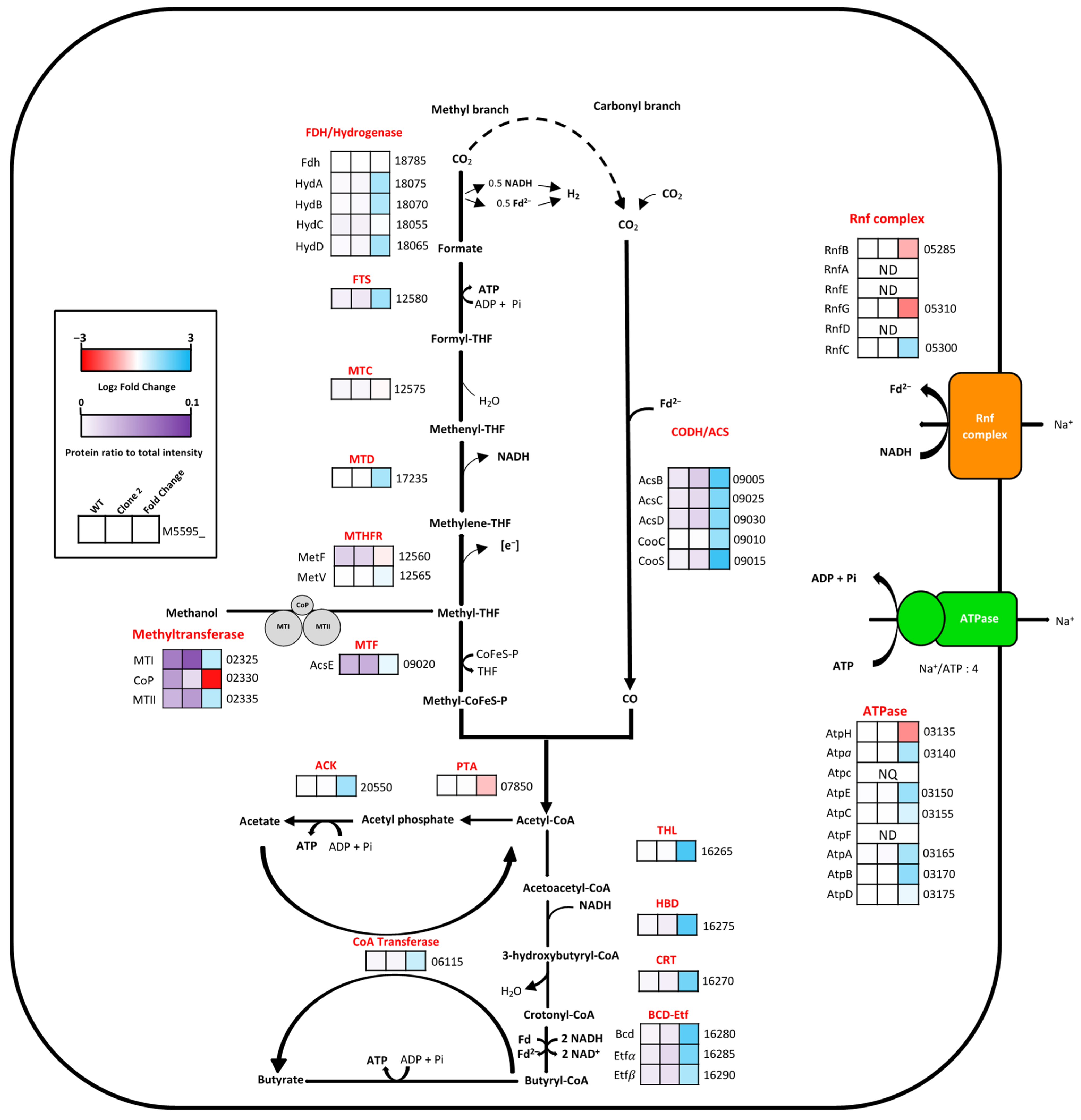
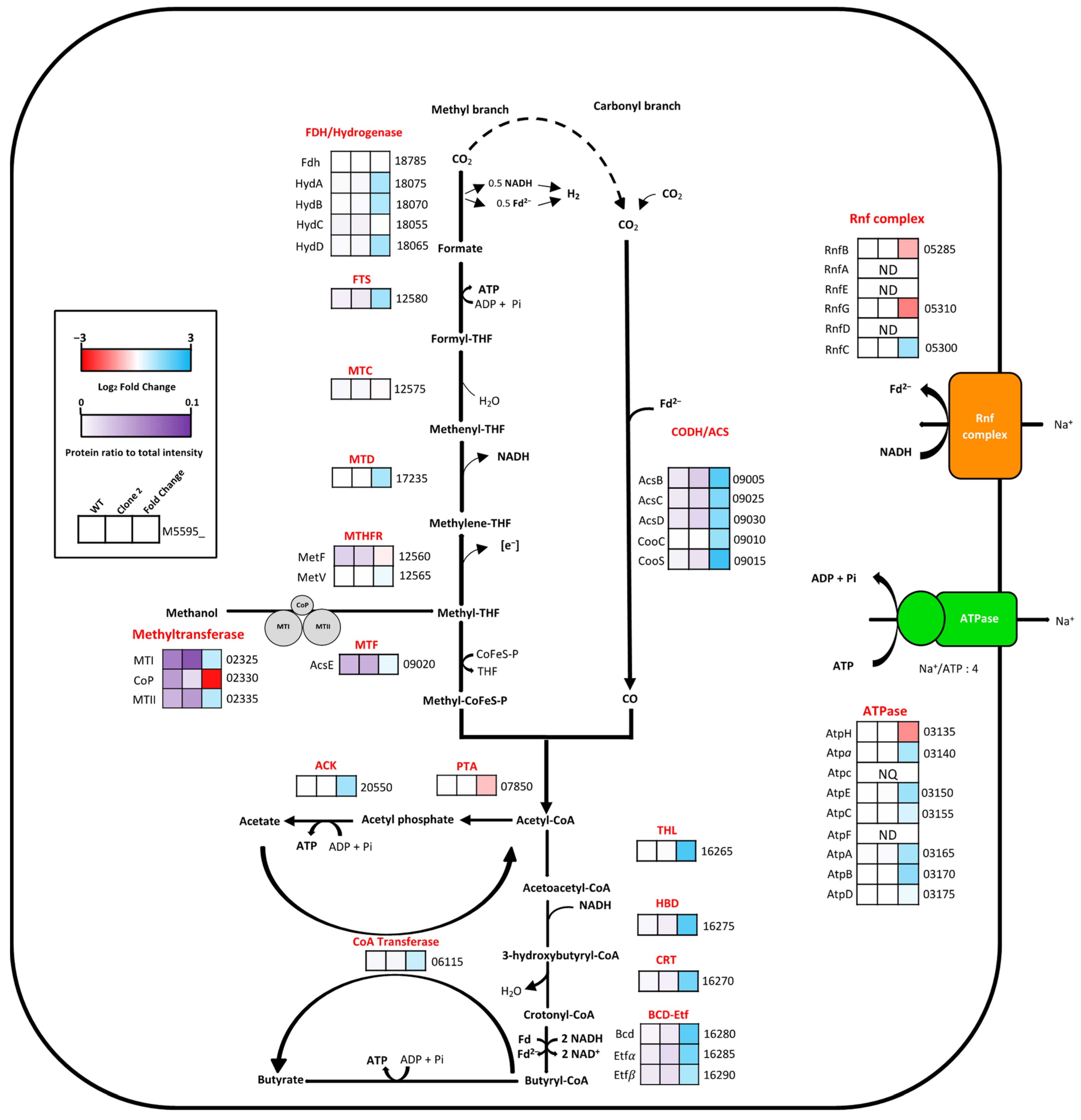
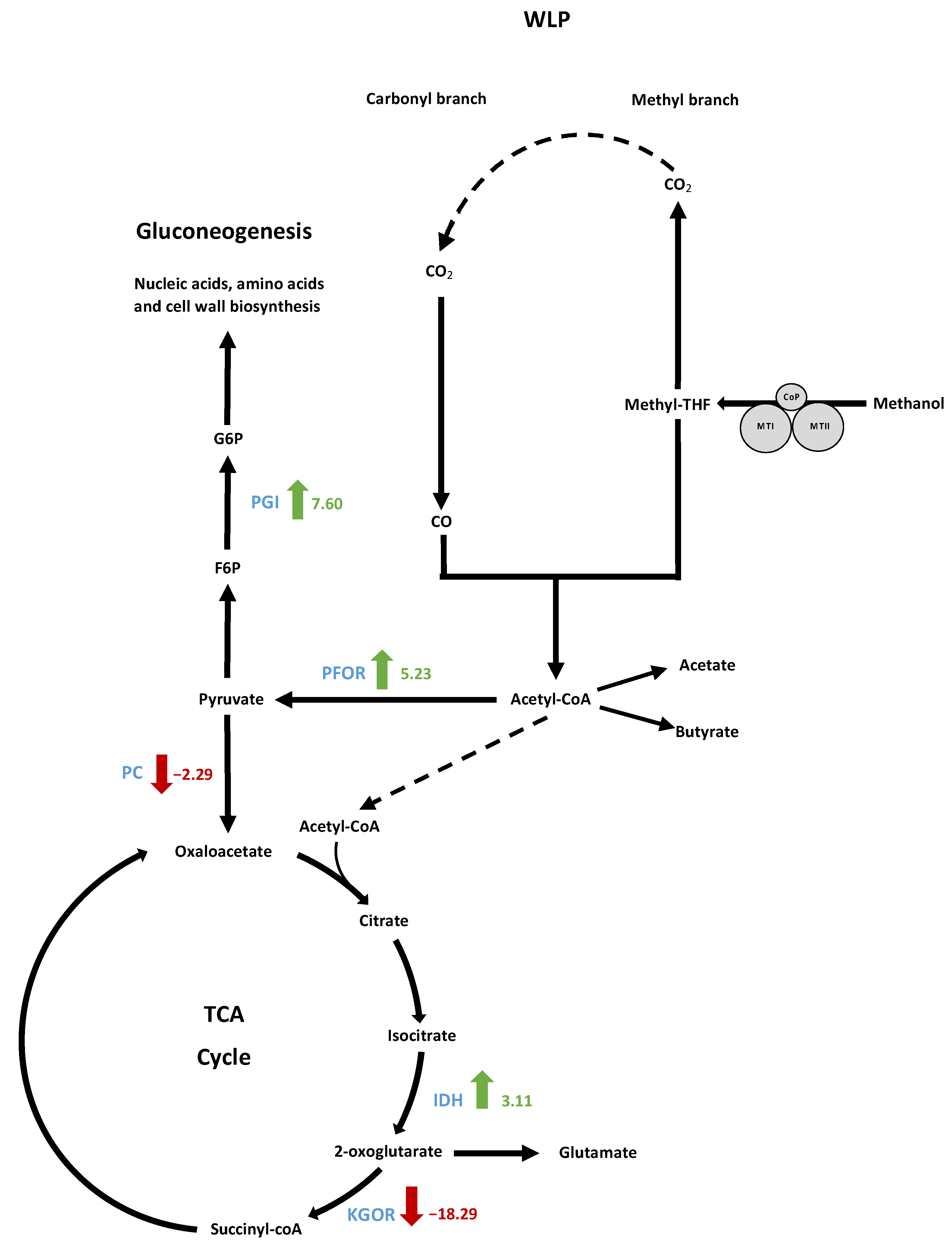
3.9.2. Analysis of the Methanol Oxidation and Wood–Ljungdahl Pathways
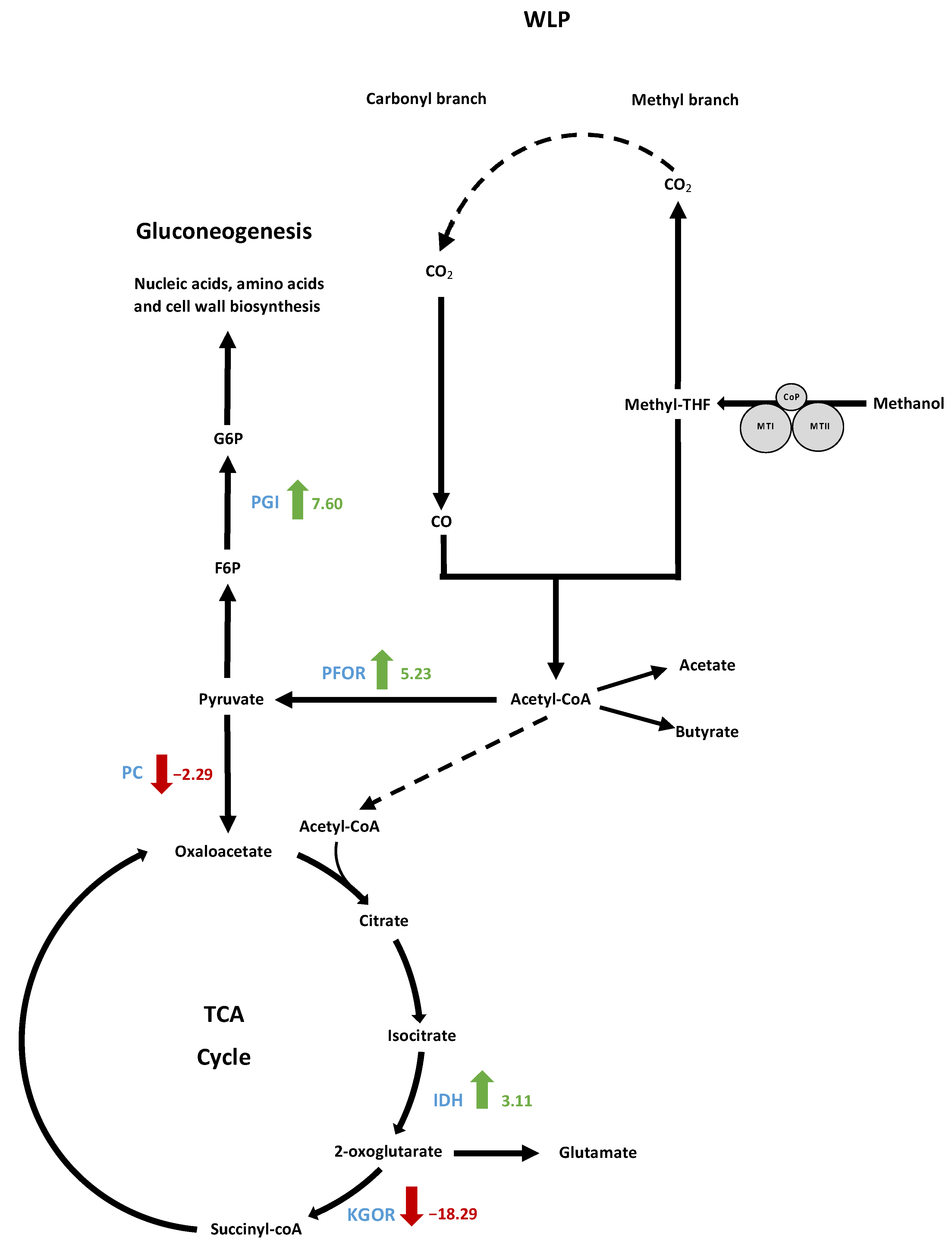
3.9.3. Analysis of Gluconeogenesis and Anaplerotic Reactions
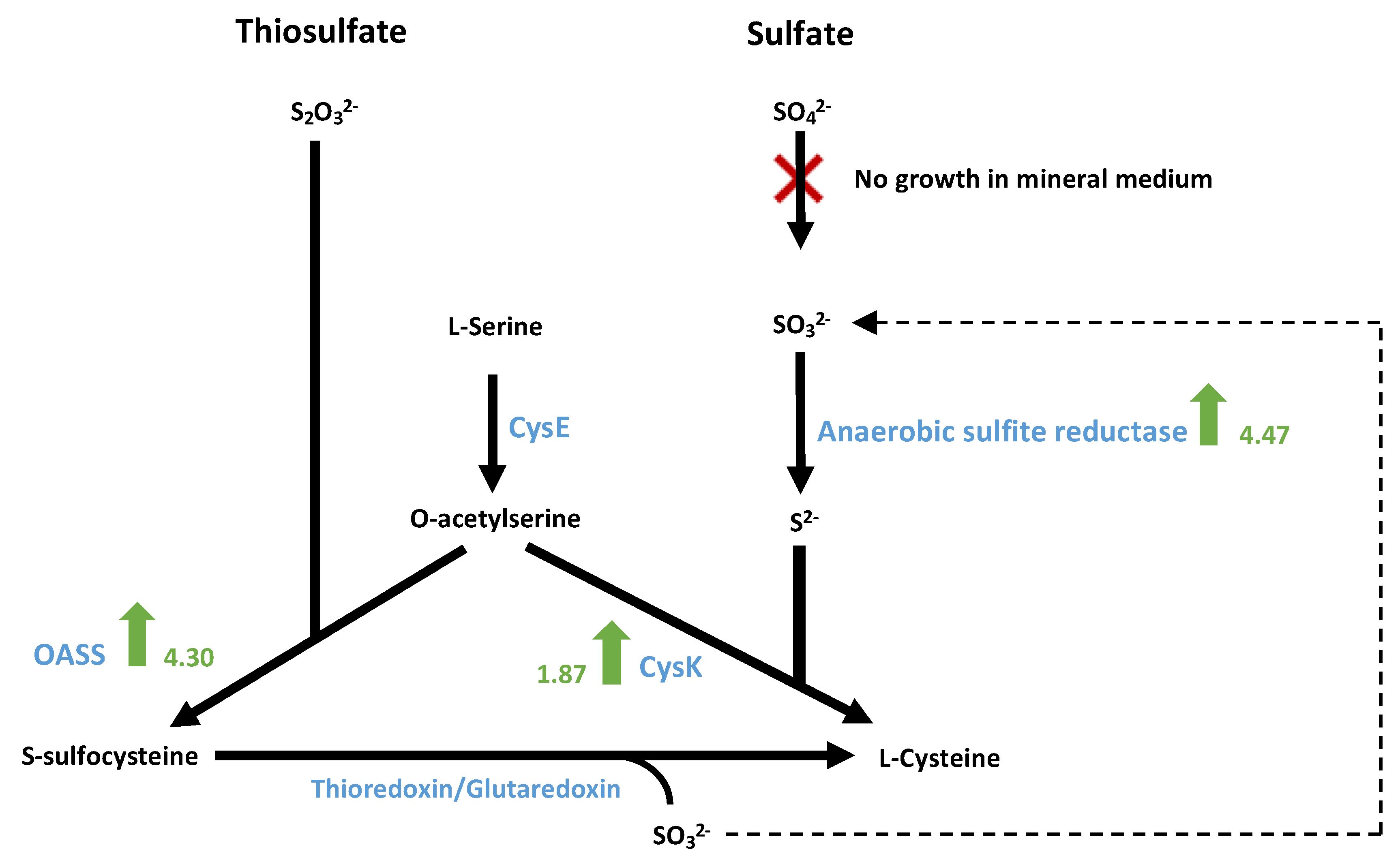
3.9.4. Sulphate Metabolism for the Biosynthesis of Sulphur-Containing Amino Acids
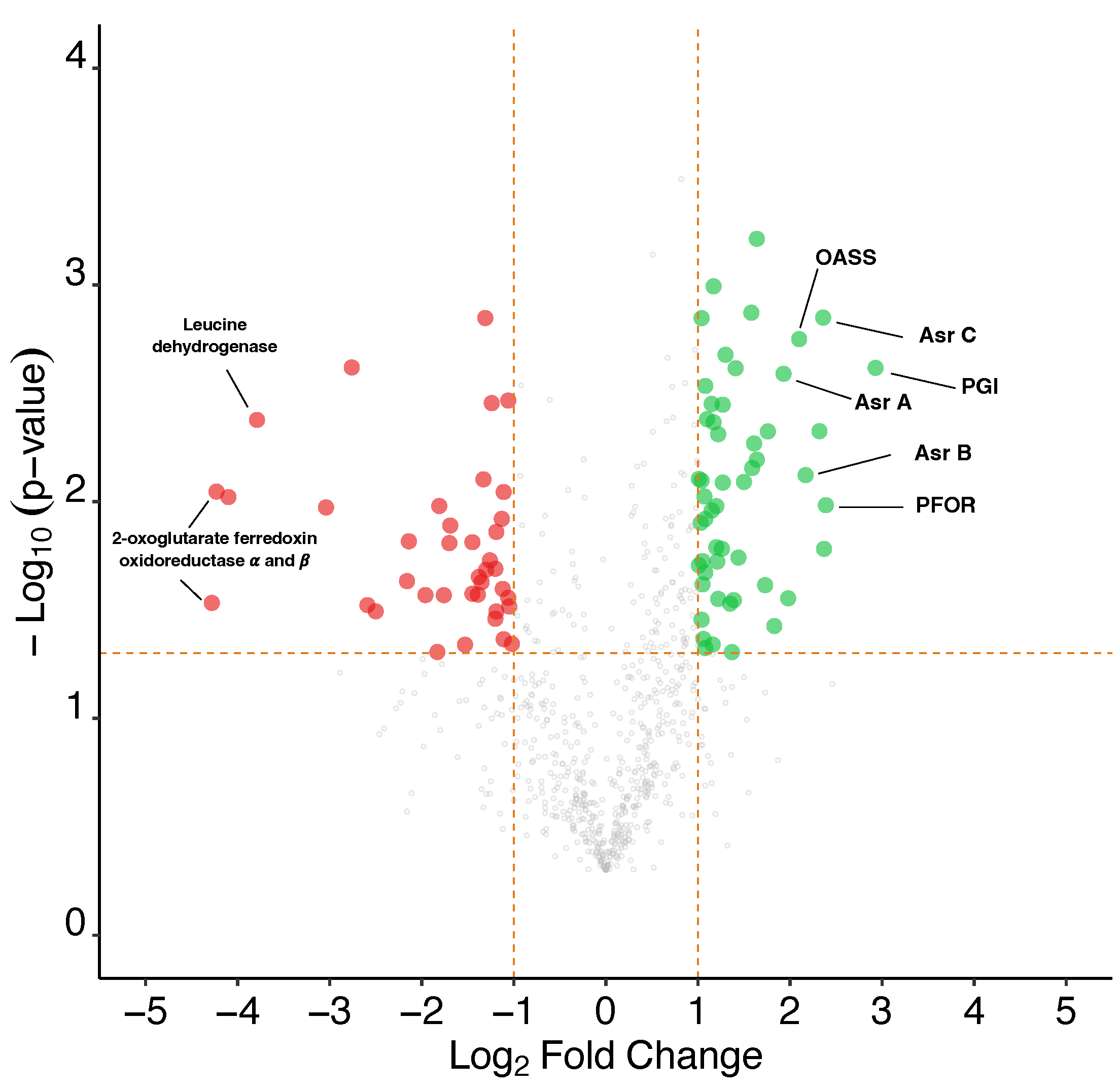
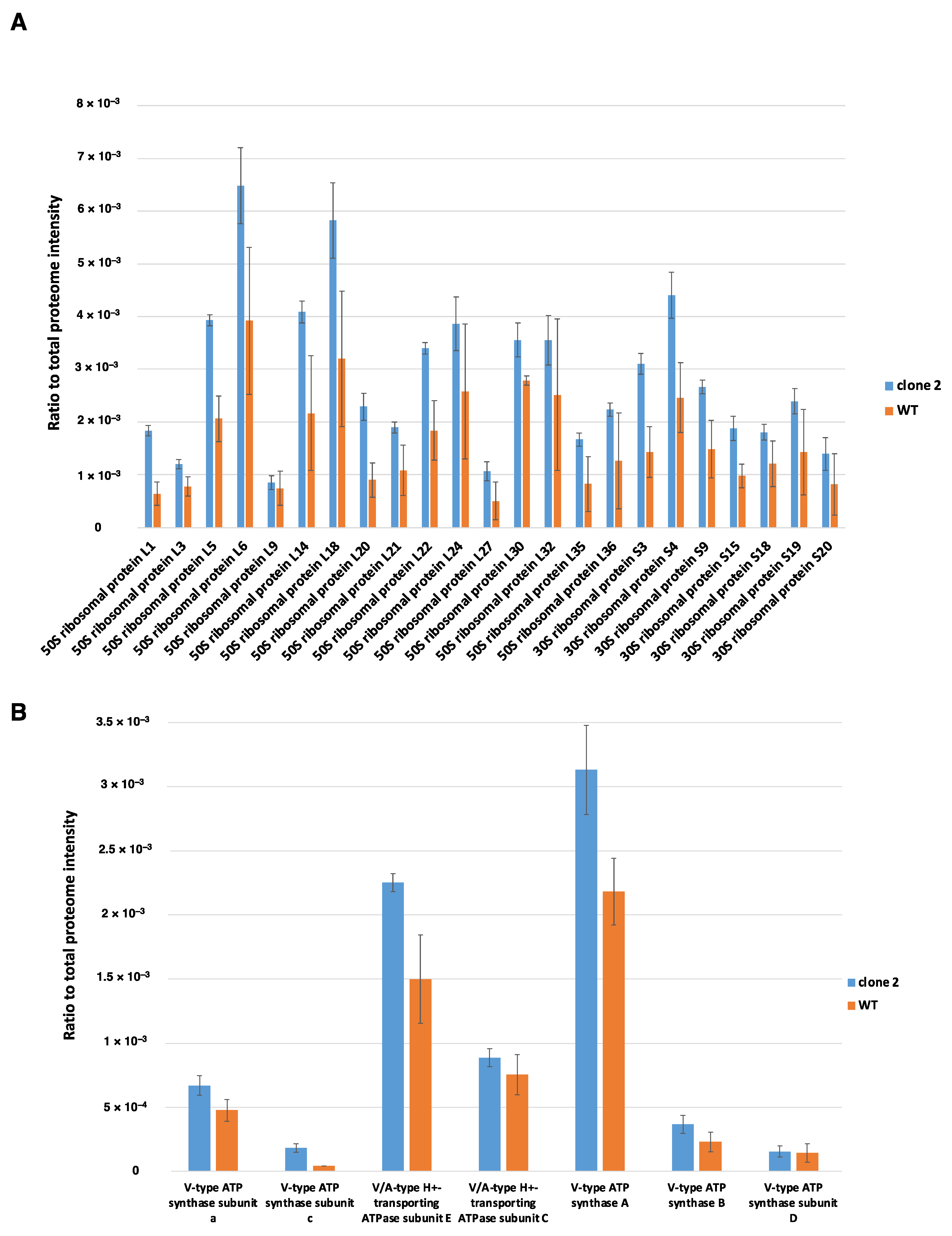
3.9.5. Other Differentially Produced Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wood, H.G.; Ragsdale, S.W.; Pezacka, E. The Acetyl-CoA Pathway of Autotrophic Growth. FEMS Microbiol. Rev. 1986, 2, 345–362. [Google Scholar] [CrossRef]
- Bengelsdorf, F.R.; Straub, M.; Dürre, P. Bacterial Synthesis Gas (Syngas) Fermentation. Environ. Technol. 2013, 34, 1639–1651. [Google Scholar] [CrossRef]
- Henstra, A.M.; Sipma, J.; Rinzema, A.; Stams, A.J.M. Microbiology of Synthesis Gas Fermentation for Biofuel Production. Curr. Opin. Biotechnol. 2007, 18, 200–206. [Google Scholar] [CrossRef]
- Dwidar, M.; Park, J.-Y.; Mitchell, R.J.; Sang, B.-I. The Future of Butyric Acid in Industry. Sci. World J. 2012, 2012, 471417. [Google Scholar] [CrossRef]
- Dürre, P. Butanol Formation from Gaseous Substrates. FEMS Microbiol. Lett. 2016, 363, fnw040. [Google Scholar] [CrossRef]
- Karlson, B.; Bellavitis, C.; France, N. Commercializing Lanza Tech, from Waste to Fuel: An Effectuation Case. J. Manag. Organ. 2021, 27, 175–196. [Google Scholar] [CrossRef]
- Bertsch, J.; Müller, V. Bioenergetic Constraints for Conversion of Syngas to Biofuels in Acetogenic Bacteria. Biotechnol. Biofuels 2015, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Cotton, C.A.; Claassens, N.J.; Benito-Vaquerizo, S.; Bar-Even, A. Renewable Methanol and Formate as Microbial Feedstocks. Curr. Opin. Biotechnol. 2020, 62, 168–180. [Google Scholar] [CrossRef]
- Giuliano, A.; Freda, C.; Catizzone, E. Techno-Economic Assessment of Bio-Syngas Production for Methanol Synthesis: A Focus on the Water–Gas Shift and Carbon Capture Sections. Bioengineering 2020, 7, 70. [Google Scholar] [CrossRef]
- Pacaud, S.; Loubiere, P.; Goma, G. Methanol Metabolism by Eubacterium limosum B2: Effects of pH and Carbon Dioxide on Growth and Organic Acid Production. Curr. Microbiol. 1985, 12, 245–250. [Google Scholar] [CrossRef]
- Loubière, P.; Gros, E.; Paquet, V.; Lindley, N.D. Kinetics and Physiological Implications of the Growth Behaviour of Eubacterium limosum on Glucose/Methanol Mixtures. Microbiology 1992, 138, 979–985. [Google Scholar] [CrossRef]
- Loubiere, P.; Pacaud, S.; Goma, G.; Lindley, N.D. The Effect of Formate on the Acidogenic Fermentation of Methanol by Eubacterium limosum. J. Gen. Appl. Microbiol. 1987, 33, 463–470. [Google Scholar] [CrossRef]
- Pacaud, S.; Loubiere, P.; Goma, G.; Lindley, N.D. Effects of Various Organic Acid Supplements on Growth Rates of Eubacterium limosum B2 on Methanol. Appl. Microbiol. Biotechnol. 1986, 24, 75–78. [Google Scholar] [CrossRef]
- Loubiere, P.; Goma, G.; Lindley, N.D. A Non-Passive Mechanism of Butyrate Excretion Operates during Acidogenic Fermentation of Methanol by Eubacterium limosum. Antonie Van Leeuwenhoek 1990, 57, 83–89. [Google Scholar] [CrossRef]
- Kremp, F.; Poehlein, A.; Daniel, R.; Müller, V. Methanol Metabolism in the Acetogenic Bacterium Acetobacterium Woodii. Environ. Microbiol. 2018, 20, 4369–4384. [Google Scholar] [CrossRef]
- Dietrich, H.M.; Kremp, F.; Öppinger, C.; Ribaric, L.; Müller, V. Biochemistry of Methanol-Dependent Acetogenesis in Eubacterium callanderi KIST612. Environ. Microbiol. 2021, 23, 4505–4517. [Google Scholar] [CrossRef]
- Wood, J.C.; Marcellin, E.; Plan, M.R.; Virdis, B. High Methanol-to-Formate Ratios Induce Butanol Production in Eubacterium limosum. Microb. Biotechnol. 2021, 15, 1542–1549. [Google Scholar] [CrossRef]
- Marinos, G.; Kaleta, C.; Waschina, S. Defining the Nutritional Input for Genome-Scale Metabolic Models: A Roadmap. PLoS ONE 2020, 15, e0236890. [Google Scholar] [CrossRef]
- Genthner, B.R.; Davis, C.L.; Bryant, M.P. Features of Rumen and Sewage Sludge Strains of Eubacterium limosum, a Methanol- and H2-CO2-Utilizing Species. Appl. Environ. Microbiol. 1981, 42, 12–19. [Google Scholar] [CrossRef]
- Richter, H.; Loftus, S.E.; Angenent, L.T. Integrating Syngas Fermentation with the Carboxylate Platform and Yeast Fermentation to Reduce Medium Cost and Improve Biofuel Productivity. Environ. Technol. 2013, 34, 1983–1994. [Google Scholar] [CrossRef]
- Dragosits, M.; Mattanovich, D. Adaptive Laboratory Evolution–Principles and Applications for Biotechnology. Microb. Cell Factories 2013, 12, 64. [Google Scholar] [CrossRef]
- Kang, S.; Song, Y.; Jin, S.; Shin, J.; Bae, J.; Kim, D.R.; Lee, J.-K.; Kim, S.; Cho, S.; Cho, B.K. Adaptive Laboratory Evolution of Eubacterium limosum ATCC 8486 on Carbon Monoxide. Front. Microbiol. 2020, 11, 402. [Google Scholar] [CrossRef] [PubMed]
- Erickson, L.E. Biomass Elemental Composition and Energy Content. Biotechnol. Bioeng. 1980, 22, 451–456. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI Prokaryotic Genome Annotation Pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. ProgressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Doellinger, J.; Schneider, A.; Hoeller, M.; Lasch, P. Sample Preparation by Easy Extraction and Digestion (SPEED)-A Universal, Rapid, and Detergent-Free Protocol for Proteomics Based on Acid Extraction. Mol. Cell. Proteom. 2020, 19, 209–222. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Kelly, W.J.; Henderson, G.; Pacheco, D.M.; Li, D.; Reilly, K.; Naylor, G.E.; Janssen, P.H.; Attwood, G.T.; Altermann, E.; Leahy, S.C. The Complete Genome Sequence of Eubacterium limosum SA11, a Metabolically Versatile Rumen Acetogen. Stand. Genom. Sci. 2016, 11, 26. [Google Scholar] [CrossRef]
- Roh, H.; Ko, H.-J.; Kim, D.; Choi, D.G.; Park, S.; Kim, S.; Chang, I.S.; Choi, I.-G. Complete Genome Sequence of a Carbon Monoxide-Utilizing Acetogen, Eubacterium limosum KIST612. J. Bacteriol. 2011, 193, 307–308. [Google Scholar] [CrossRef]
- Song, Y.; Shin, J.; Jeong, Y.; Jin, S.; Lee, J.-K.; Kim, D.R.; Kim, S.C.; Cho, S.; Cho, B.-K. Determination of the Genome and Primary Transcriptome of Syngas Fermenting Eubacterium limosum ATCC 8486. Sci. Rep. 2017, 7, 13694. [Google Scholar] [CrossRef] [Green Version]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.-J.; Buck, D.; Au, K.F. Comprehensive Comparison of Pacific Biosciences and Oxford Nanopore Technologies and Their Applications to Transcriptome Analysis. F1000Research 2017, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xiao, P.; Jiang, Y.; Dong, M.; Chen, Z.; Li, H.; Hu, Z.; Lei, A.; Wang, J. Transgenerational Epigenetic Inheritance Under Environmental Stress by Genome-Wide DNA Methylation Profiling in Cyanobacterium. Front. Microbiol. 2018, 9, 1479. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The Epigenomic Landscape of Prokaryotes. PLoS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef]
- Jensen, T.Ø.; Tellgren-Roth, C.; Redl, S.; Maury, J.; Jacobsen, S.A.B.; Pedersen, L.E.; Nielsen, A.T. Genome-Wide Systematic Identification of Methyltransferase Recognition and Modification Patterns. Nat. Commun. 2019, 10, 3311. [Google Scholar] [CrossRef] [PubMed]
- Choe, D.; Lee, J.H.; Yoo, M.; Hwang, S.; Sung, B.H.; Cho, S.; Palsson, B.; Kim, S.C.; Cho, B.-K. Adaptive Laboratory Evolution of a Genome-Reduced Escherichia coli. Nat. Commun. 2019, 10, 935. [Google Scholar] [CrossRef]
- Espinosa, M.I.; Gonzalez-Garcia, R.A.; Valgepea, K.; Plan, M.R.; Scott, C.; Pretorius, I.S.; Marcellin, E.; Paulsen, I.T.; Williams, T.C. Adaptive Laboratory Evolution of Native Methanol Assimilation in Saccharomyces Cerevisiae. Nat. Commun. 2020, 11, 5564. [Google Scholar] [CrossRef]
- Har, J.R.G.; Agee, A.; Bennett, R.K.; Papoutsakis, E.T.; Antoniewicz, M.R. Adaptive Laboratory Evolution of Methylotrophic Escherichia coli Enables Synthesis of All Amino Acids from Methanol-Derived Carbon. Appl. Microbiol. Biotechnol. 2021, 105, 869–876. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, L.; Tuyishime, P.; Liu, J.; Zhang, K.; Gao, N.; Zhang, Z.; Ni, X.; Feng, J.; Yuan, Q.; et al. Adaptive Laboratory Evolution Enhances Methanol Tolerance and Conversion in Engineered Corynebacterium glutamicum. Commun. Biol. 2020, 3, 1–15. [Google Scholar] [CrossRef]
- Mariotto, C.; Loubière, P.; Goma, G.; Lindley, N.D. Influence of Various Reducing Agents on Methylotrophic Growth and Organic Acid Production of Eubacterium limosum. Appl. Microbiol. Biotechnol. 1989, 32, 193–198. [Google Scholar] [CrossRef]
- Tremblay, P.-L.; Höglund, D.; Koza, A.; Bonde, I.; Zhang, T. Adaptation of the Autotrophic Acetogen Sporomusa ovata to Methanol Accelerates the Conversion of CO2 to Organic Products. Sci. Rep. 2015, 5, 16168. [Google Scholar] [CrossRef] [Green Version]
- Litty, D.; Müller, V. A Na+ A1AO ATP Synthase with a V-Type c Subunit in a Mesophilic Bacterium. FEBS J. 2020, 287, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Coskun, U.; Grüber, G.; Koch, M.H.J.; Godovac-Zimmermann, J.; Lemker, T.; Müller, V. Cross-Talk in the A1-ATPase from Methanosarcina mazei Gö1 Due to Nucleotide Binding. J. Biol. Chem. 2002, 277, 17327–17333. [Google Scholar] [CrossRef] [PubMed]
- Cossio, P.; Allegretti, M.; Mayer, F.; Müller, V.; Vonck, J.; Hummer, G. Bayesian Inference of Rotor Ring Stoichiometry from Electron Microscopy Images of Archaeal ATP Synthase. Microscopy 2018, 67, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Vonck, J.; Pisa, K.Y.; Morgner, N.; Brutschy, B.; Müller, V. Three-Dimensional Structure of A1A0 ATP Synthase from the Hyperthermophilic Archaeon Pyrococcus furiosus by Electron Microscopy. J. Biol. Chem. 2009, 284, 10110–10119. [Google Scholar] [CrossRef]
- Kremp, F.; Müller, V. Methanol and Methyl Group Conversion in Acetogenic Bacteria: Biochemistry, Physiology and Application. FEMS Microbiol. Rev. 2021, 45, fuaa040. [Google Scholar] [CrossRef]
- Bar-Even, A.; Flamholz, A.; Noor, E.; Milo, R. Thermodynamic Constraints Shape the Structure of Carbon Fixation Pathways. Biochim. Biophys. Acta 2012, 1817, 1646–1659. [Google Scholar] [CrossRef]
- Furdui, C.; Ragsdale, S.W. The Role of Pyruvate Ferredoxin Oxidoreductase in Pyruvate Synthesis during Autotrophic Growth by the Wood-Ljungdahl Pathway. J. Biol. Chem. 2000, 275, 28494–28499. [Google Scholar] [CrossRef]
- Pinto, R.; Harrison, J.S.; Hsu, T.; Jacobs, W.R.; Leyh, T.S. Sulfite Reduction in Mycobacteria. J. Bacteriol. 2007, 189, 6714–6722. [Google Scholar] [CrossRef]
- Kitabatake, M.; So, M.W.; Tumbula, D.L.; Söll, D. Cysteine Biosynthesis Pathway in the Archaeon Methanosarcina barkeri Encoded by Acquired Bacterial Genes? J. Bacteriol. 2000, 182, 143–145. [Google Scholar] [CrossRef]
- Hallenbeck, P.C.; Clark, M.A.; Barrett, E.L. Characterization of Anaerobic Sulfite Reduction by Salmonella typhimurium and Purification of the Anaerobically Induced Sulfite Reductase. J. Bacteriol. 1989, 171, 3008–3015. [Google Scholar] [CrossRef] [Green Version]
- Nakatani, T.; Ohtsu, I.; Nonaka, G.; Wiriyathanawudhiwong, N.; Morigasaki, S.; Takagi, H. Enhancement of Thioredoxin/Glutaredoxin-Mediated L-Cysteine Synthesis from S-Sulfocysteine Increases L-Cysteine Production in Escherichia coli. Microb. Cell Factories 2012, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.; Krzycki, J.A. Sequence and Transcript Analysis of a Novel Methanosarcina barkeri Methyltransferase II Homolog and Its Associated Corrinoid Protein Homologous to Methionine Synthase. J. Bacteriol. 1996, 178, 6599–6607. [Google Scholar] [CrossRef] [PubMed]
- Aitken, S.M.; Lodha, P.H.; Morneau, D.J.K. The Enzymes of the Transsulfuration Pathways: Active-Site Characterizations. Biochim. Biophys. Acta BBA-Proteins Proteom. 2011, 1814, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Sampson, E.M.; Bobik, T.A. Microcompartments for B12-Dependent 1,2-Propanediol Degradation Provide Protection from DNA and Cellular Damage by a Reactive Metabolic Intermediate. J. Bacteriol. 2008, 190, 2966–2971. [Google Scholar] [CrossRef]
- Liu, G.; Jiang, Y.-M.; Liu, Y.-C.; Han, L.-L.; Feng, H. A Novel DNA Methylation Motif Identified in Bacillus pumilus BA06 and Possible Roles in the Regulation of Gene Expression. Appl. Microbiol. Biotechnol. 2020, 104, 3445–3457. [Google Scholar] [CrossRef]
- Nguyen, N.-P.-T.; Raynaud, C.; Meynial-Salles, I.; Soucaille, P. Reviving the Weizmann Process for Commercial N-Butanol Production. Nat. Commun. 2018, 9, 3682. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus Tag ATCC8486 | Locus Tag B2 | Position B2 | Position ATCC8486 | Gene | Nucleotide Variation ATCC8486/B2 | Type | AA Change | Description |
|---|---|---|---|---|---|---|---|---|
| B2M23_RS00035 | M5595_17445 | 3667592 | 4431 | - | G1265T | SNP | Ala421Glu | Phage terminase |
| B2M23_RS00620 | M5595_16850 | 3547040 | 124983 | - | A514G | SNP | Tyr171His | Acyl ACP thioesterase |
| B2M23_RS00770 | M5595_16695 | 3512058 | 159965 | - | C1065T | SNP | Val355Val | Transcriptional activator |
| Intergenic region | Intergenic region | 2725372 | 946652 | - | G/A | SNP | - | - |
| Intergenic region | Intergenic region | 3264417 | 407607 | - | A/- | DEL | - | - |
| B2M23_RS06095 | M5595_11305 | 2338369 | 1333655 | cheY | A187G | SNP | Arg63Gly | Response regulator |
| B2M23_RS08865 | M5595_08530 | 1755671 | 1916353 | aspS | G1004A | SNP | Gly335Glu | Aspartate--tRNA ligase |
| B2M23_RS08970 | M5595_08425 | 1735036 | 1936988 | - | G1007T | SNP | Thr336Asp | Bacitracin export permease |
| Intergenic region | Intergenic region | 1442364 | 2231325 | - | A/C | SNP | - | Lysin riboswitch |
| B2M23_RS11215 | M5595_20825 | 4406797 | 2387123 | - | -/G | INS | - | Hypothetical protein |
| Intergenic region | M5595_20830 | 4406887 | 2387213 | - | -/A | INS | - | Hypothetical protein |
| Intergenic region | M5595_20830 | 4406916 | 2387242 | - | -/T | INS | - | Hypothetical protein |
| B2M23_RS13215 | M5595_19385 | 4089680 | 2815304 | - | G234T | SNP | Met78Ile | Mobilization protein |
| Intergenic region | M5595_00360 | 73507 | 3183052 | - | -/C | INS | - | Restriction endonuclease subunit S |
| Intergenic region | M5595_00360 | 73556 | 3183101 | - | -/TG | INS | - | Restriction endonuclease subunit S |
| Intergenic region | M5595_00360 | 73990 | 3183516 | - | -/G | INS | - | Restriction endonuclease subunit S |
| Intergenic region | Intergenic region | 367145 | 3476696 | - | T/- | DEL | - | - |
| B2M23_RS17820 | M5595_03295 | 694242 | 3803795 | ktrA | A/- | DEL | TrkA family potassium uptake protein | |
| B2M23_RS18850 | M5595_04320 | 887882 | 3997435 | pucK | C/T | SNP | Ile116Thr | Uric acid permease PucK/Xanthine permease |
| B2M23_RS18850 | M5595_04320 | 887885 | 3997438 | pucK | A/G | SNP | Gly117Asp | Uric acid permease PucK/Xanthine permease |
| B2M23_RS19910 | M5595_05415 | 1121402 | 4230955 | pbpF | T/C | SNP | Ser459Pro | Transglycosylase domain-containing protein |
| Strain | Motif | Predicted RM Type | Modified Position | Methylation Type | Fraction (%) | Number of Methylated Motifs Detected | Number of Total Motifs Detected |
|---|---|---|---|---|---|---|---|
| E. limosum B2 | GCGRAG | II | 5 | m6A | 67.6 | 2058 | 3042 |
| CAAAAAR | II | 6 | m6A | 37.3 | 1623 | 4350 | |
| CNNTAYNNNNNTCC | I | 5 | m6A | 27.9 | 293 | 1052 | |
| E. limosum B2 clone 2 | GCGRAG | II | 5 | m6A | 81 | 2464 | 3042 |
| CAAAAAR | II | 6 | m6A | 83.7 | 3642 | 4350 | |
| CNNTAYNNNNGTG | I | 5 | m6A | 79.8 | 576 | 1052 |
| Locus Tag E. limosum B2 | Position | WT | Variant | Type | 10th | 25th | 50th | 75th | C1 | C2 | C3 | Gene | Gene in 5′ of Intergenic Region | Locus tag 5′ Gene Intergenic Region | Gene in 3′ of Intergenic Region | Locus tag 3′ Gene Intergenic Region |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M5595_20825 | 4406858 | - | T | INS | 100 | 100 | 100 | 100 | 100 | 100 | 100 | Hypothetical protein | - | - | - | - |
| M5595_20830 | 4406927 | - | T | INS | 100 | 100 | 100 | 100 | 100 | 100 | Hypothetical protein | - | - | - | - | |
| M5595_00020 | 4683 | G | A | SNP | 100 | 100 | 100 | 100 | 100 | Hypothetical protein | - | - | - | - | ||
| M5595_04930 | 1017540 | C | A | SNP | 100 | 100 | 100 | Hypothetical protein | - | - | - | - | ||||
| M5595_17525 | 3682586 | A | C | SNP | 36.2 | 22 | Hypothetical protein | - | - | - | - | |||||
| - | 1193634 | A | - | DEL | 58.9 | Intergenic region | Ketol-acid reductoisomerase (3′ -> 5′) | Alpha-amylase family glycosyl hydrolase (3′ -> 5′) | ||||||||
| M5595_16535 | 3470810 | G | A | SNP | 100 | Sugar binding transcriptionnal regulator | - | - | - | - | ||||||
| M5595_12615 | 2631029 | T | C | SNP | 100 | HAD-IA family hydrolase | - | - | - | - | ||||||
| M5595_17525 | 3682598 | C | T | SNP | 31.8 | Hypothetical protein | - | - | - | - | ||||||
| - | 4262268 | C | - | DEL | 100 | Intergenic region | Threonine synthase (3′ -> 5′) | M5595_20065 | Homoserine dehydrogenase (5′ -> 3′) | M5595_20070 | ||||||
| M5595_06115 | 1274807 | T | C | SNP | 100 | Butyryl-CoA:acetate CoA-transferase | - | - | - | - | ||||||
| M5595_11550 | 2401587 | C | T | SNP | 75.5 | Prepilin peptidase | - | - | - | - | ||||||
| M5595_08805 | 1810120 | T | - | DEL | 95.9 | Hypothetical protein | - | - | - | - | ||||||
| - | 2743930 | A | - | DEL | 43.4 | Intergenic region | Helix-turn-helix transcriptional regulator (5′ -> 3′) | M5595_13160 | Lysozyme family protein (5′ -> 3′) | M5595_13165 | ||||||
| M5595_15025 | 3141664 | AT | - | DEL | 37 | Arsenate reductase family protein | - | - | - | - | ||||||
| M5595_10115 | 2083561 | A | - | DEL | 62.1 | Glycosyltransferase family 2 protein | - | - | - | - | ||||||
| - | 3378354 | T | C | SNP | 70.2 | Intergenic region | Rrf2 family transcriptional regulator (3′ -> 5′) | M5595_16095 | FtsX-like permease family protein (5′ -> 3′) | M5595_16100 | ||||||
| - | 2164707 | T | G | SNP | 70.2 | Intergenic region (riboswitch) | Leucine-rich repeat domain-containing protein (5′ -> 3′) | M5595_10385 | Energy-coupling factor ABC transporter permease (3′ -> 5′ | M5595_10390 | ||||||
| M5595_09145 | 1871776 | AACTG | - | DEL | 25 | Cysteine synthase A | - | - | - | - | ||||||
| - | 4051265 | T | - | DEL | 88.2 | Intergenic region | HPr family phosphocarrier protein (5′ -> 3′) | M5595_19190 | Aminodeoxychorismate/anthranilate synthase component II (5′ -> 3′) | M5595_19195 | ||||||
| M5595_03625 | 755658 | C | A | SNP | 35.2 | Peptidoglycan DD-metalloendopeptidase | - | - | - | - | ||||||
| - | 825607 | T | - | DEL | 46.7 | Intergenic region | PucR family transcriptional regulator (5′ -> 3′) | M5595_03980 | DUF917 domain-containing protein (5′ -> 3′) | M5595_03985 | ||||||
| M5595_00050 | 9361 | T | - | DEL | 93.3 | Hypothetical protein | - | - | - | - | ||||||
| - | 370141 | A | - | DEL | 50 | 100 | 100 | Intergenic region | ParB/RepB/Spo0J family partition protein (5′ -> 3′) | M5595_01730 | Hypothetical protein (5′ -> 3′) | M5595_01735 | ||||
| M5595_05545 | 1145373 | T | C | SNP | 100 | Nucleotide sugar dehydrogenase | - | - | - | - | ||||||
| M5595_10115 | 2082950 | A | - | DEL | 100 | Glycosyltransferase family 2 protein | - | - | - | - | ||||||
| - | 3881186 | T | C | SNP | 25 | Intergenic region | tRNA-Phe (5′ -> 3′) | M5595_18430 | Cobalamin-dependent protein (5′ -> 3′) | M5595_18435 | ||||||
| M5595_00360 | 73656 | - | T | INS | 100 | Restriction endonuclease subunit S | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pregnon, G.; Minton, N.P.; Soucaille, P. Genome Sequence of Eubacterium limosum B2 and Evolution for Growth on a Mineral Medium with Methanol and CO2 as Sole Carbon Sources. Microorganisms 2022, 10, 1790. https://doi.org/10.3390/microorganisms10091790
Pregnon G, Minton NP, Soucaille P. Genome Sequence of Eubacterium limosum B2 and Evolution for Growth on a Mineral Medium with Methanol and CO2 as Sole Carbon Sources. Microorganisms. 2022; 10(9):1790. https://doi.org/10.3390/microorganisms10091790
Chicago/Turabian StylePregnon, Guillaume, Nigel P. Minton, and Philippe Soucaille. 2022. "Genome Sequence of Eubacterium limosum B2 and Evolution for Growth on a Mineral Medium with Methanol and CO2 as Sole Carbon Sources" Microorganisms 10, no. 9: 1790. https://doi.org/10.3390/microorganisms10091790
APA StylePregnon, G., Minton, N. P., & Soucaille, P. (2022). Genome Sequence of Eubacterium limosum B2 and Evolution for Growth on a Mineral Medium with Methanol and CO2 as Sole Carbon Sources. Microorganisms, 10(9), 1790. https://doi.org/10.3390/microorganisms10091790