
SARS-CoV-2 Amino Acid Mutations Detection in Greek Patients Infected in the First Wave of the Pandemic

 ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. SARS-CoV-2 Isolates
2.2. NGS and Data Analysis
2.3. SARS-CoV-2 RNA Quantification by Reverse Transcription-Quantitative PCR (RT-qPCR)
2.4. Detection of SARS-CoV-2 Mutations in Sequences Submitted to Online Databases
2.5. Bioinformatics Analysis for Prediction of Protein Structure and Stability
3. Results
3.1. SARS-CoV-2 Genomes Used in This Study
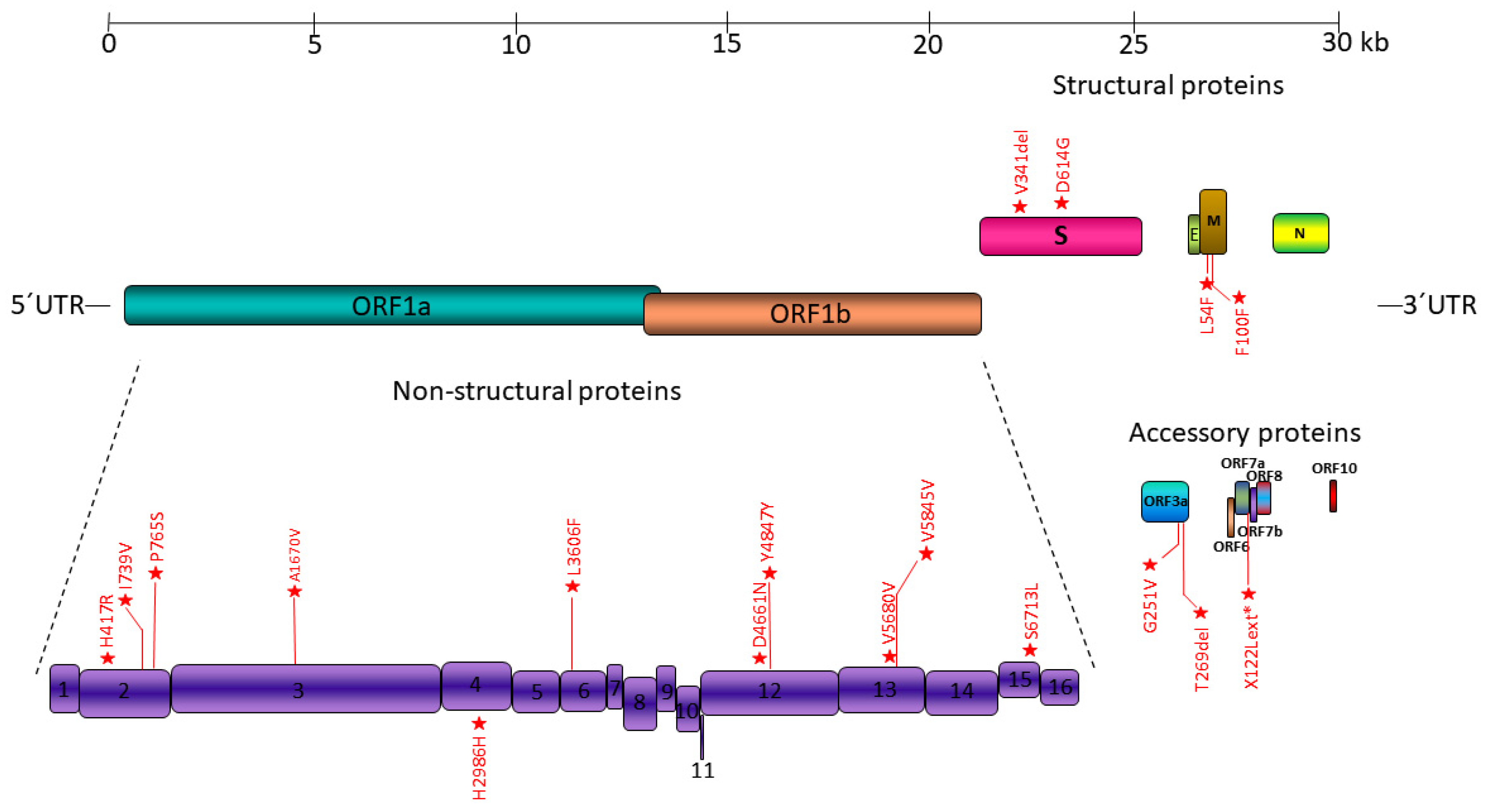
3.2. Mutation Sites in SARS-CoV-2 Genome
3.3. Most Common Mutations among the Genotypes Studied
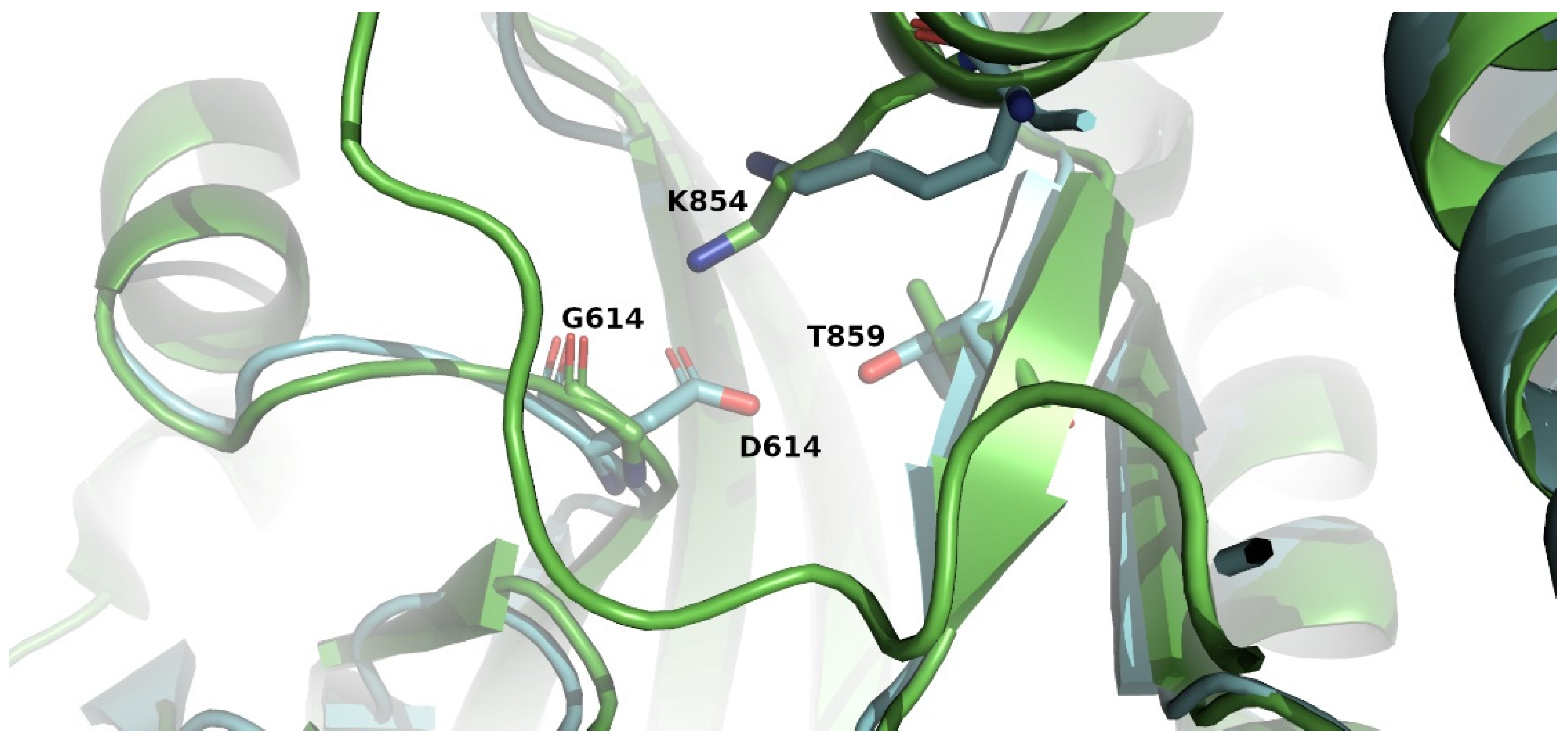
3.4. Empirical Analysis of Changes in Structure and Stability Parameters
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Bio-Med. Atenei Parm. 2020, 91, 157–160. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Naming the Coronavirus Disease (COVID-19) and the Virus that Causes It. 2020. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it (accessed on 1 May 2022).
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Testing for COVID-19. 4 October 2021. Available online: https://www.cdc.gov/coronavirus/2019-ncov/testing/index.html (accessed on 1 May 2022).
- National Institutes of Health. Clinical Spectrum of SARS-CoV-2 Infection. 19 October 2021. Available online: https://www.covid19treatmentguidelines.nih.gov/overview/clinical-spectrum/ (accessed on 1 May 2022).
- Bonaccorsi, G.; Pierri, F.; Cinelli, M.; Flori, A.; Galeazzi, A.; Porcelli, F.; Schmidt, A.L.; Valensise, C.M.; Scala, A.; Quattrociocchi, W.; et al. Economic and social consequences of human mobility restrictions under COVID-19. Proc. Natl. Acad. Sci. USA 2020, 117, 15530–15535. [Google Scholar] [CrossRef]
- Flaxman, S.; Mishra, S.; Gandy, A.; Unwin, H.J.T.; Mellan, T.A.; Coupland, H.; Whittaker, C.; Zhu, H.; Berah, T.; Eaton, J.W.; et al. Estimating the effects of non-pharmaceutical interventions on COVID-19 in Europe. Nature 2020, 584, 257–261. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 2020, 30, 1346–1351 e1342. [Google Scholar] [CrossRef]
- NCBI. Severe Acute Respiratory Syndrome Coronavirus 2 Isolate Wuhan-Hu-1, Complete Genome; NCBI: Bethesda, MD, USA, 2020. [Google Scholar]
- Zhu, G.; Zhu, C.; Zhu, Y.; Sun, F. Minireview of progress in the structural study of SARS-CoV-2 proteins. Curr. Res. Microb. Sci. 2020, 1, 53–61. [Google Scholar] [CrossRef]
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.M.; Bergeron, J.R.C. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 2020, 7, 605236. [Google Scholar] [CrossRef]
- Bhatt, P.R.; Scaiola, A.; Loughran, G.; Leibundgut, M.; Kratzel, A.; Meurs, R.; Dreos, R.; O’Connor, K.M.; McMillan, A.; Bode, J.W.; et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science 2021, 372, 1306–1313. [Google Scholar] [CrossRef]
- Redondo, N.; Zaldivar-Lopez, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 708264. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280 e278. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Áine O’Toole, E.S.; Rambaut, A. Lineage List. Available online: https://cov-lineages.org/lineage_list.html (accessed on 1 May 2022).
- Cella, E.; Benedetti, F.; Fabris, S.; Borsetti, A.; Pezzuto, A.; Ciotti, M.; Pascarella, S.; Ceccarelli, G.; Zella, D.; Ciccozzi, M.; et al. SARS-CoV-2 Lineages and Sub-Lineages Circulating Worldwide: A Dynamic Overview. Chemotherapy 2021, 66, 3–7. [Google Scholar] [CrossRef]
- O’Toole, Á.; Hill, V.; Pybus, O.G.; Watts, A.; Bogoch, I.I.; Khan, K.; Messina, J.P.; Tegally, H.; Lessells, R.R.; Giandhari, J.; et al. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2 with grinch. Wellcome Open Res. 2021, 6, 121. [Google Scholar] [CrossRef]
- SARS-CoV-2 Variant Classifications and Definitions. 10 March 2022. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 1 May 2022).
- Chan, J.F.; Yuan, S.; Kok, K.H.; To, K.K.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.; Poon, R.W.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Hui, D.; Wu, A.; Chan, P.; Cameron, P.; Joynt, G.M.; Ahuja, A.; Yung, M.Y.; Leung, C.B.; To, K.F.; et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986–1994. [Google Scholar] [CrossRef]
- Peiris, J.S.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Hoque, M.N.; Islam, M.R.; Akter, S.; Rubayet Ul Alam, A.S.M.; Siddique, M.A.; Saha, O.; Rahaman, M.M.; Sultana, M.; Crandall, K.A.; et al. Epitope-based chimeric peptide vaccine design against S, M and E proteins of SARS-CoV-2, the etiologic agent of COVID-19 pandemic: An in silico approach. PeerJ 2020, 8, e9572. [Google Scholar] [CrossRef] [PubMed]
- Domingo, P.; Mur, I.; Pomar, V.; Corominas, H.; Casademont, J.; de Benito, N. The four horsemen of a viral Apocalypse: The pathogenesis of SARS-CoV-2 infection (COVID-19). EBioMedicine 2020, 58, 102887. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.; Fang, Y.; Liu, J.; Ye, Q.; Ding, L. SARS-CoV-2 spike L452R mutation increases Omicron variant fusogenicity and infectivity as well as host glycolysis. Signal Transduct. Target. Ther. 2022, 7, 76. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, B.; Vazquez-Sirvent, L.; Soria, M.E.; Minguez, P.; Salar-Vidal, L.; Garcia-Crespo, C.; Gallego, I.; Avila, A.; Llorens, C.; Soriano, B.; et al. Vaccine-breakthrough infections with SARS-CoV-2 Alpha mirror mutations in Delta Plus, Iota and Omicron. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Weng, S.; Zhou, H.; Ji, C.; Li, L.; Han, N.; Yang, R.; Shang, J.; Wu, A. Conserved Pattern and Potential Role of Recurrent Deletions in SARS-CoV-2 Evolution. Microbiol. Spectr. 2022, 10, e02191-21. [Google Scholar] [CrossRef]
- Wu, H.; Xing, N.; Meng, K.; Fu, B.; Xue, W.; Dong, P.; Tang, W.; Xiao, Y.; Liu, G.; Luo, H.; et al. Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host Microbe 2021, 29, 1788–1801 e1786. [Google Scholar] [CrossRef]
- Laamarti, M.; Alouane, T.; Kartti, S.; Chemao-Elfihri, M.W.; Hakmi, M.; Essabbar, A.; Laamarti, M.; Hlali, H.; Bendani, H.; Boumajdi, N.; et al. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. PLoS ONE 2020, 15, e0240345. [Google Scholar] [CrossRef]
- Schrors, B.; Riesgo-Ferreiro, P.; Sorn, P.; Gudimella, R.; Bukur, T.; Rosler, T.; Lower, M.; Sahin, U. Large-scale analysis of SARS-CoV-2 spike-glycoprotein mutants demonstrates the need for continuous screening of virus isolates. PLoS ONE 2021, 16, e0249254. [Google Scholar] [CrossRef]
- Meredith, L.W.; Hamilton, W.L.; Warne, B.; Houldcroft, C.J.; Hosmillo, M.; Jahun, A.S.; Curran, M.D.; Parmar, S.; Caller, L.G.; Caddy, S.L.; et al. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: A prospective genomic surveillance study. Lancet. Infect. Dis. 2020, 20, 1263–1271. [Google Scholar] [CrossRef]
- Zekri, A.N.; Mohanad, M.; Hafez, M.M.; Soliman, H.K.; Hassan, Z.K.; Abouelhoda, M.; Amer, K.E.; Seadawy, M.G.; Ahmed, O.S. Genome sequencing of SARS-CoV-2 in a cohort of Egyptian patients revealed mutation hotspots that are related to clinical outcomes. Biochim. Biophys. Acta. Mol. Basis Dis. 2021, 1867, 166154. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- O’Toole, A.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Omata, M. Detection of R.1 lineage severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with spike protein W152L/E484K/G769V mutations in Japan. PLoS Pathog. 2021, 17, e1009619. [Google Scholar] [CrossRef]
- Charre, C.; Ginevra, C.; Sabatier, M.; Regue, H.; Destras, G.; Brun, S.; Burfin, G.; Scholtes, C.; Morfin, F.; Valette, M.; et al. Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation. Virus Evol. 2020, 6, veaa075. [Google Scholar] [CrossRef]
- Laimer, J.; Hiebl-Flach, J.; Lengauer, D.; Lackner, P. MAESTROweb: A web server for structure-based protein stability prediction. Bioinformatics 2016, 32, 1414–1416. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ochoa-Montano, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. A Publ. Protein Soc. 2021, 30, 60–69. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Fariselli, P.; Martelli, P.L.; Savojardo, C.; Casadio, R. INPS: Predicting the impact of non-synonymous variations on protein stability from sequence. Bioinformatics 2015, 31, 2816–2821. [Google Scholar] [CrossRef] [Green Version]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef] [PubMed]
- Eskier, D.; Suner, A.; Oktay, Y.; Karakulah, G. Mutations of SARS-CoV-2 nsp14 exhibit strong association with increased genome-wide mutation load. PeerJ 2020, 8, e10181. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Omotoso, O.E.; Olugbami, J.O.; Gbadegesin, M.A. Assessment of intercontinents mutation hotspots and conserved domains within SARS-CoV-2 genome. Infect. Genet. Evol. 2021, 96, 105097. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Pongor, S.; Gyorffy, B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int. J. Antimicrob. Agents 2021, 57, 106272. [Google Scholar] [CrossRef]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e819. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751 e738. [Google Scholar] [CrossRef]
- Li, J.; Wang, H.; Mao, L.; Yu, H.; Yu, X.; Sun, Z.; Qian, X.; Cheng, S.; Chen, S.; Chen, J.; et al. Rapid genomic characterization of SARS-CoV-2 viruses from clinical specimens using nanopore sequencing. Sci. Rep. 2020, 10, 17492. [Google Scholar] [CrossRef]
- Martin, J.; Klapsa, D.; Wilton, T.; Zambon, M.; Bentley, E.; Bujaki, E.; Fritzsche, M.; Mate, R.; Majumdar, M. Tracking SARS-CoV-2 in Sewage: Evidence of Changes in Virus Variant Predominance during COVID-19 Pandemic. Viruses 2020, 12, 1144. [Google Scholar] [CrossRef]
- Sekulic, M.; Harper, H.; Nezami, B.G.; Shen, D.L.; Sekulic, S.P.; Koeth, A.T.; Harding, C.V.; Gilmore, H.; Sadri, N. Molecular Detection of SARS-CoV-2 Infection in FFPE Samples and Histopathologic Findings in Fatal SARS-CoV-2 Cases. Am. J. Clin. Pathol. 2020, 154, 190–200. [Google Scholar] [CrossRef]
- Umair, M.; Ikram, A.; Salman, M.; Khurshid, A.; Alam, M.; Badar, N.; Suleman, R.; Tahir, F.; Sharif, S.; Montgomery, J.; et al. Whole-genome sequencing of SARS-CoV-2 reveals the detection of G614 variant in Pakistan. PLoS ONE 2021, 16, e0248371. [Google Scholar] [CrossRef]
- Ugurel, O.M.; Ata, O.; Turgut-Balik, D. An updated analysis of variations in SARS-CoV-2 genome. Turk. J. Biol. Turk Biyol. Derg. 2020, 44, 157–167. [Google Scholar] [CrossRef]
- Li, T.; Liu, D.; Yang, Y.; Guo, J.; Feng, Y.; Zhang, X.; Cheng, S.; Feng, J. Phylogenetic supertree reveals detailed evolution of SARS-CoV-2. Sci. Rep. 2020, 10, 22366. [Google Scholar] [CrossRef]
- Leary, S.; Gaudieri, S.; Parker, M.D.; Chopra, A.; James, I.; Pakala, S.; Alves, E.; John, M.; Lindsey, B.B.; Keeley, A.J.; et al. Generation of a novel SARS-CoV-2 sub-genomic RNA due to the R203K/G204R variant in nucleocapsid: Homologous recombination has potential to change SARS-CoV-2 at both protein and RNA level. bioRxiv 2021. [Google Scholar] [CrossRef]
- Omotoso, O.E.; Babalola, A.D.; Matareek, A. Mutational hotspots and conserved domains of SARS-CoV-2 genome in African population. Beni-Suef Univ. J. Basic Appl. Sci. 2021, 10, 11. [Google Scholar] [CrossRef]
- Hanifehnezhad, A.; Kehribar, E.S.; Oztop, S.; Sheraz, A.; Kasirga, S.; Ergunay, K.; Onder, S.; Yilmaz, E.; Engin, D.; Oguzoglu, T.C.; et al. Characterization of local SARS-CoV-2 isolates and pathogenicity in IFNAR(-/-) mice. Heliyon 2020, 6, e05116. [Google Scholar] [CrossRef]
- Nzivo, M.M.; Budambula, N.L.M. Mutations and Epidemiology of SARS-CoV-2 Compared to Selected Corona Viruses during the First Six Months of the COVID-19 Pandemic: A Review. J. Pure Appl. Microbiol. 2021, 15, 524–533. [Google Scholar] [CrossRef]
- van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef]
- Liu, S.; Shen, J.; Fang, S.; Li, K.; Liu, J.; Yang, L.; Hu, C.D.; Wan, J. Genetic spectrum and distinct evolution patterns of SARS-CoV-2. Front. Microbiol. 2020, 11, 593548. [Google Scholar] [CrossRef]
- Alouane, T.; Laamarti, M.; Essabbar, A.; Hakmi, M.; Bouricha, E.M.; Chemao-Elfihri, M.W.; Kartti, S.; Boumajdi, N.; Bendani, H.; Laamarti, R.; et al. Genomic Diversity and Hotspot Mutations in 30,983 SARS-CoV-2 Genomes: Moving Toward a Universal Vaccine for the “Confined Virus”? Pathogens 2020, 9, 829. [Google Scholar] [CrossRef]
- Patro, L.P.P.; Sathyaseelan, C.; Uttamrao, P.P.; Rathinavelan, T. Global variation in SARS-CoV-2 proteome and its implication in pre-lockdown emergence and dissemination of 5 dominant SARS-CoV-2 clades. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2021, 93, 104973. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Ewert, W.; Günther, S.; Miglioli, F.; Falke, S.; Reinke, P.Y.A.; Niebling, S.; Günther, C.; Han, H.; Srinivasan, V.; Brognaro, H.; et al. Hydrazones and Thiosemicarbazones Targeting Protein-Protein-Interactions of SARS-CoV-2 Papain-like Protease. Front. Chem. 2022, 10, 832431. [Google Scholar] [CrossRef]
- Oostra, M.; Hagemeijer, M.C.; van Gent, M.; Bekker, C.P.; te Lintelo, E.G.; Rottier, P.J.; de Haan, C.A. Topology and membrane anchoring of the coronavirus replication complex: Not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J. Virol. 2008, 82, 12392–12405. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Wang, H.; Gao, Y.; Liu, Q.; Mu, A.; Ji, W.; Yan, L.; Zhu, Y.; Zhu, C.; et al. Structural Basis for RNA Replication by the SARS-CoV-2 Polymerase. Cell 2020, 182, 417–428.e413. [Google Scholar] [CrossRef]
- Frazier, M.N.; Dillard, L.B.; Krahn, J.M.; Perera, L.; Williams, J.G.; Wilson, I.M.; Stewart, Z.D.; Pillon, M.C.; Deterding, L.J.; Borgnia, M.J.; et al. Characterization of SARS2 Nsp15 nuclease activity reveals it’s mad about U. Nucleic Acids Res. 2021, 49, 10136–10149. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.-h.; et al. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ejikemeuwa, A.; Gerzanich, V.; Nasr, M.; Tang, Q.; Simard, J.M.; Zhao, R.Y. Understanding the Role of SARS-CoV-2 ORF3a in Viral Pathogenesis and COVID-19. Front. Microbiol. 2022, 13, 854567. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.M.; Sorum, B.; Mali, S.S.; Hoel, C.M.; Sridharan, S.; Remis, J.P.; Toso, D.B.; Kotecha, A.; Bautista, D.M.; Brohawn, S.G. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 573–582. [Google Scholar] [CrossRef]
- Lu, W.; Zheng, B.J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545. [Google Scholar] [CrossRef] [Green Version]
- McClenaghan, C.; Hanson, A.; Lee, S.J.; Nichols, C.G. Coronavirus Proteins as Ion Channels: Current and Potential Research. Front. Immunol. 2020, 11, 573339. [Google Scholar] [CrossRef]
- Bianchi, M.; Benvenuto, D.; Giovanetti, M.; Angeletti, S.; Ciccozzi, M.; Pascarella, S. Sars-CoV-2 Envelope and Membrane Proteins: Structural Differences Linked to Virus Characteristics? BioMed Res. Int. 2020, 2020, 4389089. [Google Scholar] [CrossRef]
- de Haan, C.A.M.; Vennema, H.; Rottier, P.J.M. Assembly of the Coronavirus Envelope: Homotypic Interactions between the M Proteins. J. Virol. 2000, 74, 4967–4978. [Google Scholar] [CrossRef]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef]
- Arndt, A.L.; Larson, B.J.; Hogue, B.G. A Conserved Domain in the Coronavirus Membrane Protein Tail Is Important for Virus Assembly. J. Virol. 2010, 84, 11418–11428. [Google Scholar] [CrossRef] [Green Version]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. A Combined Transmembrane Topology and Signal Peptide Prediction Method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.C.F.; Anderson, D.E.; Young, B.E.; Linster, M.; Zhu, F.; Jayakumar, J.; Zhuang, Y.; Kalimuddin, S.; Low, J.G.H.; Tan, C.W.; et al. Discovery and Genomic Characterization of a 382-Nucleotide Deletion in ORF7b and ORF8 during the Early Evolution of SARS-CoV-2. Mbio 2020, 11, e01610-20. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef]
- Guo, C.; Tsai, S.J.; Ai, Y.; Li, M.; Pekosz, A.; Cox, A.; Atai, N.; Gould, S.J. The D614G Mutation Enhances the Lysosomal Trafficking of SARS-CoV-2 Spike. bioRxiv 2020. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Zarate, S.; Eberl, S.; Gladue, D.P.; Novella, I.; Borca, M.V. Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 During the 2020 COVID-19 Pandemic. Front. Microbiol. 2020, 11, 550674. [Google Scholar] [CrossRef]
- Wu, S.; Tian, C.; Liu, P.; Guo, D.; Zheng, W.; Huang, X.; Zhang, Y.; Liu, L. Effects of SARS-CoV-2 mutations on protein structures and intraviral protein-protein interactions. J. Med. Virol. 2021, 93, 2132–2140. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Redondo, R.; Nam, H.H.; Roberts, S.C.; Simons, L.M.; Jennings, L.J.; Qi, C.; Achenbach, C.J.; Hauser, A.R.; Ison, M.G.; Hultquist, J.F.; et al. A clade of SARS-CoV-2 viruses associated with lower viral loads in patient upper airways. EBioMedicine 2020, 62, 103112. [Google Scholar] [CrossRef] [PubMed]
- Voloch, C.M.; da Silva Francisco, R., Jr.; de Almeida, L.G.P.; Brustolini, O.J.; Cardoso, C.C.; Gerber, A.L.; Guimaraes, A.P.C.; Leitao, I.C.; Mariani, D.; Ota, V.A.; et al. Intra-host evolution during SARS-CoV-2 prolonged infection. Virus Evol. 2021, 7, veab078. [Google Scholar] [CrossRef] [PubMed]
- Salehi, N.; Amiri-Yekta, A.; Totonchi, M. Profiling of Initial Available SARS-CoV-2 Sequences from Iranian Related COVID-19 Patients. Cell J. 2020, 22, 148–150. [Google Scholar] [CrossRef]
- Nassir, A.A.; Musanabaganwa, C.; Mwikarago, I. Mutation Landscape of Sars Cov2 In Africa. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tan, K.K.; Tan, J.Y.; Wong, J.E.; Teoh, B.T.; Tiong, V.; Abd-Jamil, J.; Nor’e, S.S.; Khor, C.S.; Johari, J.; Yaacob, C.N.; et al. Emergence of B.1.524(G) SARS-CoV-2 in Malaysia during the third COVID-19 epidemic wave. Sci. Rep. 2021, 11, 22105. [Google Scholar] [CrossRef]
- Puenpa, J.; Suwannakarn, K.; Chansaenroj, J.; Nilyanimit, P.; Yorsaeng, R.; Auphimai, C.; Kitphati, R.; Mungaomklang, A.; Kongklieng, A.; Chirathaworn, C.; et al. Molecular epidemiology of the first wave of severe acute respiratory syndrome coronavirus 2 infection in Thailand in 2020. Sci. Rep. 2020, 10, 16602. [Google Scholar] [CrossRef]
- Kim, J.M.; Park, S.Y.; Lee, D.; Kim, J.S.; Park, Y.; Gwack, J.; Kim, M.Y.; Song, D.H.; Jeong, S.T.; Chung, Y.S.; et al. Genomic investigation of the coronavirus disease-2019 outbreak in the Republic of Korea. Sci. Rep. 2021, 11, 6009. [Google Scholar] [CrossRef]
- Manuto, L.; Grazioli, M.; Spitaleri, A.; Fontana, P.; Bianco, L.; Bertolotti, L.; Bado, M.; Mazzotti, G.; Bianca, F.; Onelia, F.; et al. Rapid SARS-CoV-2 Intra-Host and Within-Household Emergence of Novel Haplotypes. Viruses 2022, 14, 399. [Google Scholar] [CrossRef]
- Gao, R.; Zu, W.; Liu, Y.; Li, J.; Li, Z.; Wen, Y.; Wang, H.; Yuan, J.; Cheng, L.; Zhang, S.; et al. Quasispecies of SARS-CoV-2 revealed by single nucleotide polymorphisms (SNPs) analysis. Virulence 2021, 12, 1209–1226. [Google Scholar] [CrossRef]
- Singh, D.; Yi, S.V. On the origin and evolution of SARS-CoV-2. Exp. Mol. Med. 2021, 53, 537–547. [Google Scholar] [CrossRef]
- El-Shabasy, R.M.; Nayel, M.A.; Taher, M.M.; Abdelmonem, R.; Shoueir, K.R.; Kenawy, E.R. Three waves changes, new variant strains, and vaccination effect against COVID-19 pandemic. Int. J. Biol. Macromol. 2022, 204, 161–168. [Google Scholar] [CrossRef]
- Pang, X.; Li, P.; Zhang, L.; Que, L.; Dong, M.; Xie, B.; Wang, Q.; Wei, Y.; Xie, X.; Li, L.; et al. Emerging Severe Acute Respiratory Syndrome Coronavirus 2 Mutation Hotspots Associated With Clinical Outcomes and Transmission. Front. Microbiol. 2021, 12, 3047. [Google Scholar] [CrossRef]
- Groves, D.C.; Rowland-Jones, S.L.; Angyal, A. The D614G mutations in the SARS-CoV-2 spike protein: Implications for viral infectivity, disease severity and vaccine design. Biochem. Biophys. Res. Commun. 2021, 538, 104–107. [Google Scholar] [CrossRef]
- Voss, J.D.; Skarzynski, M.; McAuley, E.M.; Maier, E.J.; Gibbons, T.; Fries, A.C.; Chapleau, R.R. Variants in SARS-CoV-2 associated with mild or severe outcome. Evol. Med. Public Health 2021, 9, 267–275. [Google Scholar] [CrossRef]
- Murata, T.; Sakurai, A.; Suzuki, M.; Komoto, S.; Ide, T.; Ishihara, T.; Doi, Y. Shedding of Viable Virus in Asymptomatic SARS-CoV-2 Carriers. mSphere 2021, 6, e00019-21. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Hozumi, Y.; Yin, C.; Wei, G.W. Decoding Asymptomatic COVID-19 Infection and Transmission. J. Phys. Chem. Lett. 2020, 11, 10007–10015. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Ilmain, J.K.; Guo, X.; Bhabha, G.; tenOever, B.R.; Sanjana, N.E. The Spike D614G mutation increases SARS-CoV-2 infection of multiple human cell types. eLife 2021, 10, e65365. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H., 3rd; Leist, S.R.; Schafer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Koyama, T.; Weeraratne, D.; Snowdon, J.L.; Parida, L. Emergence of Drift Variants That May Affect COVID-19 Vaccine Development and Antibody Treatment. Pathogens 2020, 9, 324. [Google Scholar] [CrossRef]
- Weissman, D.; Alameh, M.G.; de Silva, T.; Collini, P.; Hornsby, H.; Brown, R.; LaBranche, C.C.; Edwards, R.J.; Sutherland, L.; Santra, S.; et al. D614G Spike Mutation Increases SARS CoV-2 Susceptibility to Neutralization. Cell Host Microbe 2021, 29, 23–31.e24. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.; Bruneau, N.; Fasce, R.; Martin, H.S.; Balanda, M.; Bustos, P.; Ulloa, S.; Mora, J.; Ramirez, E. Neutralization of alpha, gamma, and D614G SARS-CoV-2 variants by CoronaVac vaccine-induced antibodies. J. Med. Virol. 2022, 94, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Su, C.M.; Wang, L.; Yoo, D. Activation of NF-kappaB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci. Rep. 2021, 11, 13464. [Google Scholar] [CrossRef] [PubMed]
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S.; Gilbert, J.A. SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 2020, 5, e00266-20. [Google Scholar] [CrossRef]
- Kostaki, E.G.; Pavlopoulos, G.A.; Verrou, K.M.; Ampatziadis-Michailidis, G.; Harokopos, V.; Hatzis, P.; Moulos, P.; Siafakas, N.; Pournaras, S.; Hadjichristodoulou, C.; et al. Molecular Epidemiology of SARS-CoV-2 in Greece Reveals Low Rates of Onward Virus Transmission after Lifting of Travel Restrictions Based on Risk Assessment during Summer 2020. mSphere 2021, 6, e0018021. [Google Scholar] [CrossRef]
- Spanakis, N.; Kassela, K.; Dovrolis, N.; Bampali, M.; Gatzidou, E.; Kafasi, A.; Froukala, E.; Stavropoulou, A.; Lilakos, K.; Veletza, S.; et al. A main event and multiple introductions of SARS-CoV-2 initiated the COVID-19 epidemic in Greece. J. Med. Virol. 2021, 93, 2899–2907. [Google Scholar] [CrossRef]
- Cosar, B.; Karagulleoglu, Z.Y.; Unal, S.; Ince, A.T.; Uncuoglu, D.B.; Tuncer, G.; Kilinc, B.R.; Ozkan, Y.E.; Ozkoc, H.C.; Demir, I.N.; et al. SARS-CoV-2 Mutations and their Viral Variants. Cytokine Growth Factor Rev. 2022, 63, 10–22. [Google Scholar] [CrossRef]
- Pechlivanis, N.; Tsagiopoulou, M.; Maniou, M.C.; Togkousidis, A.; Mouchtaropoulou, E.; Chassalevris, T.; Chaintoutis, S.C.; Petala, M.; Kostoglou, M.; Karapantsios, T.; et al. Detecting SARS-CoV-2 lineages and mutational load in municipal wastewater and a use-case in the metropolitan area of Thessaloniki, Greece. Sci. Rep. 2022, 12, 2659. [Google Scholar] [CrossRef]
- Halvatsiotis, P.; Vassiliu, S.; Koulouvaris, P.; Chatzantonaki, K.; Asonitis, K.; Charvalos, E.; Siatelis, A.; Houhoula, D. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Mutational Pattern in the Fourth Pandemic Phase in Greece. Curr. Issues Mol. Biol. 2022, 44, 329–335. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Sample Name | Accession Number | Age | Gender # | Viral Load Log (−ΔΔCt) | Lineage | Amino Acid (Nucleotide) Mutations |
|---|---|---|---|---|---|---|
| 1851-S45 | SRR19213734 | 23 | M | 3.89 | A | S_D614G (A23403G) |
| 4405-S34 | SRR19215536 | 42 | M | 1.70 | A | S_D614G (A23403G), ORF7a_X122Lext * (G27758T) |
| 2384-S29 | SRR19215599 | 45 | F | 4.13 | A | S_D614G (A23403G) |
| 3125-S32 | SRR19215604 | 40 | M | 2.12 | B.39 | ORF1ab_H417R or Nsp2_H237R (A1515G), ORF1ab_H2986H or Nsp4_H223H (C9223T), ORF1ab_L3606F or Nsp6_L37F (G11083T), ORF1ab_D4661N or Nsp12_D269N (G14245A), ORF1ab_Y4847Y or Nsp12_Y455Y (C14805T), ORF1ab_V5845V or Nsp13_V521V (A17799G), ORF3a_G251V (G26144T) |
| 3396-S31 | SRR19215602 | 59 | M | 3.81 | B.40 | ORF1ab_I739V or Nsp2_I559V (A2480G), ORF1ab_P765S or Nsp2_P585S (C2558T), ORF1ab_Y4847Y or Nsp12_Y455Y (C14805T), ORF1ab_S6713L or Nsp15_S261L (C20402T), ORF3a_G251V (G26144T) |
| 9096-S37 | SRR19215566 | 42 | M | −0.69 | A | ORF1ab_A1670V or Nsp3_A852V (C3064T), S_F342fs # (GTT22583G), S_D614G (A23403G), M_L54F (C26682T), M_F100F (C26822T) |
| 9097-S38 | SRR19215601 | 37 | F | −0.35 | B.40 | ORF1ab_I739V or Nsp2_I559V (A2480G), ORF1ab_P765S or Nsp2_P585S (C2558T), ORF1ab_L3606F or Nsp6_L37F (G11083T), ORF1ab_Y4847Y or Nsp12_Y455Y (C14805T), ORF3a_T271del (G26199-ACT) |
| 0524-S39 | SRR19215600 | 39 | F | −1.22 | A | ORF1ab_L3606F or Nsp6_L37F (G11083T), ORF1ab_Y4847Y or Nsp12_Y455Y (C14805T), ORF1ab_V5680V or Nsp13_V356V (C17304A), S_D614G (A23403G) |
| 2098-S40 | SRR19215598 | 26 | M | −1.37 | B.39 | ORF1ab_H417R or Nsp2_H237R (A1515G), ORF1ab_H2986H or Nsp4_H223H (C9223T), ORF1ab_L3606F or Nsp6_L37F (G11083T), ORF1ab_D4661N or Nsp12_D269N (G14245A), ORF1ab_Y4847Y or Nsp12_Y455Y (C14805T), ORF1ab_V5845V or Nsp13_V521V (A17799G) |
| 6642-S30 | SRR19215603 | 59 | M | 4.56 | B.40 | ORF1ab_I739V or Nsp2_I559V (A2480G), ORF1ab_P765S or Nsp2_P585S (C2558T), ORF1ab_Y4847Y or Nsp12_Y455Y (C14805T), ORF1ab_S6713L or Nsp15_S261L (C20402T), ORF3a_G251V (G26144T) |
| Mutations | ORF1ab Mutations | MutType | C1 | C2 | C3 | Ref-Codon | Mut-Codon | Count | Proportion |
|---|---|---|---|---|---|---|---|---|---|
| Nsp2_H237R | H417R | nonsyn | 1514 | 1515 | 1516 | cAt | cGt | 1219 | 0.000233 |
| Nsp2_I559V | I739V | nonsyn | 2480 | 2481 | 2482 | Att | Gtt | 3029 | 0.000579 |
| Nsp2_P585S | P765S | nonsyn | 2558 | 2559 | 2560 | Cca | Tca | 3215 | 0.000615 |
| Nsp3_A852V | A1670V | nonsyn | 5273 | 5274 | 5275 | gCa | gTa | 1428 | 0.000273 |
| Nsp4_H223H | H2986H | syn | 9221 | 9222 | 9223 | caC | caT | 7684 | 0.00147 |
| Nsp6_L37F | L3606F | nonsyn | 11,081 | 11,082 | 11,083 | ttG | ttT | 133,400 | 0.025514 |
| Nsp12_D269N | D4661N | nonsyn | 14,245 | 14,246 | 14,247 | Gat | Aat | 1456 | 0.000278 |
| Nsp12_Y455Y | Y4847Y | syn | 14,803 | 14,804 | 14,805 | taC | taT | 76,696 | 0.014669 |
| Nsp13_V356V | V5680V | syn | 17,302 | 17,303 | 17,304 | gtC | gtA | 136 | 0.000026 |
| Nsp13_V521V | V5845V | syn | 17,797 | 17,798 | 17,799 | gtA | gtG | 3079 | 0.000589 |
| Nsp15_S261L | S6713L | nonsyn | 20,401 | 20,402 | 20,403 | tCa | tTa | 5484 | 0.001049 |
| S_V341del | del | 22,583 | 22,584 | 22,585 | gTT | g-- | 13 | 0.000002 | |
| S_D614G | nonsyn | 23,402 | 23,403 | 23,404 | gAt | gGt | 5,182,511 | 0.991216 | |
| ORF3a_G251V | nonsyn | 26,143 | 26,144 | 26,145 | gGt | gTt | 8069 | 0.001543 | |
| M_F100F | syn | 26,820 | 26,821 | 26,822 | ttC | ttT | 8300 | 0.001587 | |
| ORF7a_X122L | nonsyn | 27,757 | 27,758 | 27,759 | tGa | tTa | 2104 | 0.000402 |
| Protein | Mutation | Protein Structure | Dynamut2 (ΔΔGStability) | SDM (ΔΔGpred.) | MAESTROweb (ΔΔGpred.) |
|---|---|---|---|---|---|
| Nsp2 | H417R (H237R) # | 7MSW | −0.16 | +0.07 | −0.020 (0.902) |
| Nsp2 | I739V & P765S (I559V & P585S) | 7MSW | −0.28 | −2.13 & 0.46 | +0.035 (0.902) |
| Nsp3 | A1670V (A852V) | 7QCM | −0.58 | +1.16 | +0.010 (0.923) |
| Nsp12 | D4661N (D269N) | 7C2K | −0.21 | −0.11 | +0.095 (0.862) |
| Nsp15 | S6713L (S261L) | 7N06 | +0.28 | −0.3 | +0.006 (0.877) |
| ORF3a | G251V | D-I-Tasser model | −1.53 | +0.31 | +0.473 (0.845) |
| M protein | L54F | D-I-Tasser model | −0.78 | −1.31 | +2.230 (0.825) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassilaki, N.; Papadimitriou, K.; Ioannidis, A.; Papandreou, N.C.; Milona, R.S.; Iconomidou, V.A.; Chatzipanagiotou, S. SARS-CoV-2 Amino Acid Mutations Detection in Greek Patients Infected in the First Wave of the Pandemic. Microorganisms 2022, 10, 1430. https://doi.org/10.3390/microorganisms10071430
Vassilaki N, Papadimitriou K, Ioannidis A, Papandreou NC, Milona RS, Iconomidou VA, Chatzipanagiotou S. SARS-CoV-2 Amino Acid Mutations Detection in Greek Patients Infected in the First Wave of the Pandemic. Microorganisms. 2022; 10(7):1430. https://doi.org/10.3390/microorganisms10071430
Chicago/Turabian StyleVassilaki, Niki, Konstantinos Papadimitriou, Anastasios Ioannidis, Nikos C. Papandreou, Raphaela S. Milona, Vassiliki A. Iconomidou, and Stylianos Chatzipanagiotou. 2022. "SARS-CoV-2 Amino Acid Mutations Detection in Greek Patients Infected in the First Wave of the Pandemic" Microorganisms 10, no. 7: 1430. https://doi.org/10.3390/microorganisms10071430
APA StyleVassilaki, N., Papadimitriou, K., Ioannidis, A., Papandreou, N. C., Milona, R. S., Iconomidou, V. A., & Chatzipanagiotou, S. (2022). SARS-CoV-2 Amino Acid Mutations Detection in Greek Patients Infected in the First Wave of the Pandemic. Microorganisms, 10(7), 1430. https://doi.org/10.3390/microorganisms10071430