
Analysis of Microbiota Persistence in Quebec’s Terroir Cheese Using a Metabarcoding Approach

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Sequencing Data and Taxonomic Assignation
3.2. Choice of the Best Targets for Metabarcoding
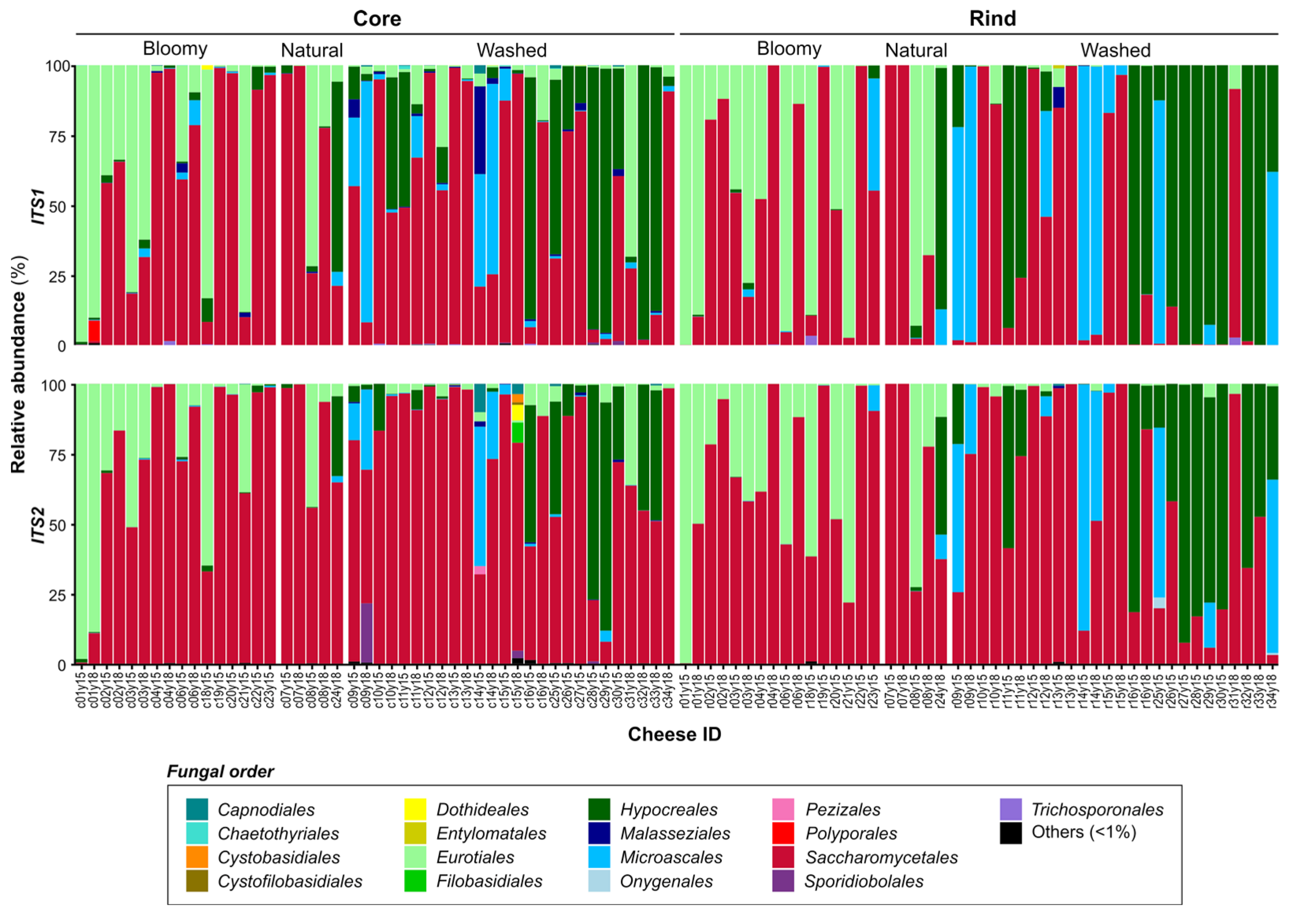
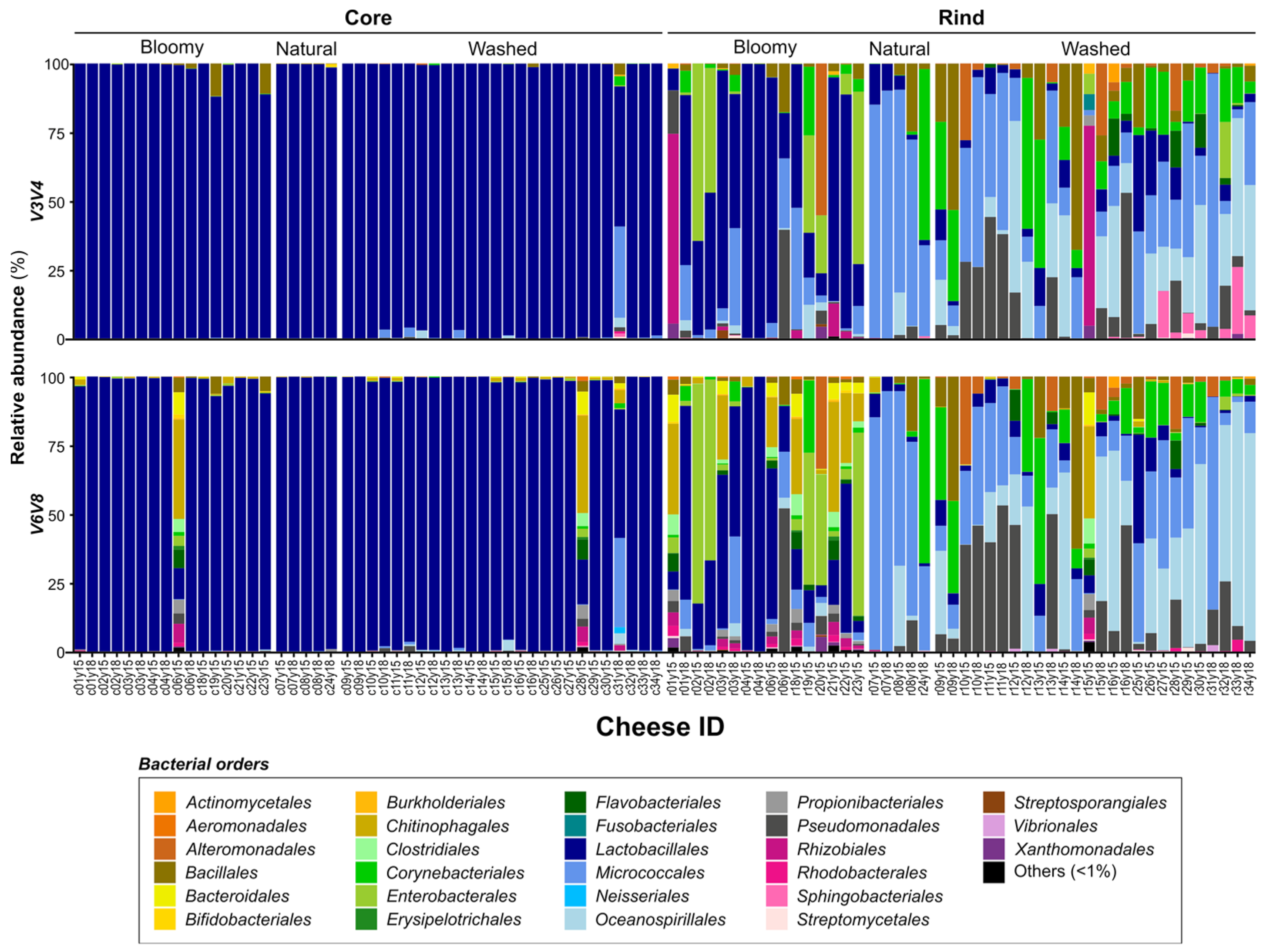
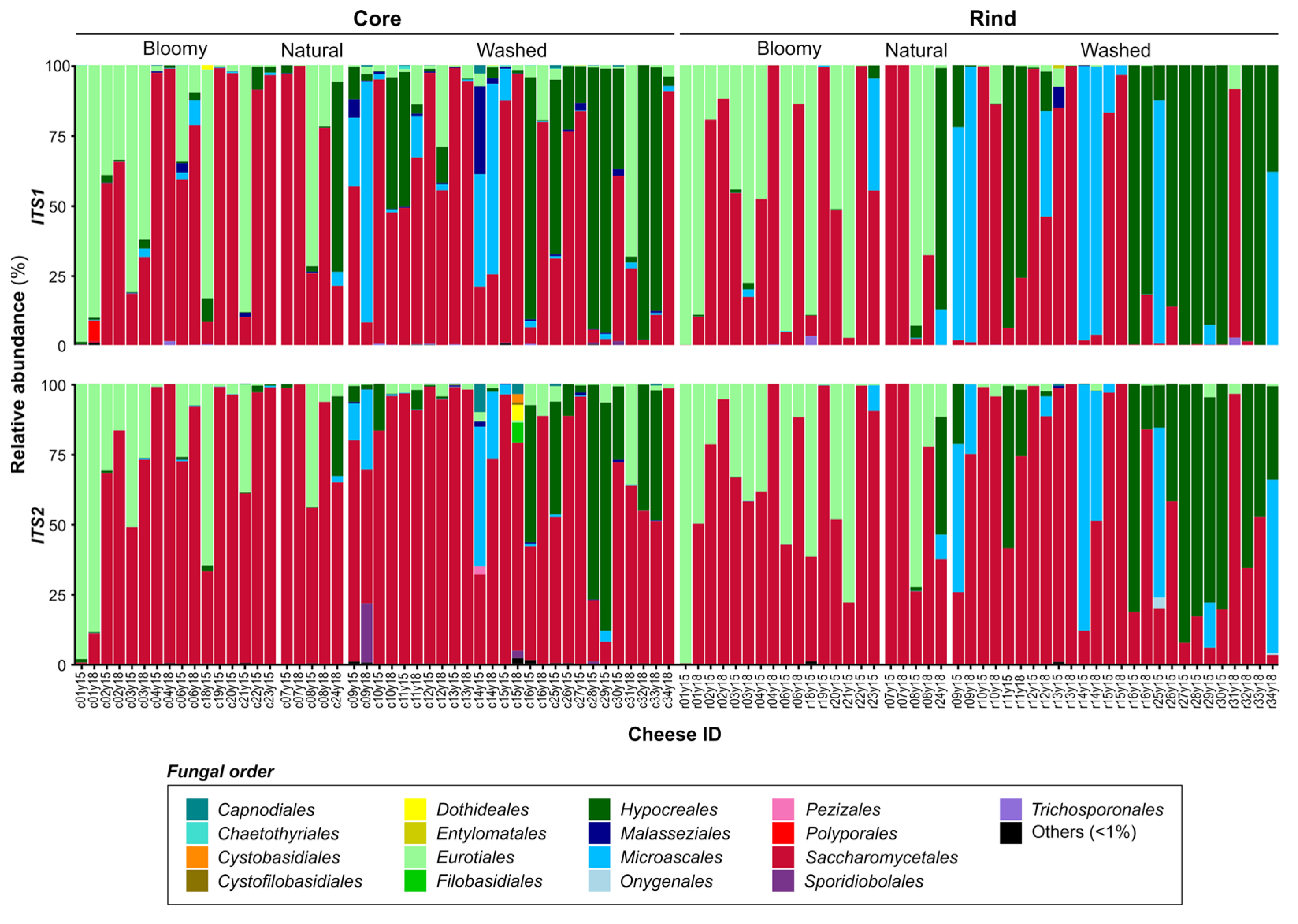
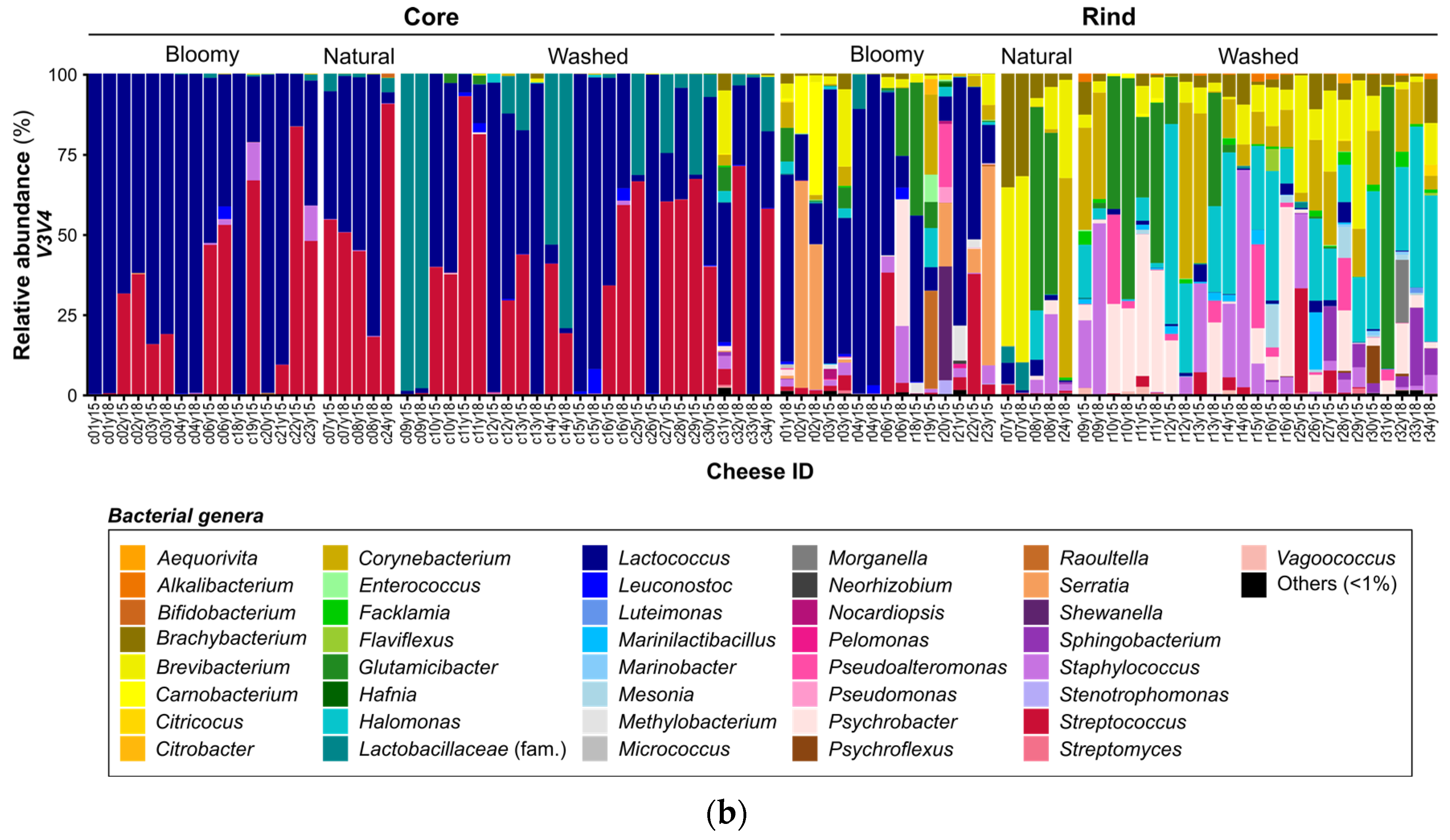
3.3. Microbial Landscape of Quebec’s Terroir Cheeses
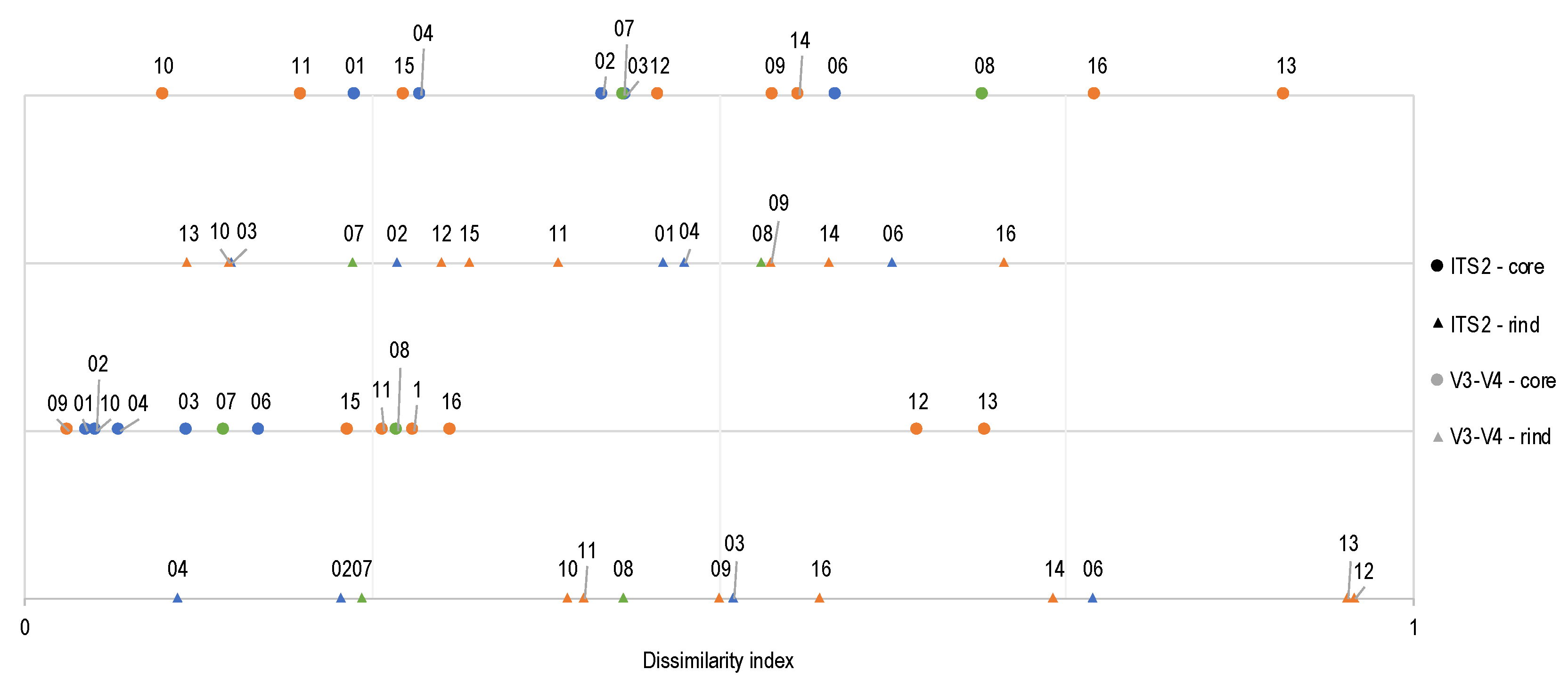
3.4. Assessing the Stability of Quebec’s Terroir Cheese Ecosystems
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Sullivan, D.J.; Cotter, P.D.; O’Sullivan, O.; Giblin, L.; McSweeney, P.L.; Sheehan, J.J. Temporal and spatial differences in microbial composition during the manufacture of a continental-type cheese. Appl. Environ. Microbiol. 2015, 81, 2525–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amenu, B.; Deeth, H.C. The impact of milk composition on Cheddar cheese manufacture. Aust. J. Dairy Technol. 2007, 62, 171–180. [Google Scholar]
- Daviau, C.; Famelart, M.-H.; Pierre, A.; Goudédranche, H.; Maubois, J.-L. Rennet coagulation of skim milk and curd drainage: Effect of pH, casein concentration, ionic strength and heat treatment. Le Lait 2000, 80, 397–415. [Google Scholar] [CrossRef] [Green Version]
- Fenelon, M.A.; Guinee, T.P. The effect of milk fat on Cheddar cheese yield and its prediction, using modifications of the Van Slyke cheese yield formula. J. Dairy Sci. 1999, 82, 2287–2299. [Google Scholar] [CrossRef]
- Quigley, L.; O’Sullivan, O.; Beresford, T.P.; Ross, R.P.; Fitzgerald, G.F.; Cotter, P.D. High-throughput sequencing for detection of subpopulations of bacteria not previously associated with artisanal cheeses. Appl. Environ. Microbiol. 2012, 78, 5717–5723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, B.E.; Button, J.E.; Santarelli, M.; Dutton, R.J. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 2014, 158, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Mills, D.A. Facility-specific “house” microbiome drives microbial landscapes of artisan cheesemaking plants. Appl. Environ. Microbiol. 2013, 79, 5214–5223. [Google Scholar] [CrossRef] [Green Version]
- Sperber, W.H. Introduction to the microbiological spoilage of foods and beverages. In Compendium of the Microbiological Spoilage of Foods and Beverages, 1st ed.; Sperber, W., Doyle, M., Eds.; Springer: New York, NY, USA, 2009; pp. 1–40. [Google Scholar] [CrossRef]
- Lavoie, K.; Touchette, M.; St-Gelais, D.; Labrie, S. Characterization of the fungal microflora in raw milk and specialty cheeses of the province of Québec. Dairy Sci. Technol. 2012, 92, 455–468. [Google Scholar] [CrossRef] [Green Version]
- Delcenserie, V.; Taminiau, B.; Delhalle, L.; Nezer, C.; Doyen, P.; Crevecoeur, S.; Roussey, D.; Korsak, N.; Daube, G. Microbiota characterization of a Belgian protected designation of origin cheese, Herve cheese, using metagenomic analysis. J. Dairy Sci. 2014, 97, 6046–6056. [Google Scholar] [CrossRef] [Green Version]
- Dugat-Bony, E.; Garnier, L.; Denonfoux, J.; Ferreira, S.; Sarthou, A.S.; Bonnarme, P.; Irlinger, F. Highlighting the microbial diversity of 12 French cheese varieties. Int. J. Food Microbiol. 2016, 238, 265–273. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, F.; Shi, X.; Wang, B.; Li, K.; Li, B.; Zhuge, B. Dynamic correlations between microbiota succession and flavor development involved in the ripening of Kazak artisanal cheese. Food Res. Int. 2018, 105, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Alegria, A.; Szczesny, P.; Mayo, B.; Bardowski, J.; Kowalczyk, M. Biodiversity in Oscypek, a traditional Polish cheese, determined by culture-dependent and -independent approaches. Appl. Environ. Microbiol. 2012, 78, 1890–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding, C. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzicalupo, A.L.; Bálint, M.; Schmitt, I. Comparison of ITS1 and ITS2 rDNA in 454 sequencing of hyperdiverse fungal communities. Fungal Ecol. 2013, 6, 102–109. [Google Scholar] [CrossRef]
- Blaalid, R.; Kumar, S.; Nilsson, R.H.; Abarenkov, K.; Kirk, P.M.; Kauserud, H. ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol. Ecol. Resour. 2013, 13, 218–224. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, F.; Laiola, M.; Blaiotta, G.; Ercolini, D. Different amplicon targets for sequencing-based studies of fungal diversity. Appl. Environ. Microbiol. 2017, 83, e00905–e00917. [Google Scholar] [CrossRef] [Green Version]
- Mbareche, H.; Veillette, M.; Bilodeau, G.J.; Duchaine, C. An amplicon-based sequencing approach for the study of aeromycology. J. Xenobiotics 2018, 8, 8–7810. [Google Scholar] [CrossRef] [Green Version]
- Mbareche, H.; Veillette, M.; Bilodeau, G.J.; Duchaine, C. Comparison of the performance of ITS1 and ITS2 as barcodes in amplicon-based sequencing of bioaerosols. PeerJ 2020, 8, e8523. [Google Scholar] [CrossRef] [Green Version]
- Mello, A.; Napoli, C.; Murat, C.; Morin, E.; Marceddu, G.; Bonfante, P. ITS-1 versus ITS-2 pyrosequencing: A comparison of fungal populations in truffle grounds. Mycologia 2011, 103, 1184–1193. [Google Scholar] [CrossRef] [Green Version]
- Monard, C.; Gantner, S.; Stenlid, J. Utilizing ITS1 and ITS2 to study environmental fungal diversity using pyrosequencing. FEMS Microbiol. Ecol. 2013, 84, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Poirier, S.; Rué, O.; Coeuret, G.; Champomier-Vergès, M.-C.; Loux, V.; Chaillou, S. Detection of an amplification bias associated to Leuconostocaceae family with a universal primer routinely used for monitoring microbial community structures within food products. BMC Res. Notes 2018, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- Ceugniez, A.; Taminiau, B.; Coucheney, F.; Jacques, P.; Delcenserie, V.; Daube, G.; Drider, D. Use of a metagenetic approach to monitor the bacterial microbiota of “Tomme d’Orchies” cheese during the ripening process. Int. J. Food Microbiol. 2017, 247, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Alessandria, V.; Ferrocino, I.; De Filippis, F.; Fontana, M.; Rantsiou, K.; Ercolini, D.; Cocolin, L. Microbiota of an italian Grana-like cheese during manufacture and ripening, unraveled by 16S rRNA-based approaches. Appl. Environ. Microbiol. 2016, 82, 3988–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, W.; Wik, M.; Vogensen, F.; Lillevang, S.; Abu Al-Soud, W.; Sørensen, S.; Jakobsen, M. Characterization of bacterial populations in Danish raw milk cheeses made with different starter cultures by denaturing gradient gel electrophoresis and pyrosequencing. Int. Dairy J. 2011, 21, 142–148. [Google Scholar] [CrossRef]
- Riquelme, C.; Camara, S.; Dapkevicius, M.; Vinuesa, P.; da Silva, C.C.; Malcata, F.X.; Rego, O.A. Characterization of the bacterial biodiversity in Pico cheese (an artisanal Azorean food). Int. J. Food Microbiol. 2015, 192, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Chamberland, J.; Lessard, M.-H.; Doyen, A.; Labrie, S.; Pouliot, Y. A sequencing approach targeting the 16S rRNA gene unravels the biofilm composition of spiral-wound membranes used in the dairy industry. Dairy Sci. Technol. 2017, 96, 827–843. [Google Scholar] [CrossRef] [Green Version]
- Ceugniez, A.; Taminiau, B.; Coucheney, F.; Jacques, P.; Delcenserie, V.; Daube, G.; Drider, D. Fungal diversity of “Tomme d’Orchies” cheese during the ripening process as revealed by a metagenomic study. Int. J. Food Microbiol. 2017, 258, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Comeau, A.M.; Li, W.K.; Tremblay, J.E.; Carmack, E.C.; Lovejoy, C. Arctic Ocean microbial community structure before and after the 2007 record sea ice minimum. PLoS ONE 2011, 6, e27492. [Google Scholar] [CrossRef]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Polme, S.; Riit, T.; Liiv, I.; Koljalg, U.; Kisand, V.; Nilsson, R.H.; Hildebrand, F.; et al. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 2015, 10, 1–43. [Google Scholar] [CrossRef]
- Chamberland, J.; Messier, T.; Dugat-Bony, E.; Lessard, M.-H.; Labrie, S.; Doyen, A.; Pouliot, Y. Influence of feed temperature to biofouling of ultrafiltration membrane during skim milk processing. Int. Dairy J. 2019, 93, 99–105. [Google Scholar] [CrossRef]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics 2018, 34, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Mahé, F.; Rognes, T.; Quince, C.; de Vargas, C.; Dunthorn, M. Swarm: Robust and fast clustering method for amplicon-based studies. PeerJ 2014, 2, e593. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjørnsgaard Aas, A.; Davey, M.L.; Kauserud, H. ITS all right mama: Investigating the formation of chimeric sequences in the ITS 2 region by DNA metabarcoding analyses of fungal mock communities of different complexities. Mol. Ecol. Resour. 2017, 17, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, H.; Zhan, Y. Characterization of microbial community composition and pathogens risk assessment in typical Italian-style salami by high-throughput sequencing technology. Food Sci. Biotechnol. 2018, 27, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Lanzén, A.; Curtis, T.P.; Davenport, R.J.; Hall, N.; Head, I.M.; Read, L.F.; Sloan, W.T. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods 2009, 6, 639–641. [Google Scholar] [CrossRef]
- Chamberland, J.; Beaulieu-Carbonneau, G.; Lessard, M.H.; Labrie, S.; Bazinet, L.; Doyen, A.; Pouliot, Y. Effect of membrane material chemistry and properties on biofouling susceptibility during milk and cheese whey ultrafiltration. J. Membr. Sci. 2017, 542, 208–216. [Google Scholar] [CrossRef]
- Alper, I.; Frenette, M.; Labrie, S. Ribosomal DNA polymorphisms in the yeast Geotrichum candidum. Fungal Biol. 2011, 115, 1259–1269. [Google Scholar] [CrossRef]
- Tedersoo, L.; Ramirez, K.S.; Nilsson, R.H.; Kaljuvee, A.; Koljalg, U.; Abarenkov, K. Standardizing metadata and taxonomic identification in metabarcoding studies. Gigascience 2015, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef]
- Taylor, D.L.; Walters, W.A.; Lennon, N.J.; Bochicchio, J.; Krohn, A.; Caporaso, J.G.; Pennanen, T. Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for Illumina amplicon sequencing. Appl. Environ. Microbiol. 2016, 82, 7217–7226. [Google Scholar] [CrossRef] [Green Version]
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an environmental DNA barcode for fungi: An in silico approach reveals potential PCR biases. BMC Microbiol. 2010, 10, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogier, J.-C.; Son, O.; Gruss, A.; Tailliez, P.; Delacroix-Buchet, A. Identification of the bacterial microflora in dairy products by temporal temperature gradient gel electrophoresis. Appl. Environ. Microbiol. 2002, 68, 3691–3701. [Google Scholar] [CrossRef] [Green Version]
- Leclercq-Perlat, M.N.; Buono, F.; Lambert, D.; Latrille, E.; Spinnler, H.E.; Corrieu, G. Controlled production of Camembert-type cheeses. Part I: Microbiological and physicochemical evolutions. J. Dairy Res. 2004, 71, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Gobbetti, M.; Burzigotti, R.; Smacchi, E.; Corsetti, A.; De Angelis, M. Microbiology and biochemistry of gorgonzola cheese during ripening. Int. Dairy J. 1997, 7, 519–529. [Google Scholar] [CrossRef]
- Giraffa, G.; Rossetti, L.; Mucchetti, G.; Addeo, F.; Neviani, E. Influence of the temperature gradient on the growth of thermophilic Lactobacilli used as natural starters in Grana cheese. J. Dairy Sci. 1998, 81, 31–36. [Google Scholar] [CrossRef]
- Almena-Aliste, M.; Mietton, B. Cheese classification, characterization, and categorization: A global perspective. Microbiol. Spectr. 2014, 2, CM-0003-2012. [Google Scholar] [CrossRef] [Green Version]
- Broadbent, J.R.; Cai, H.; Larsen, R.L.; Hughes, J.E.; Welker, D.L.; De Carvalho, V.G.; Tompkins, T.A.; Ardo, Y.; Vogensen, F.; De Lorentiis, A.; et al. Genetic diversity in proteolytic enzymes and amino acid metabolism among Lactobacillus helveticus strains. J. Dairy Sci. 2011, 94, 4313–4328. [Google Scholar] [CrossRef]
- Parente, E.; Cogan, T.M.; Powell, I.B. Chapter 8—Starter Cultures: General Aspects. In Cheese, 4th ed.; McSweeney, P.L.H., Fox, P.F., Cotter, P.D., Everett, D.W., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 201–226. [Google Scholar]
- Irlinger, F.; Helinck, S.; Jany, J.L. Chapter 11—Secondary and Adjunct Cultures. In Cheese, 4th ed.; McSweeney, P.L.H., Fox, P.F., Cotter, P.D., Everett, D.W., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 273–300. [Google Scholar]
- Auclair, J.; Accolas, J.P. Use of thermophilic lactic starters in the dairy industry. Antonie Leeuwenhoek 1983, 49, 313–326. [Google Scholar] [CrossRef]
- Lawrence, R.C.; Creamer, L.K.; Gilles, J. Texture development during cheese ripening. J. Dairy Sci. 1987, 70, 1748–1760. [Google Scholar] [CrossRef]
- Lessard, M.H.; Belanger, G.; St-Gelais, D.; Labrie, S. The composition of Camembert cheese ripening cultures modulates both mycelial growth and appearance. Appl. Environ. Microbiol. 2012, 78, 1813–1819. [Google Scholar] [CrossRef] [Green Version]
- Irlinger, F.; Layec, S.; Helinck, S.; Dugat-Bony, E. Cheese rind microbial communities: Diversity, composition and origin. FEMS Microbiol. Lett. 2015, 362, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mounier, J.; Rea, M.C.; O’Connor, P.M.; Fitzgerald, G.F.; Cogan, T.M. Growth characteristics of Brevibacterium, Corynebacterium, Microbacterium, and Staphylococcus spp. isolated from surface-ripened cheese. Appl. Environ. Microbiol. 2007, 73, 7732–7739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, N.M.; Brown, R.; Goodfellow, M.; Ward, A.C.; Beresford, T.P.; Simpson, P.J.; Fox, P.F.; Cogan, T.M. Corynebacterium mooreparkense sp. nov. and Corynebacterium casei sp. nov., isolated from the surface of a smear-ripened cheese. Int. J. Syst. Evol. Microbiol. 2001, 51, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Rea, M.C.; Gorges, S.; Gelsomino, R.; Brennan, N.M.; Mounier, J.; Vancanneyt, M.; Scherer, S.; Swings, J.; Cogan, T.M. Stability of the biodiversity of the surface consortia of Gubbeen, a red-smear cheese. J. Dairy Sci. 2007, 90, 2200–2210. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Goerges, S.; Mounier, J.; Rea, M.C.; Gelsomino, R.; Heise, V.; Beduhn, R.; Cogan, T.M.; Vancanneyt, M.; Scherer, S. Commercial ripening starter microorganisms inoculated into cheese milk do not successfully establish themselves in the resident microbial ripening consortia of a South german red smear cheese. Appl. Environ. Microbiol. 2008, 74, 2210–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feurer, C.; Vallaeys, T.; Corrieu, G.; Irlinger, F. Does smearing inoculum reflect the bacterial composition of the smear at the end of the ripening of a French soft, red-smear cheese? J. Dairy Sci. 2004, 87, 3189–3197. [Google Scholar] [CrossRef]
- Irlinger, F.; Mounier, J. Microbial interactions in cheese: Implications for cheese quality and safety. Curr. Opin. Biotechnol. 2009, 20, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Quijada, N.M.; Mann, E.; Wagner, M.; Rodriguez-Lazaro, D.; Hernandez, M.; Schmitz-Esser, S. Autochthonous facility-specific microbiota dominates washed-rind Austrian hard cheese surfaces and its production environment. Int. J. Food Microbiol. 2018, 267, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Kodama, K.; Yasuda, H.; Okamoto-Kainuma, A.; Koizumi, K.; Yamasato, K. Presence of halophilic and alkaliphilic lactic acid bacteria in various cheeses. Lett. Appl. Microbiol. 2007, 44, 308–313. [Google Scholar] [CrossRef]
- Monnet, C.; Landaud, S.; Bonnarme, P.; Swennen, D. Growth and adaptation of microorganisms on the cheese surface. FEMS Microbiol. Lett. 2015, 362, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mounier, J.; Goerges, S.; Gelsomino, R.; Vancanneyt, M.; Vandemeulebroecke, K.; Hoste, B.; Brennan, N.M.; Scherer, S.; Swings, J.; Fitzgerald, G.F.; et al. Sources of the adventitious microflora of a smear-ripened cheese. J. Appl. Microbiol. 2006, 101, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Mounier, J.; Coton, M.; Irlinger, F.; Landaud, S.; Bonnarme, P. Chapter 38—Smear-ripened cheeses. In Cheese, 4th ed.; McSweeney, P.L.H., Fox, P.F., Cotter, P.D., Everett, D.W., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 955–996. [Google Scholar]
- Spinnler, H.E. Chapter 36—Surface mold-ripened cheeses. In Cheese, 4th ed.; McSweeney, P.L.H., Fox, P.F., Cotter, P.D., Everett, D.W., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 911–928. [Google Scholar]
- Cotter, P.D.; Beresford, T.P. Chapter 15—Microbiome changes during ripening. In Cheese, 4th ed.; McSweeney, P.L.H., Fox, P.F., Cotter, P.D., Everett, D.W., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 389–409. [Google Scholar]
- Bachmann, H.P.; Bobst, C.; Bütikofer, U.; Casey, M.G.; Dalla Torre, M.; Fröhlich-Wyder, M.T.; Fürst, M. Occurrence and significance of Fusarium domesticum alias Anticollanti on smear-ripened cheeses. LWT-Food Sci. Technol. 2005, 38, 399–407. [Google Scholar] [CrossRef]
- Marcellino OSB, S.N.; Benson, D.R. The good, the bad, and the ugly: Tales of mold-ripened cheese. Microbiol. Spectr. 2013, 1, CM-0005-0012. [Google Scholar] [CrossRef] [Green Version]
- Bothast, R.J.; Lancaster, E.B.; Hesseltine, C.W. Scopulariopsis brevicaulis: Effect of pH and substrate on growth. Eur. J. Appl. Microbiol. Biotechnol. 1975, 1, 55–66. [Google Scholar] [CrossRef]
- Houbraken, J.; Samson, R.A. Current taxonomy and identification of foodborne fungi. Curr. Opin. Food Sci. 2017, 17, 84–88. [Google Scholar] [CrossRef]
- Woudenberg, J.H.C.; Meijer, M.; Houbraken, J.; Samson, R.A. Scopulariopsis and scopulariopsis-like species from indoor environments. Stud. Mycol. 2017, 88, 1–35. [Google Scholar] [CrossRef]
- Geiser, D.M.; Aoki, T.; Bacon, C.W.; Baker, S.E.; Bhattacharyya, M.K.; Brandt, M.E.; Brown, D.W.; Burgess, L.W.; Chulze, S.; Coleman, J.J. One fungus, one name: Defining the genus Fusarium in a scientifically robust way that preserves longstanding use. Phytopathology 2013, 103, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.R.; Somerfield, P.J.; Chapman, M.G. On resemblance measures for ecological studies, including taxonomic dissimilarities and a zero-adjusted Bray–Curtis coefficient for denuded assemblages. J. Exp. Mar. Biol. Ecol. 2006, 330, 55–80. [Google Scholar] [CrossRef]
- Quigley, L.; McCarthy, R.; O’Sullivan, O.; Beresford, T.P.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C.; Cotter, P.D. The microbial content of raw and pasteurized cow milk as determined by molecular approaches. J. Dairy Sci. 2013, 96, 4928–4937. [Google Scholar] [CrossRef]
- Dolci, P.; Alessandria, V.; Rantsiou, K.; Bertolino, M.; Cocolin, L. Microbial diversity, dynamics and activity throughout manufacturing and ripening of Castelmagno PDO cheese. Int. J. Food Microbiol. 2010, 143, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.M.; Westall, S.; Jespersen, L. Microbial succession of Debaryomyces hansenii strains during the production of Danish surfaced-ripened cheeses. J. Dairy Sci. 2002, 85, 478–486. [Google Scholar] [CrossRef]
- Bockelmann, W.; Willems, K.P.; Neve, H.; Heller, K.H. Cultures for the ripening of smear cheeses. Int. Dairy J. 2005, 15, 719–732. [Google Scholar] [CrossRef]
- Mounier, J.; Gelsomino, R.; Goerges, S.; Vancanneyt, M.; Vandemeulebroecke, K.; Hoste, B.; Scherer, S.; Swings, J.; Fitzgerald, G.F.; Cogan, T.M. Surface microflora of four smear-ripened cheeses. Appl. Environ. Microbiol. 2005, 71, 6489–6500. [Google Scholar] [CrossRef] [Green Version]
- Bertuzzi, A.S.; Walsh, A.M.; Sheehan, J.J.; Cotter, P.D.; Crispie, F.; McSweeney, P.L.H.; Kilcawley, K.N.; Rea, M.C. Omics-based insights into flavor development and microbial succession within surface-ripened cheese. MSystems 2018, 3, e00211–e00217. [Google Scholar] [CrossRef] [Green Version]
- Cogan, T.M.; Goerges, S.; Gelsomino, R.; Larpin, S.; Hohenegger, M.; Bora, N.; Jamet, E.; Rea, M.C.; Mounier, J.; Vancanneyt, M.; et al. Biodiversity of the Surface Microbial Consortia from Limburger, Reblochon, Livarot, Tilsit, and Gubbeen Cheeses. Microbiol. Spectr. 2014, 2, CM-0010-2012. [Google Scholar] [CrossRef] [Green Version]
- Büchl, N.R.; Seiler, H. Yeasts and molds | Yeasts in milk and dairy products. In Encyclopedia of Dairy Sciences, 2nd ed.; Fuquay, J.W., Ed.; Academic Press: San Diego, CA, USA, 2011; pp. 744–753. [Google Scholar]
- Buerth, C.; Tielker, D.; Ernst, J.F. Candida utilis and Cyberlindnera (Pichia) jadinii: Yeast relatives with expanding applications. Appl. Microbiol. Biotechnol. 2016, 100, 6981–6990. [Google Scholar] [CrossRef]
- Butinar, L.; Santos, S.; Spencer-Martins, I.; Oren, A.; Gunde-Cimerman, N. Yeast diversity in hypersaline habitats. FEMS Microbiol. Lett. 2005, 244, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Breuer, U.; Harms, H. Debaryomyces hansenii—An extremophilic yeast with biotechnological potential. Yeast 2006, 23, 415–437. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Region | Bacteria | Fungi | ||
|---|---|---|---|---|
| V3–V4 | V6–V8 | ITS1 | ITS2 | |
| Nb. reads sequenced 1 | 4,505,810 | 6,156,446 | 5,207,694 | 5,746,122 |
| Nb. trimmed and assembled sequences | 3,904,825 | 5,482,054 | 4,931,241 | 5,675,978 |
| Nb. non-chimeric OTU sequences 1 | 2,588,028 | 3,451,507 | 4,683,355 | 5,096,678 |
| Non-chimeric sequences | 66.28% | 62.96% | 94.97% | 89.79% |
| Nb. non-chimeric OTUs 1 | 108 | 154 | 67 | 78 |
| Nb. genera assigned 1/Nb. shared genera assignation | 57/ 37 | 83/ 37 | 34/ 18 | 35/ 18 |
| Nb. abundant genera 2/Nb. shared genera assignation | 22/ 17 | 20/ 17 | 9/ 6 | 9/ 6 |
| Cheese Section | Rind Type | Bacteria | Fungi | ||
|---|---|---|---|---|---|
| Richness 1 | α-Diversity 1 | Richness | α-Diversity | ||
| Cheese core | Bloomy | 16 ± 11 | 1.497 ± 0.517 | 23 ± 6 | 2.020 ± 0.829 |
| Natural | 16 ± 7 | 1.783 ± 0.430 | 21 ± 4 | 2.251 ± 0.932 | |
| Washed | 24 ± 18 | 1.794 ± 0.722 | 24 ± 6 | 2.317 ± 0.800 | |
| Cheese rind | Bloomy | 32 ± 8 | 3.026 ± 1.683 | 19 ± 10 | 1.745 ± 0.496 |
| Natural | 34 ± 16 | 2.732 ± 0.372 | 18 ± 10 | 2.039 ± 1.139 | |
| Washed | 36 ± 10 | 5.045 ± 2.223 | 17 ± 5 | 1.974 ± 0.737 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raymond-Fleury, A.; Lessard, M.-H.; Chamberland, J.; Pouliot, Y.; Dugat-Bony, E.; Turgeon, S.L.; St-Gelais, D.; Labrie, S. Analysis of Microbiota Persistence in Quebec’s Terroir Cheese Using a Metabarcoding Approach. Microorganisms 2022, 10, 1381. https://doi.org/10.3390/microorganisms10071381
Raymond-Fleury A, Lessard M-H, Chamberland J, Pouliot Y, Dugat-Bony E, Turgeon SL, St-Gelais D, Labrie S. Analysis of Microbiota Persistence in Quebec’s Terroir Cheese Using a Metabarcoding Approach. Microorganisms. 2022; 10(7):1381. https://doi.org/10.3390/microorganisms10071381
Chicago/Turabian StyleRaymond-Fleury, Annick, Marie-Hélène Lessard, Julien Chamberland, Yves Pouliot, Eric Dugat-Bony, Sylvie L. Turgeon, Daniel St-Gelais, and Steve Labrie. 2022. "Analysis of Microbiota Persistence in Quebec’s Terroir Cheese Using a Metabarcoding Approach" Microorganisms 10, no. 7: 1381. https://doi.org/10.3390/microorganisms10071381
APA StyleRaymond-Fleury, A., Lessard, M.-H., Chamberland, J., Pouliot, Y., Dugat-Bony, E., Turgeon, S. L., St-Gelais, D., & Labrie, S. (2022). Analysis of Microbiota Persistence in Quebec’s Terroir Cheese Using a Metabarcoding Approach. Microorganisms, 10(7), 1381. https://doi.org/10.3390/microorganisms10071381