
Use of Flavin-Related Cellular Autofluorescence to Monitor Processes in Microbial Biotechnology



Abstract
1. Introduction
2. Materials and Methods
2.1. Microorganisms and Cultivation
2.2. Sample Tubes Preparation for Fluorescence Analysis
2.3. Propidium Iodide Staining Essay for Flow Cytometry
2.4. BODIPY 493/503 Staining Essay
2.5. Flow Cytometry
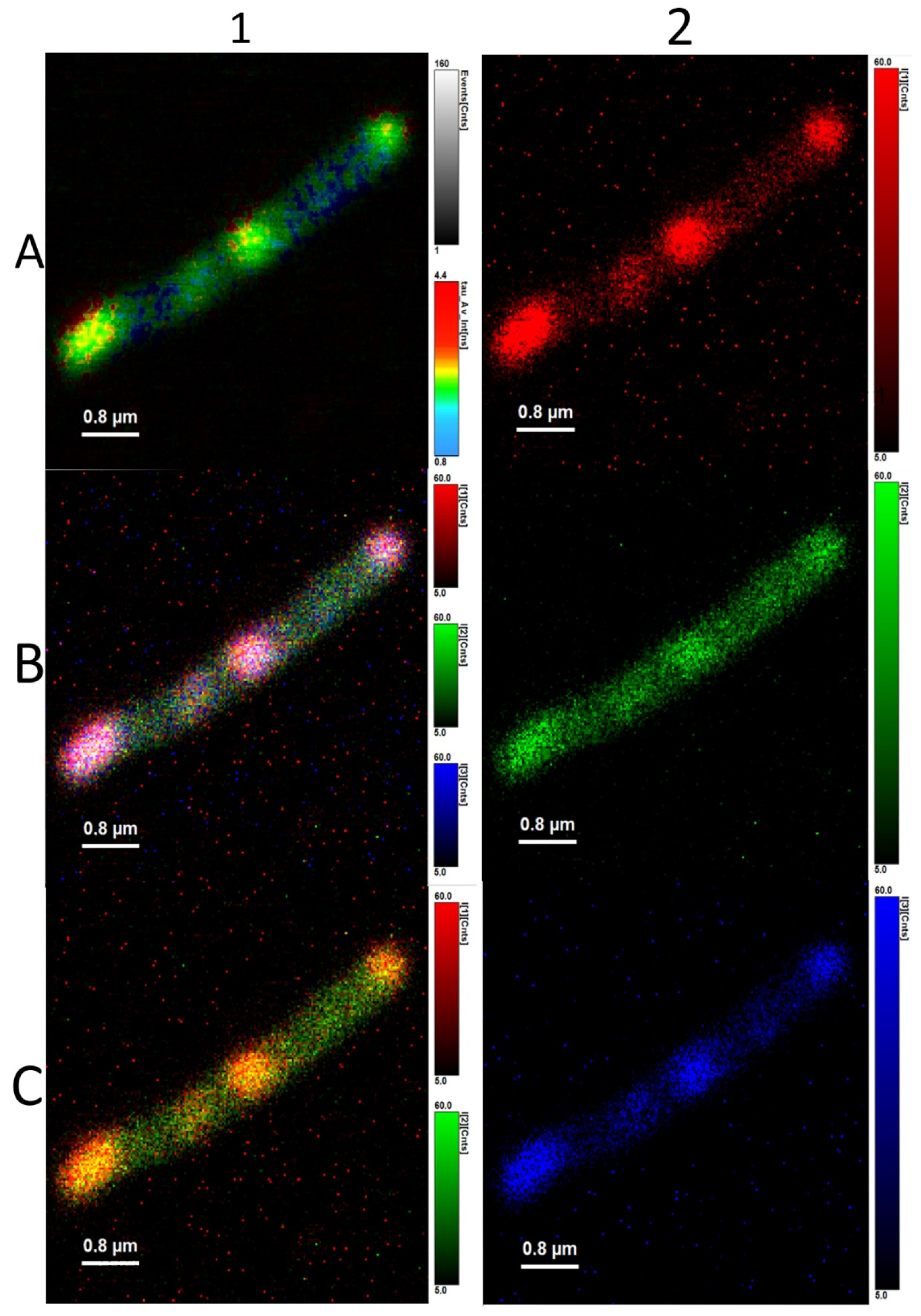
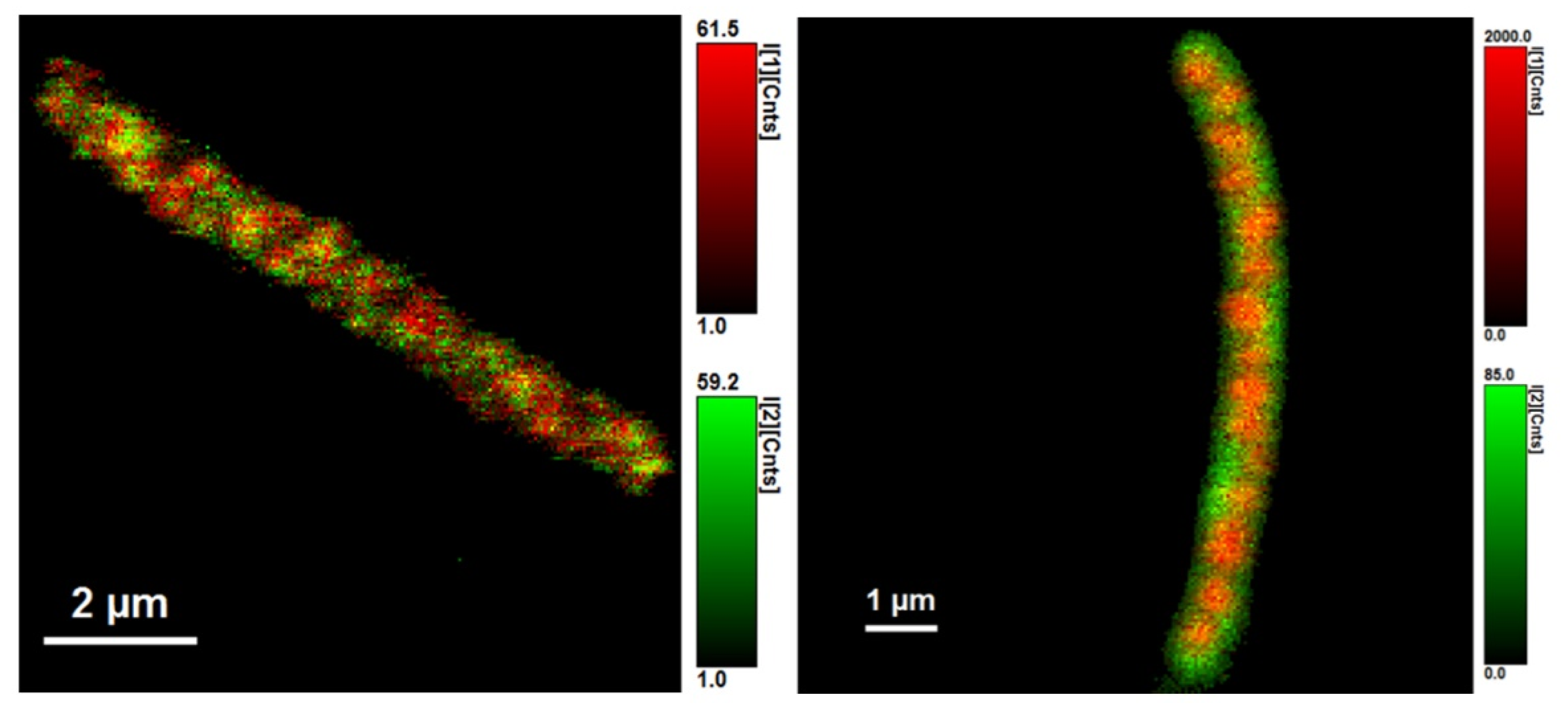
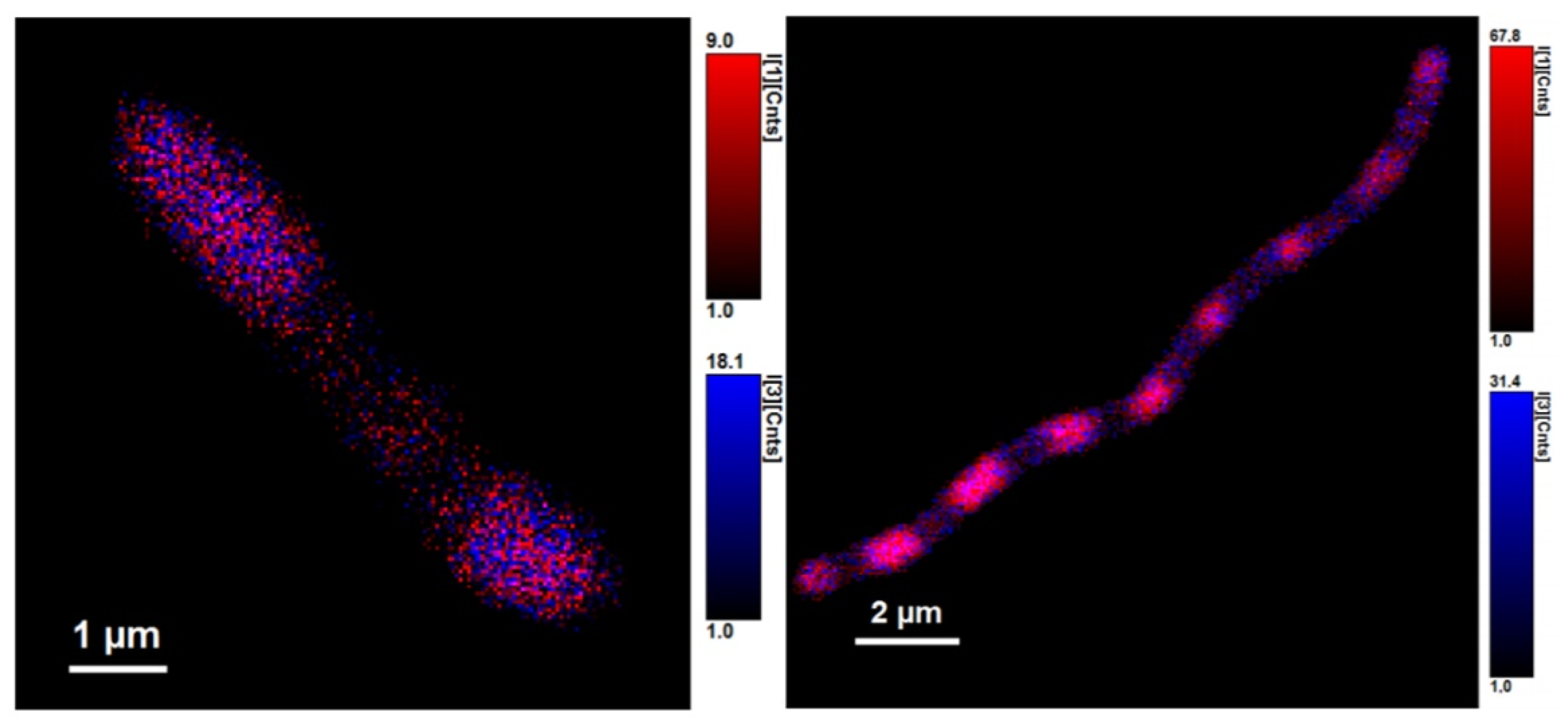
2.6. Fluorescence Microscopy, FLIM
3. Results
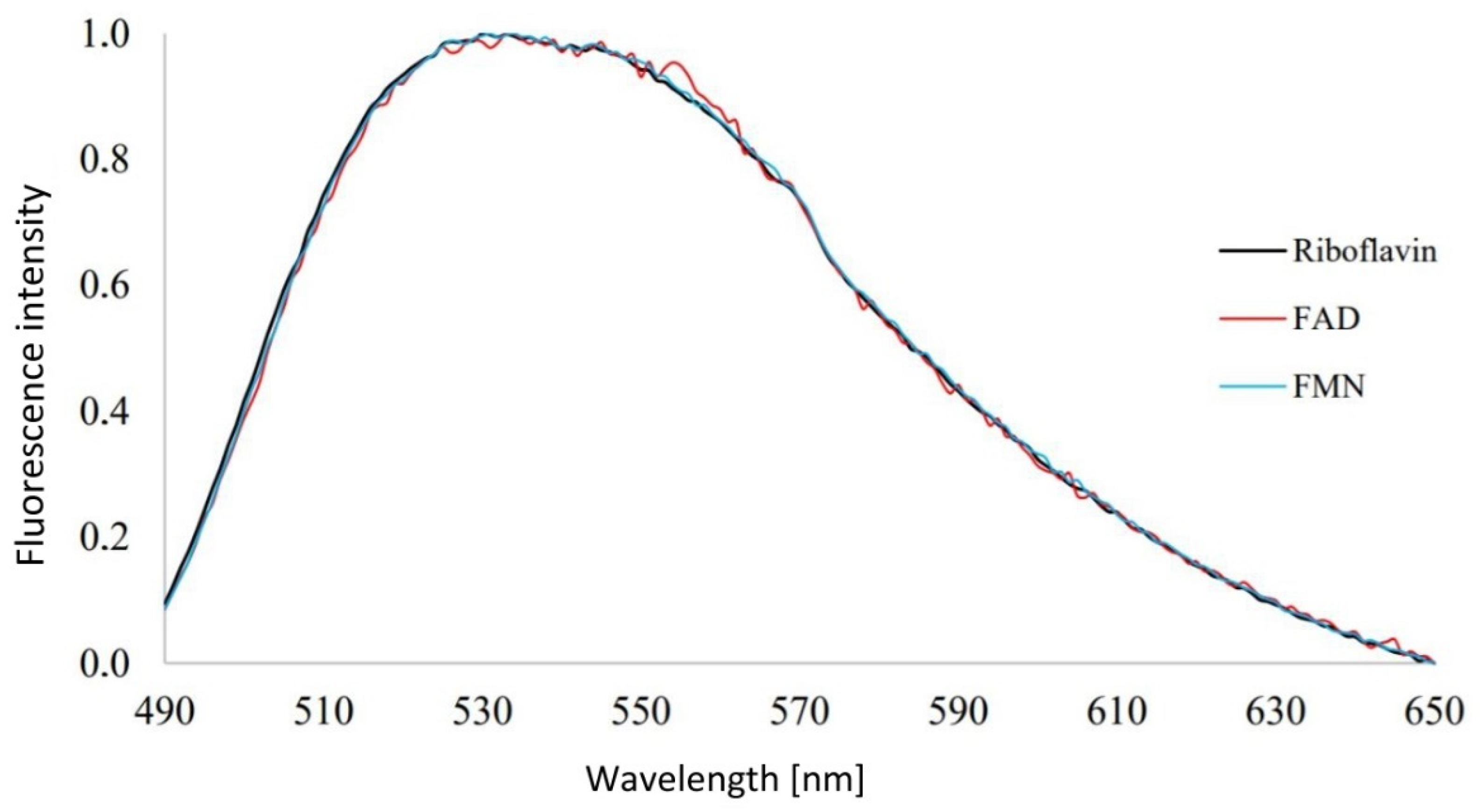
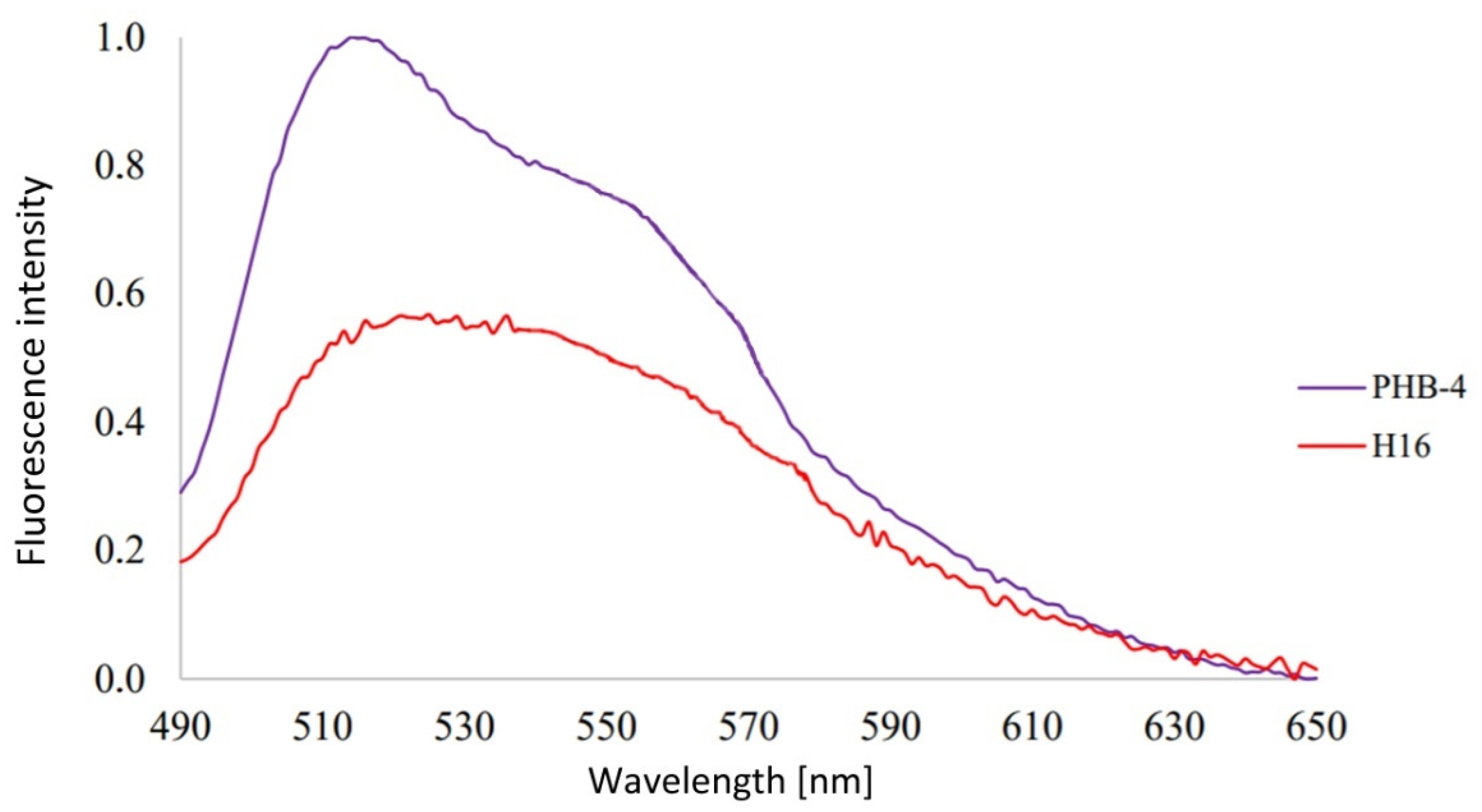
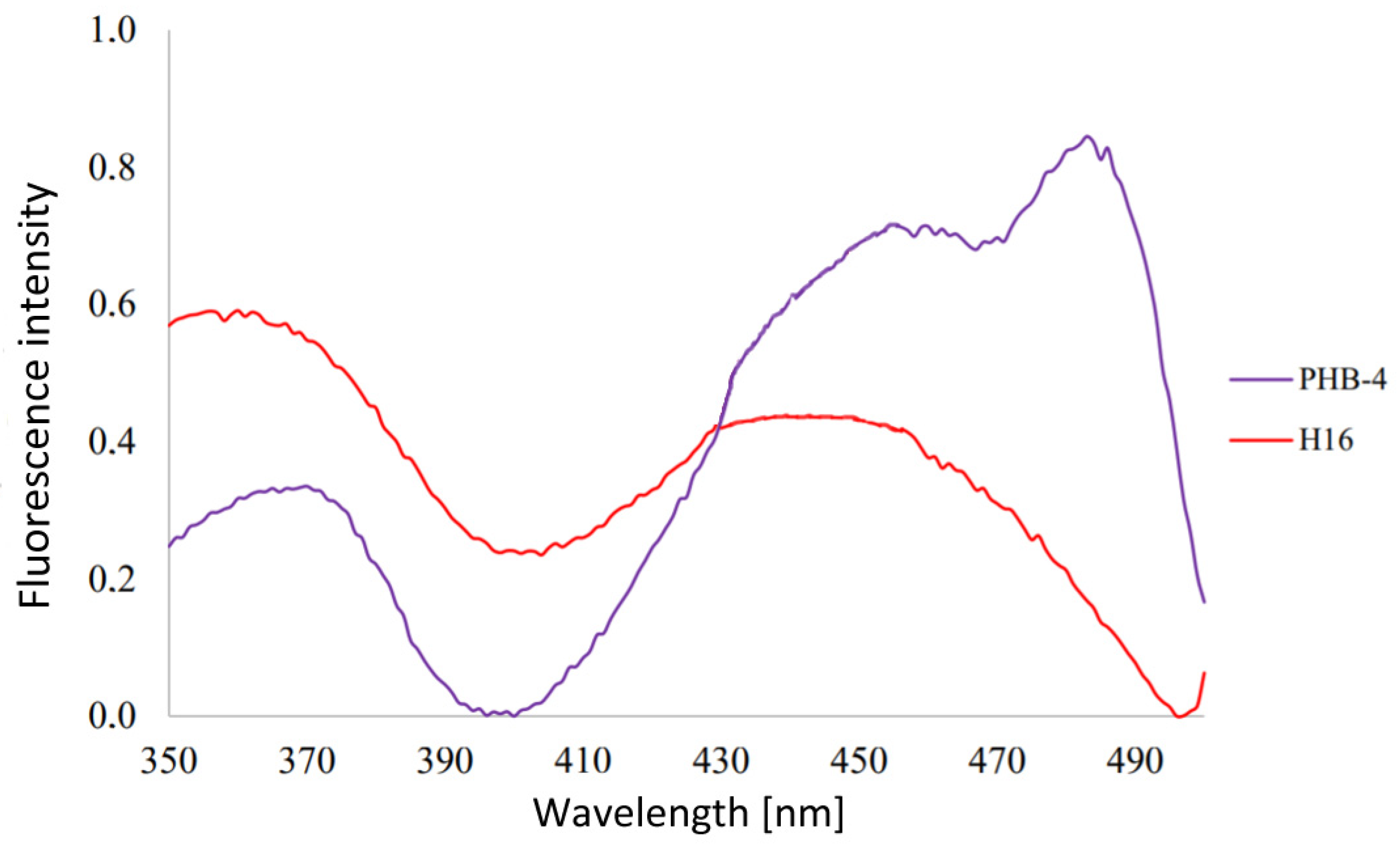
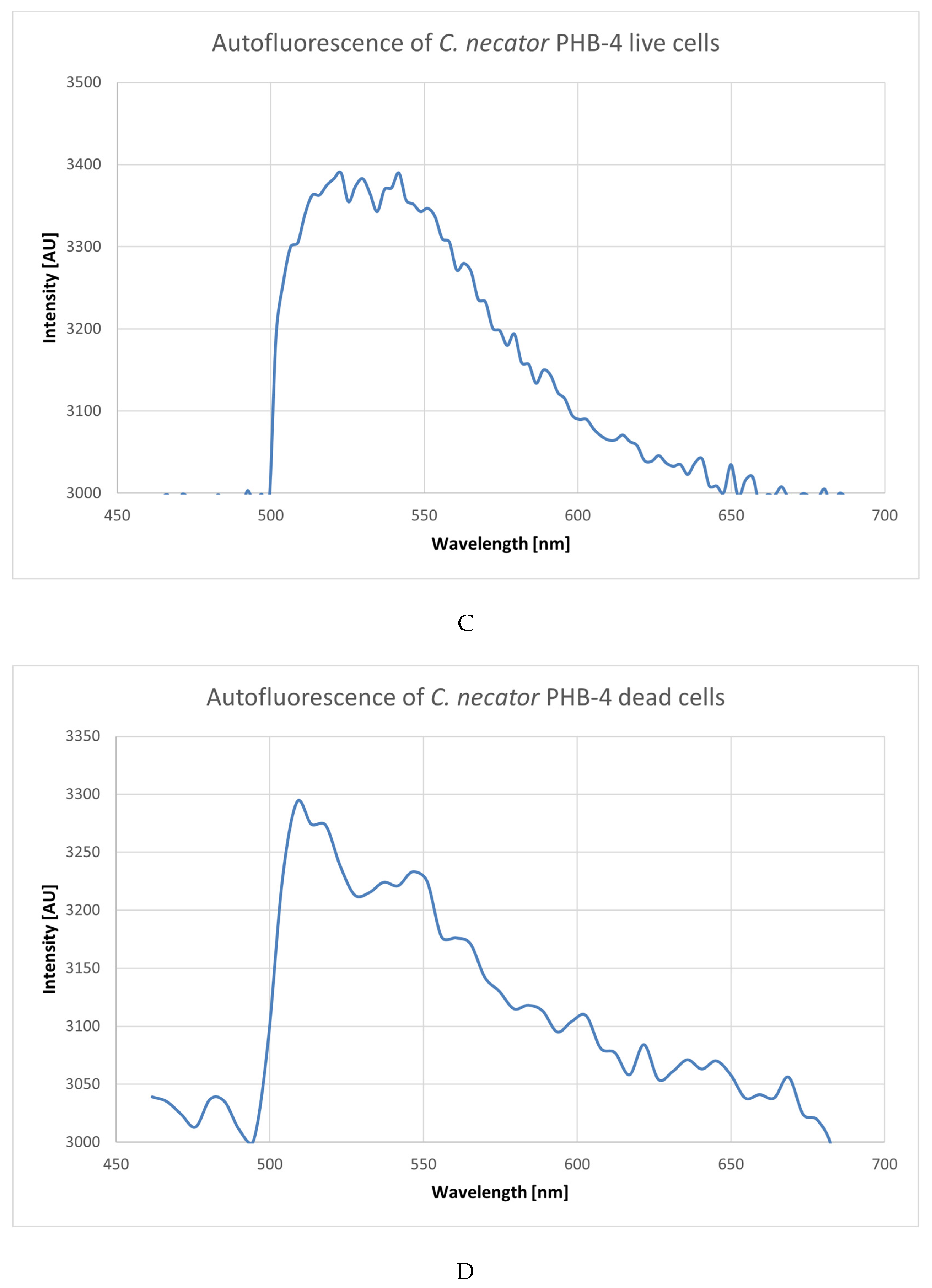
3.1. Analysis of Emission Spectra of Flavins and Whole Bacterial Cells
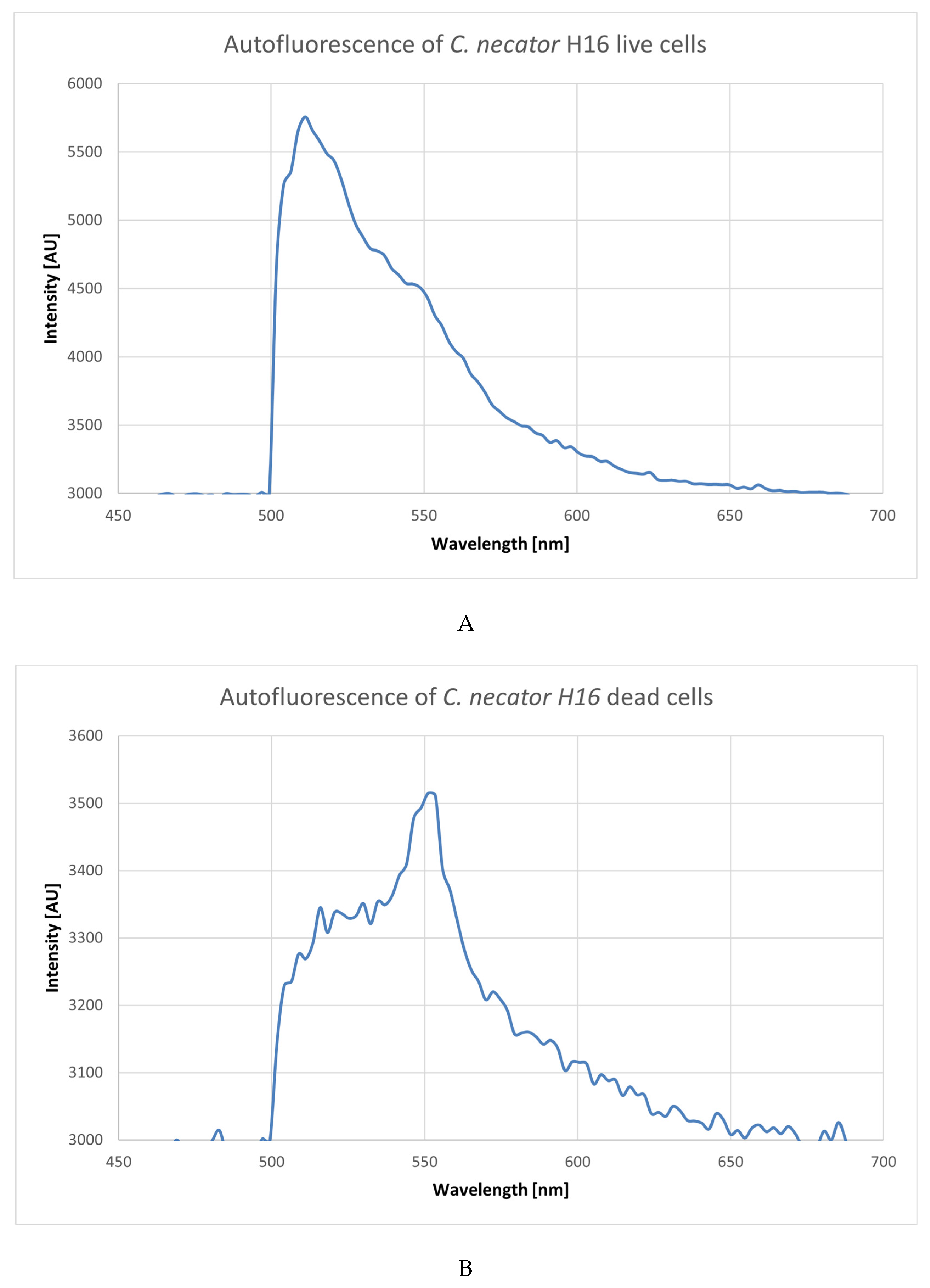
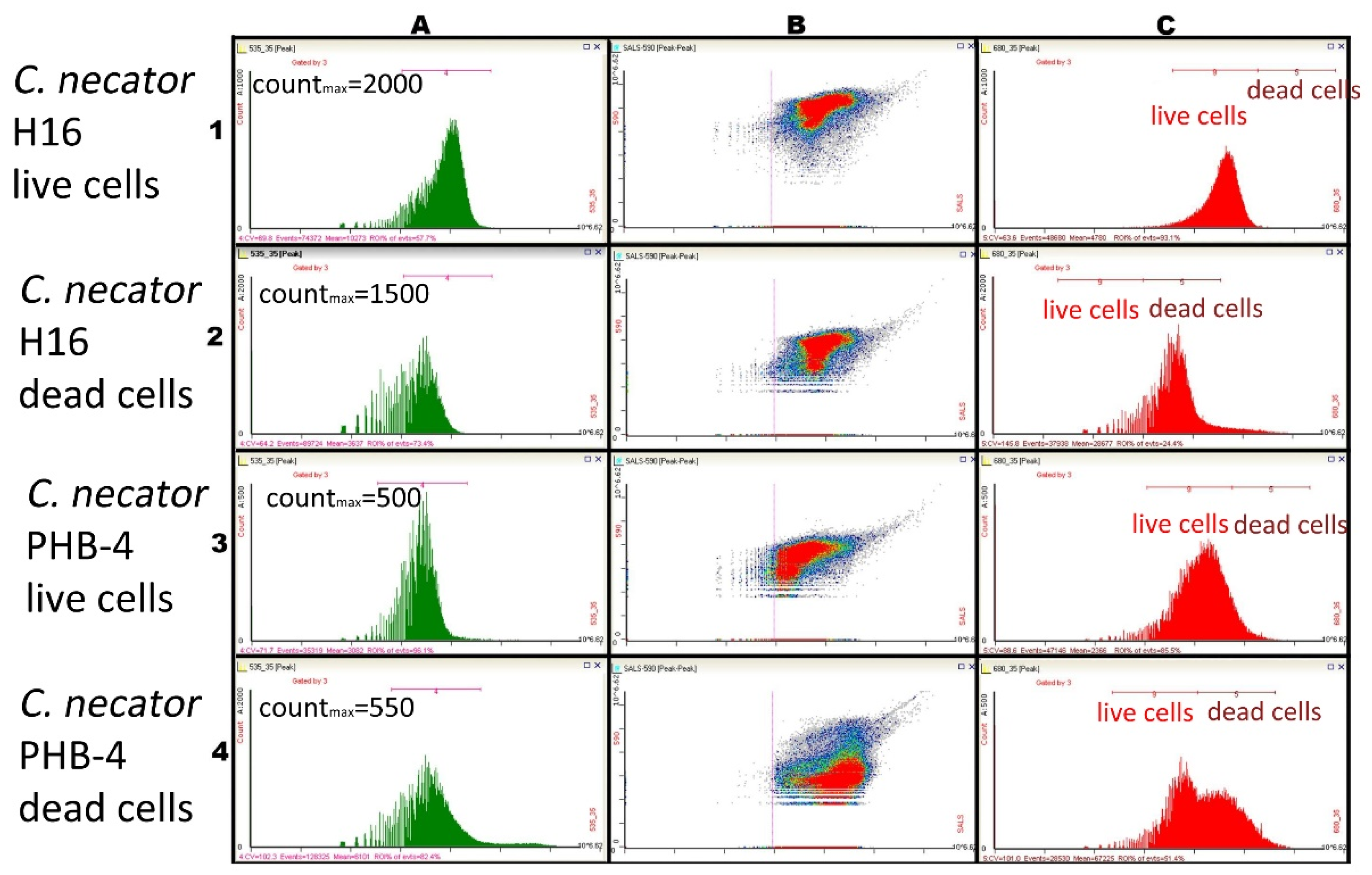
3.2. Using Green Autofluorescence to Monitor the Viability of Prokaryotic Cell Cultures
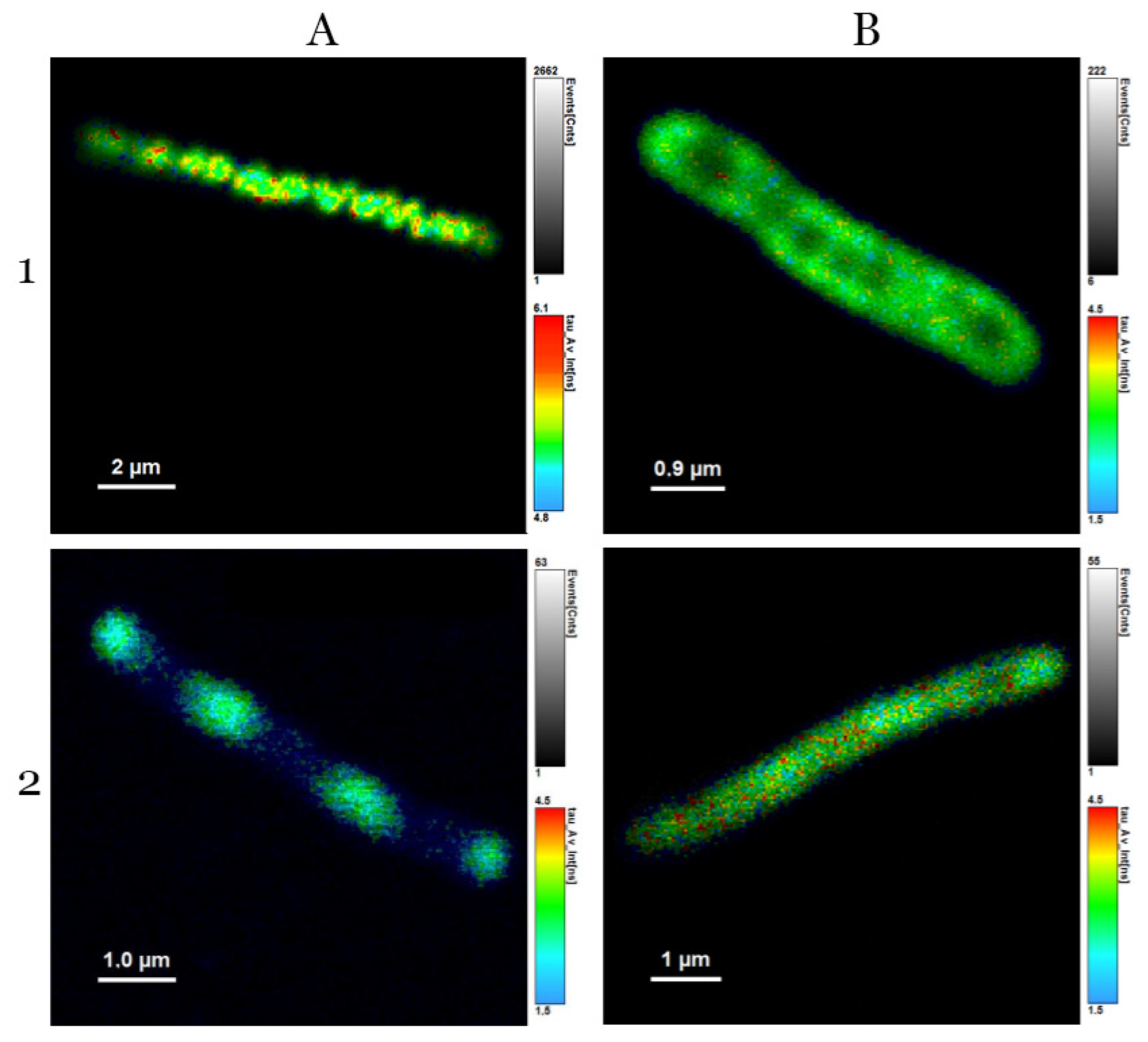
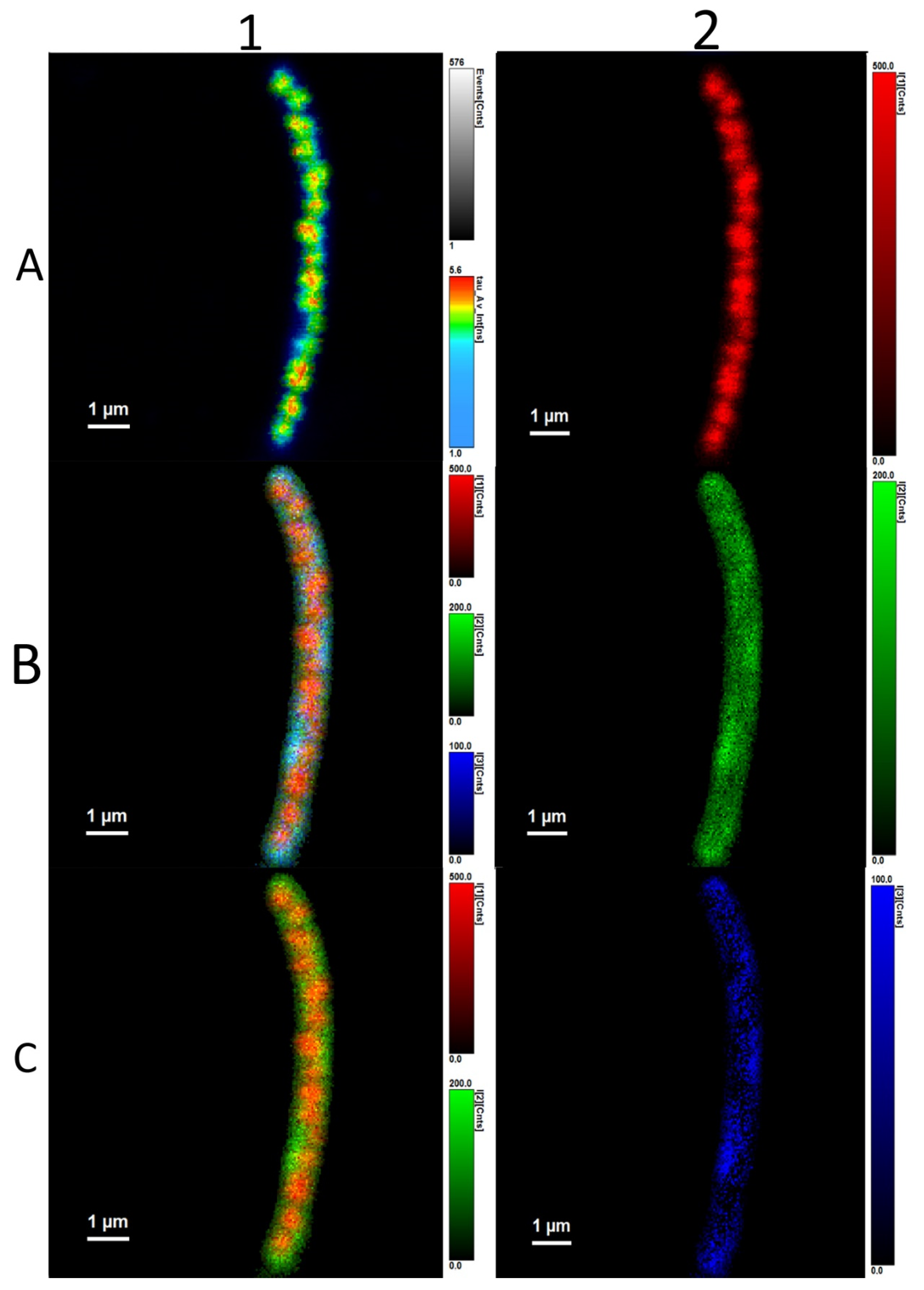
3.3. Analysis of Average Fluorescence Lifetimes
4. Discussion
4.1. Analysis of Emission Spectra of Flavins and Whole Bacterial Cells
4.2. Using Green Autofluorescence to Monitor the Viability of Prokaryotic Cell Cultures
4.3. Analysis of Average Fluorescence Lifetimes
4.4. Flavin-PHAs Hypothesis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yuan, G.-C.; Cai, L.; Elowitz, M.; Enver, T.; Fan, G.; Guo, G.; Irizarry, R.; Kharchenko, P.; Kim, J.; Orkin, S.; et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017, 18, 84. [Google Scholar] [CrossRef]
- Croce, A.; Bottiroli, G. Autofluorescence spectroscopy and imaging: A tool for biomedical research and diagnosis. Eur. J. Histochem. 2014, 58, 2461. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, Y.; Zhu, S.; Huang, T.; Wu, L.; Yan, X. Detection and Quantification of Bacterial Autofluorescence at the Single-Cell Level by a Laboratory-Built High-Sensitivity Flow Cytometer. Anal. Chem. 2012, 84, 1526–1532. [Google Scholar] [CrossRef] [PubMed]
- Surre, J.; Saint-Ruf, C.; Collin, V.; Orenga, S.; Ramjeet, M.; Matic, I. Strong increase in the autofluorescence of cells signals struggle for survival. Sci. Rep. 2018, 8, 12088. [Google Scholar] [CrossRef] [PubMed]
- Pincus, Z.; Mazer, T.C.; Slack, F.J. Autofluorescence as a measure of senescence in C. elegans: Look to red, not blue or green. Aging 2016, 8, 889–898. [Google Scholar] [CrossRef]
- Christian, L.; Laflamme, C.; Verreault, D.; Lavigne, S.; Trudel, L.; Ho, J.; Duchaine, C. Autofluorescence as a viability marker for detection of bacterial spores. Front. Biosci. 2005, 10, 1647–1653. [Google Scholar] [CrossRef]
- Bao, N.; Jagadeesan, B.; Bhunia, A.; Yao, Y.; Lu, C. Quantification of bacterial cells based on autofluorescence on a microfluidic platform. J. Chromatogr. A 2008, 1181, 153–158. [Google Scholar] [CrossRef]
- Schulze, K.; López, D.A.; Tillich, U.M.; Frohme, M. A simple viability analysis for unicellular cyanobacteria using a new autofluorescence assay, automated microscopy, and ImageJ. BMC Biotechnol. 2011, 11, 118. [Google Scholar] [CrossRef]
- Bhartia, R.; Salas, E.C.; Hug, W.F.; Reid, R.D.; Lane, A.L.; Edwards, K.J.; Nealson, K.H. Label-Free Bacterial Imaging with Deep-UV-Laser-Induced Native Fluorescence. Appl. Environ. Microbiol. 2010, 76, 7231–7237. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Datta, R.; Gratton, E.; Hochbaum, A.I. Metabolic fingerprinting of bacteria by fluorescence lifetime imaging microscopy. Sci. Rep. 2017, 7, 3743. [Google Scholar] [CrossRef]
- Ghukasyan, V.V.; Kao, F.J. Monitoring cellular metabolism with fluorescence lifetime of reduced nicotinamide adenine dinucleotide. J. Phys. Chem. C 2009, 113, 11532–11540. [Google Scholar] [CrossRef]
- Skala, M.C.; Riching, K.M.; Gendron-Fitzpatrick, A.; Eickhoff, J.; Eliceiri, K.W.; White, J.G.; Ramanujam, N. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc. Natl. Acad. Sci. USA 2007, 104, 19494–19499. [Google Scholar] [CrossRef]
- Cramm, R. Genomic View of Energy Metabolism in Ralstonia eutropha H16. J. Mol. Microbiol. Biotechnol. 2009, 16, 38–52. [Google Scholar] [CrossRef]
- Koller, M.; Bona, R.; Braunegg, G.; Hermann, C.; Horvat, P.; Kroutil, M.; Martinz, J.; Neto, J.; Pereira, A.L.; Varila, P. Production of Polyhydroxyalkanoates from Agricultural Waste and Surplus Materials. Biomacromolecules 2005, 6, 561–565. [Google Scholar] [CrossRef]
- Nyström, T. The trials and tribulations of growth arrest. Trends Microbiol. 1995, 3, 131–136. [Google Scholar] [CrossRef]
- Penzer, G.R. Molecular emission spectroscopy (Fluorescence and Phosphorescence). In An Introduction to Spectroscopy for Biochemists; Brown, S.B., Ed.; Academic Press: London, UK, 1980; pp. 70–114. [Google Scholar]
- Li, M.; Wilkins, M. Flow cytometry for quantitation of polyhydroxybutyrate production by Cupriavidus necator using alkaline pretreated liquor from corn stover. Bioresour. Technol. 2019, 295, 122254. [Google Scholar] [CrossRef]
- Saranya, V.; Poornimakkani; Krishnakumari, M.S.; Suguna, P.; Binuramesh, C.; Abirami, P.; Rajeswari, V.; Ramachandran, K.B.; Shenbagarathai, R. Quantification of Intracellular Polyhydroxyalkanoates by Virtue of Personalized Flow Cytometry Protocol. Curr. Microbiol. 2012, 65, 589–594. [Google Scholar] [CrossRef]
- Karmann, S.; Follonier, S.; Bassas-Galia, M.; Panke, S.; Zinn, M. Robust at-line quantification of poly(3-hydroxyalkanoate) biosynthesis by flow cytometry using a BODIPY 493/503-SYTO 62 double-staining. J. Microbiol. Methods 2016, 131, 166–171. [Google Scholar] [CrossRef]
- Wolfbeiss, O.S. The fluorescence of organic natural products. In Molecular Fluorescence Spectroscopy. Methods and Applications; Part I; Schulman, S.G., Ed.; John Wiley & Sons: New York, NY, USA, 1985; pp. 167–370. [Google Scholar]
- Peter, M. UV-Visible Spectroscopy as a Tool to Study Flavoproteins. In Flavoprotein Protocols; Chapman, S.K., Reid, G.A., Eds.; Humana Press: Totowa, NJ, USA, 1999; pp. 1–8. ISBN 1-59259-266-X. [Google Scholar] [CrossRef]
- Mihalcescu, I.; Van-Melle Gateau, M.; Chelli, B.; Pinel, C.; Ravanat, J.-L. Green autofluorescence, a double edged monitoring tool for bacterial growth and activity in micro-plates. Phys. Biol. 2015, 12, 066016. [Google Scholar] [CrossRef]
- Kotaki, A.; Yagi, K. Fluorescence Properties of Flavins in Various Solvents. J. Biochem. 1970, 68, 509–516. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, L.; Ge, J.; He, Y. Ultrasensitive determination for flavin coenzyme by using a ZnO nanorod photoelectrode in a four-electrode system. Mikrochim. Acta 2017, 184, 2333–2339. [Google Scholar] [CrossRef]
- Valle, L.; Vieyra, F.E.M.; Borsarelli, C.D. Hydrogen-bonding modulation of excited-state properties of flavins in a model of aqueous confined environment. Photochem. Photobiol. Sci. 2012, 11, 1051–1061. [Google Scholar] [CrossRef]
- Galbán, J.; Sanz-Vicente, I.; Navarro, J.; De Marcos, S. The intrinsic fluorescence of FAD and its application in analytical chemistry: A review. Methods Appl. Fluoresc. 2016, 4, 42005. [Google Scholar] [CrossRef]
- Albani, J.R. Fluorophores: Descriptions and Properties. In Structure and Dynamics of Macromolecules: Absorption and Fluorescence Studies; Elsevier: Amsterdam, The Netherlands, 2004; pp. 99–140. [Google Scholar] [CrossRef]
- Schmid, J.; Hoenes, K.; Vatter, P.; Hessling, M. Antimicrobial Effect of Visible Light—Photoinactivation of Legionella rubrilucens by Irradiation at 450, 470, and 620 nm. Antibiotics 2019, 8, 187. [Google Scholar] [CrossRef]
- Crocker, L.; Fruk, L. Flavin Conjugated Polydopamine Nanoparticles Displaying Light-Driven Monooxygenase Activity. Front. Chem. 2019, 7, 278. [Google Scholar] [CrossRef]
- Pan, Y.-L. Detection and characterization of biological and other organic-carbon aerosol particles in atmosphere using fluorescence. J. Quant. Spectrosc. Radiat. Transf. 2015, 150, 12–35. [Google Scholar] [CrossRef]
- Chen, R.; Wu, R.; Zhang, G.; Gao, Y.; Xiao, L.; Jia, S. Electron Transfer-Based Single Molecule Fluorescence as a Probe for Nano-Environment Dynamics. Sensors 2014, 14, 2449–2467. [Google Scholar] [CrossRef]
- Barrio, J.R.; Tolman, G.L.; Leonard, N.J.; Spencer, R.D.; Weber, G. Flavin 1, N 6 -ethenoadenine dinucleotide: Dynamic and static quenching of fluorescence. Proc. Natl. Acad. Sci. USA 1973, 70, 941–943. [Google Scholar] [CrossRef]
- Islam, S.; Honma, M.; Nakabayashi, T.; Kinjo, M.; Ohta, N. pH Dependence of the Fluorescence Lifetime of FAD in Solution and in Cells. Int. J. Mol. Sci. 2013, 14, 1952–1963. [Google Scholar] [CrossRef]
- Berg, P.A.V.D.; van Hoek, A.; Visser, A.J. Evidence for a Novel Mechanism of Time-Resolved Flavin Fluorescence Depolarization in Glutathione Reductase. Biophys. J. 2004, 87, 2577–2586. [Google Scholar] [CrossRef][Green Version]
- Slaninova, E.; Sedlacek, P.; Mravec, F.; Mullerova, L.; Samek, O.; Koller, M.; Hesko, O.; Kucera, D.; Marova, I.; Obruca, S. Light scattering on PHA granules protects bacterial cells against the harmful effects of UV radiation. Appl. Microbiol. Biotechnol. 2018, 102, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dai, T.; Gu, Y. Antimicrobial blue light inactivation of Neisseria gonorrhoeae. In Light-Based Diagnosis and Treatment of Infectious Diseases; SPIE: San Francisco, CA, USA, 2018; ISBN 9781510614437. [Google Scholar] [CrossRef]
- Li, G.; Glusac, K.D. Light-triggered proton and electron transfer in flavin cofactors. J. Phys. Chem. A 2008, 112, 4573–4583. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, P.; Schmidt-Emrich, S.; Maniura-Weber, K.; Ren, Q. Critical aspects of using bacterial cell viability assays with the fluorophores SYTO9 and propidium iodide. BMC Microbiol. 2015, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.; Hong, Y.; Danavall, D.; Howard-Jones, M.; Gibson, D.; Frischer, M.; Verity, P. Distinguishing between living and nonliving bacteria: Evaluation of the vital stain propidium iodide and its combined use with molecular probes in aquatic samples. J. Microbiol. Methods 1998, 32, 225–236. [Google Scholar] [CrossRef]
- Boulos, L.; Prévost, M.; Barbeau, B.; Coallier, J.; Desjardins, R. LIVE/DEAD®BacLight™: Application of a new rapid staining method for direct enumeration of viable and total bacteria in drinking water. J. Microbiol. Methods 1999, 37, 77–86. [Google Scholar] [CrossRef]
- Rosenberg, M.; Azevedo, N.F.; Ivask, A. Propidium iodide staining underestimates viability of adherent bacterial cells. Sci. Rep. 2019, 9, 6483. [Google Scholar] [CrossRef]
- Shi, L.; Günther, S.; Hübschmann, T.; Wick, L.Y.; Harms, H.; Müller, S. Limits of propidium iodide as a cell viability indicator for environmental bacteria. Cytom. Part A 2007, 71, 592–598. [Google Scholar] [CrossRef]
- Davey, H.M.; Hexley, P. Red but not dead? Membranes of stressed Saccharomyces cerevisiae are permeable to propidium iodide. Environ. Microbiol. 2010, 13, 163–171. [Google Scholar] [CrossRef]
- Gião, M.S.; Wilks, S.A.; Azevedo, N.F.; Vieira, M.J.; Keevil, C.W. Validation of SYTO 9/Propidium Iodide Uptake for Rapid Detection of Viable but Noncultivable Legionella pneumophila. Microb. Ecol. 2008, 58, 56–62. [Google Scholar] [CrossRef]
- Pletnev, S.; Pletneva, N.V.; Souslova, E.A.; Chudakov, D.; Lukyanov, S.; Wlodawer, A.; Dauter, Z.; Pletnev, V. Structural basis for bathochromic shift of fluorescence in far-red fluorescent proteins eqFP650 and eqFP670. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1088–1097. [Google Scholar] [CrossRef]
- Spahn, C.; Glaesmann, M.; Grimm, J.B.; Ayala, A.; Lavis, L.D.; Heilemann, M. A toolbox for multiplexed super-resolution imaging of the E. coli nucleoid and membrane using novel PAINT labels. Sci. Rep. 2018, 8, 14768. [Google Scholar] [CrossRef]
- Manzo, N.; Di Luccia, B.; Isticato, R.; D’Apuzzo, E.; De Felice, M.; Ricca, E. Pigmentation and Sporulation Are Alternative Cell Fates in Bacillus pumilus SF214. PLoS ONE 2013, 8, e62093. [Google Scholar] [CrossRef]
- Wang, J.; Bakken, L. Screening of Soil Bacteria for Poly-β-Hydroxybutyric Acid Production and Its Role in the Survival of Starvation. Microb. Ecol. 1998, 35, 94–101. [Google Scholar] [CrossRef]
- Martin, K. (Ed.) Recent Advances in Biotechnology; Bentham Science Publishers: Sharjah, United Arab Emirates, 2016; ISBN 9781681083735. [Google Scholar]
- García-Torreiro, M.; López-Abelairas, M.; Lu-Chau, T.A.; Lema, J. Application of flow cytometry for monitoring the production of poly(3-hydroxybutyrate) by Halomonas boliviensis. Biotechnol. Prog. 2016, 33, 276–284. [Google Scholar] [CrossRef]
- Karmann, S.; Panke, S.; Zinn, M. The Bistable Behaviour of Pseudomonas putida KT2440 during PHA Depolymerization under Carbon Limitation. Bioengineering 2017, 4, 58. [Google Scholar] [CrossRef]
- Bhagowati, P.; Pradhan, S.; Dash, H.R.; Das, S. Production, optimization and characterization of polyhydroxybutyrate, a biodegradable plastic by Bacillus spp. Biosci. Biotechnol. Biochem. 2015, 79, 1454–1463. [Google Scholar] [CrossRef]
- Biernacki, M.; Marzec, M.; Roick, T.; Pätz, R.; Baronian, K.; Bode, R.; Kunze, G. Enhancement of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) accumulation in Arxula adeninivorans by stabilization of production. Microb. Cell Factories 2017, 16, 144. [Google Scholar] [CrossRef]
- Kacmar, J.; Carlson, R.; Balogh, S.J.; Srienc, F. Staining and quantification of poly-3-hydroxybutyrate inSaccharomyces cerevisiae andCupriavidus necator cell populations using automated flow cytometry. Cytom. Part A 2005, 69, 27–35. [Google Scholar] [CrossRef]
- Kadouri, D.; Jurkevitch, E.; Okon, Y.; Castro-Sowinski, S. Ecological and Agricultural Significance of Bacterial Polyhydroxyalkanoates. Crit. Rev. Microbiol. 2005, 31, 55–67. [Google Scholar] [CrossRef]
- Jendrossek, D.; Handrick, R. Microbial Degradation of Polyhydroxyalkanoates. Annu. Rev. Microbiol. 2002, 56, 403–432. [Google Scholar] [CrossRef]
- Winnacker, M. Polyhydroxyalkanoates: Recent Advances in Their Synthesis and Applications. Eur. J. Lipid Sci. Technol. 2019, 121, 1900101. [Google Scholar] [CrossRef]
- Chan, R.T.; Marçal, H.; Ahmed, T.; Russell, R.A.; Holden, P.J.; Foster, L.J.R. Poly(ethylene glycol)-modulated cellular biocompatibility of polyhydroxyalkanoate films. Polym. Int. 2013, 62, 884–892. [Google Scholar] [CrossRef]
- Wahl, A.; Schuth, N.; Pfeiffer, D.; Nussberger, S.; Jendrossek, D. PHB granules are attached to the nucleoid via PhaM in Ralstonia eutropha. BMC Microbiol. 2012, 12, 262. [Google Scholar] [CrossRef]
- Pfeiffer, D.; Wahl, A.; Jendrossek, D. Identification of a multifunctional protein, PhaM, that determines number, surface to volume ratio, subcellular localization and distribution to daughter cells of poly(3-hydroxybutyrate), PHB, granules in Ralstonia eutropha H16. Mol. Microbiol. 2011, 82, 936–951. [Google Scholar] [CrossRef]
- Durner, R.; Witholt, B.; Egli, T. Accumulation of Poly[(R)-3-hydroxyalkanoates] in Pseudomonas oleovorans during growth with octanoate in continuous culture at different dilution rates. Appl. Environ. Microbiol. 2000, 66, 3408–3414. [Google Scholar] [CrossRef]
- Trotsenko, Y.A. Biosynthesis of Poly(3-Hydroxybutyrate) and Poly(3-Hydroxybutyrate-co-3-Hydroxyvalerate) and Its Regulation in Bacteria. Microbiology 2000, 69, 635–645. [Google Scholar] [CrossRef]
- Lee, S.Y. Bacterial polyhydroxyalkanoates. Biotechnol. Bioeng. 1996, 49, 1–14. [Google Scholar] [CrossRef]
- Stubbe, J.; Tian, J.; He, A.; Sinskey, A.J.; Lawrence, A.G.; Liu, P. NONTEMPLATE-DEPENDENT POLYMERIZATION PROCESSES: Polyhydroxyalkanoate Synthases as a Paradigm. Annu. Rev. Biochem. 2005, 74, 433–480. [Google Scholar] [CrossRef]
- Ruiz, J.A.; López, N.I.; Fernández, R.O.; Méndez, B.S. Polyhydroxyalkanoate Degradation Is Associated with Nucleotide Accumulation and Enhances Stress Resistance and Survival of Pseudomonas oleovorans in Natural Water Microcosms. Appl. Environ. Microbiol. 2001, 67, 225–230. [Google Scholar] [CrossRef]
- Roohi; Zaheer, M.R.; Kuddus, M. PHB (poly-β-hydroxybutyrate) and its enzymatic degradation. Polym. Adv. Technol. 2017, 29, 30–40. [Google Scholar] [CrossRef]
- Höfer, P.; Vermette, P.; Groleau, D. Introducing a new Bioengineered Bug: Methylobacterium extorquens tuned as a microbial bioplastic factory. Bioeng. Bugs 2011, 2, 71–79. [Google Scholar] [CrossRef]
- Kocharin, K.; Chen, Y.; Siewers, V.; Nielsen, J. Engineering of acetyl-CoA metabolism for the improved production of polyhydroxybutyrate in Saccharomyces cerevisiae. AMB Express 2012, 2, 52. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate dehydrogenase—Assembly, regulation and role in human disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, H.; Xia, Y.; Kang, Z.; Qi, Q. Complete PHB mobilization in Escherichia coli enhances the stress tolerance: A potential biotechnological application. Microb. Cell Factories 2009, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Delfino, I.; Esposito, R.; Portaccio, M.; Lepore, M. Dynamical and structural properties of flavin adenine dinucleotide in aqueous solutions and bound to free and sol–gel immobilized glucose oxidase. J. Sol Gel Sci. Technol. 2016, 82, 239–252. [Google Scholar] [CrossRef]
- Hussain, S.; Malik, A.H.; Iyer, P.K. FRET-assisted selective detection of flavins via cationic conjugated polyelectrolyte under physiological conditions. J. Mater. Chem. B 2016, 4, 4439–4446. [Google Scholar] [CrossRef] [PubMed]
- Hanko, E.; Minton, N.; Malys, N. Characterisation of a 3-hydroxypropionic acid-inducible system from Pseudomonas putida for orthogonal gene expression control in Escherichia coli and Cupriavidus necator. Sci. Rep. 2017, 7, 1724. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | A1 [kCnts] | A2 [kCnts] | τ1 [ns] | τ2 [ns] | A2/A1 | |
|---|---|---|---|---|---|---|
| Cupriavidus necator H16 | 1 | 0.105 ± 0.004 | 0.197 ± 0.007 | 4.17 ± 0.02 | 1.29 ± 0.06 | 1.9 |
| 2 | 0.452 ± 0.008 | 0.775 ± 0.023 | 5.04 ± 0.04 | 1.13 ± 0.04 | 1.7 | |
| 3 | 0.262 ± 0.009 | 0378 ± 0.016 | 4.54 ± 0.05 | 1.22 ± 0.03 | 2.2 | |
| 4 | 0.279 ± 0.015 | 0.700 ± 0.008 | 4.60 ± 0.12 | 122 ± 0.04 | 2.5 | |
| 5 | 0.320 ± 0.020 | 0.751 ± 0.012 | 4.30 ± 0.11 | 1.15 ± 0.05 | 2.3 | |
| 6 | 0.034 ± 0.003 | 0.082 ± 0.004 | 3.80 ± 0.11 | 0.92 ± 0.05 | 2.4 | |
| 7 | 0.061 ± 0.005 | 0.103 ± 0.003 | 4.l0 ± 0.13 | 1.14 ± 0.07 | 1.7 | |
| Cupriavidus necator PHB-4 | 1 | 0359 ± 0.008 | 1.270 ± 0.015 | 4.09 ± 0.04 | 1.12 ± 0.02 | 3.5 |
| 2 | 0.147 ± 0.006 | 0.479 ± 0.012 | 3.80 ± 0.06 | 1.11 ± 0.03 | 3.3 | |
| 3 | 0.594 ± 0.012 | 2.420 ± 0.027 | 3.61 ± 0.05 | 1.07 ± 0.01 | 4.1 | |
| 4 | 0.740 ± 0.020 | 3.210 ± 0.037 | 3.38 ± 0.03 | 1.06 ± 0.02 | 4.3 | |
| 5 | 0.072 ± 0.003 | 0.220 ± 0.007 | 3.65 ± 0.07 | 0.88 ± 0.03 | 3.1 | |
| 6 | 0.052 ± 0.006 | 0.161 ± 0.006 | 4.00 ± 0.22 | 0.90 ± 0.06 | 3.1 | |
| 7 | 0.288 ± 0.015 | 0.979 ± 0.008 | 4.33 ± 0.06 | 1.18 ± 0.03 | 3.4 |
| Sample | A1 [kCnts] | A2 [kCnts] | τ1 [ns] | τ2 [ns] | A2/A1 | |
|---|---|---|---|---|---|---|
| CN H16 | 1 | 6.000 ± 0.018 | 0.549 ± 0.023 | 5.06 ± 0.01 | 1.50 ± 0.15 | 0.09 |
| 2 | 5.240 ± 0.022 | 0.571 ± 0.017 | 5.11 ± 0.01 | 1.22 ± 0.10 | 0.11 | |
| 3 | 6.470 ± 0.028 | 0.596 ± 0.022 | 5.19 ± 0.01 | 1.22 ± 0.09 | 0.009 | |
| CN PHB-4 | 1 | 0.412 ± 0.019 | 0.476 ± 0.008 | 5.08 ± 0.08 | 1.40 ± 0.11 | 1.15 |
| 2 | 0.273 ± 0.005 | 0.339 ± 0.007 | 5.07 ± 0.07 | 1.29 ± 0.03 | 1.24 | |
| 3 | 0.295 ± 0.016 | 0.364 ± 0.016 | 5.10 ± 0.03 | 1.18 ± 0.08 | 1.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müllerová, L.; Marková, K.; Obruča, S.; Mravec, F. Use of Flavin-Related Cellular Autofluorescence to Monitor Processes in Microbial Biotechnology. Microorganisms 2022, 10, 1179. https://doi.org/10.3390/microorganisms10061179
Müllerová L, Marková K, Obruča S, Mravec F. Use of Flavin-Related Cellular Autofluorescence to Monitor Processes in Microbial Biotechnology. Microorganisms. 2022; 10(6):1179. https://doi.org/10.3390/microorganisms10061179
Chicago/Turabian StyleMüllerová, Lucie, Kateřina Marková, Stanislav Obruča, and Filip Mravec. 2022. "Use of Flavin-Related Cellular Autofluorescence to Monitor Processes in Microbial Biotechnology" Microorganisms 10, no. 6: 1179. https://doi.org/10.3390/microorganisms10061179
APA StyleMüllerová, L., Marková, K., Obruča, S., & Mravec, F. (2022). Use of Flavin-Related Cellular Autofluorescence to Monitor Processes in Microbial Biotechnology. Microorganisms, 10(6), 1179. https://doi.org/10.3390/microorganisms10061179