Abstract
Genomic characterization of circulating influenza type-A viruses (IAVs) directs the selection of appropriate vaccine formulations and early detection of potentially pandemic virus strains. However, longitudinal data on the genomic evolution and transmission of IAVs in Africa are scarce, limiting Africa’s benefits from potential influenza control strategies. We searched seven databases: African Journals Online, Embase, Global Health, Google Scholar, PubMed, Scopus, and Web of Science according to the PRISMA guidelines for studies that sequenced and/or genomically characterized Africa IAVs. Our review highlights the emergence and diversification of IAVs in Africa since 1993. Circulating strains continuously acquired new amino acid substitutions at the major antigenic and potential N-linked glycosylation sites in their hemagglutinin proteins, which dramatically affected vaccine protectiveness. Africa IAVs phylogenetically mixed with global strains forming strong temporal and geographical evolution structures. Phylogeographic analyses confirmed that viral migration into Africa from abroad, especially South Asia, Europe, and North America, and extensive local viral mixing sustained the genomic diversity, antigenic drift, and persistence of IAVs in Africa. However, the role of reassortment and zoonosis remains unknown. Interestingly, we observed substitutions and clades and persistent viral lineages unique to Africa. Therefore, Africa’s contribution to the global influenza ecology may be understated. Our results were geographically biased, with data from 63% (34/54) of African countries. Thus, there is a need to expand influenza surveillance across Africa and prioritize routine whole-genome sequencing and genomic analysis to detect new strains early for effective viral control.
1. Introduction
Influenza type-A viruses (IAVs) cause potentially deadly respiratory infections among humans globally [1]. The evolution of IAVs is punctuated by gene exchanges (reassortment) between avian, swine, and human IAVs responsible for the past global influenza pandemics: the 1918-H1N1, 1957-H2N2, 1968-H3N2, and the 2009-H1N1pdm09 that claimed millions of lives [1,2,3,4]. The effects of the pandemics are further worsened by seasonal influenza infections estimated to cause excess mortality at a rate of 4.0–8.8 per 100,000 individuals globally, with the highest rates (2.8–16.5 per 100,000 individuals) in sub-Saharan Africa [5].
The influenza type-A viral whole-genome consists of eight single-stranded RNA gene segments coding for two immune-stimulating surfaces (hemagglutinin-HA and neuraminidase-NA), one transmembrane, and five internal proteins [6]. There are 18 HA and 11 NA subtypes of IAVs, but frequently H1-H3 and N1-N2 viruses have been sampled among humans [7].
Genomic characterization of circulating IAVs to understand their evolution and how novel viruses emerge and spread among populations is important for effective viral control and prevention. However, the rapid and continuous genomic changes through mutations (genes) or substitutions (proteins) [8], reassortment, and rarely recombination [9] make the predictions very difficult. Amino acid substitutions at the major antigenic sites of the IAV H1 (Sa, Sb, Ca1, Ca2, and Cb) and H3 proteins (A, B, C, D, and E) [10,11,12], and the NA proteins [13,14] cause the IAVs to drift away from vaccine-mediated immunity. IAVs also escape host immunity by gaining new N-linked glycosylation sites, Asn-X-Ser/Thr, where X is any amino acid except proline, in their HA and NA proteins [15,16]. When a sugar molecule attaches to the amide nitrogen on the Asn, the HA antigenic and NA catalytic sites become masked from binding to host antibodies and protected, respectively [15]. Substitutions in the internal genes of IAVs also result in new viruses with altered pathogenicity, virulence, transmissibility, and reduced sensitivity to antiviral drugs and vaccines [17,18,19].
Modern sequencing technologies, bioinformatic and phylogenetic-based analyses, and epidemiological and evolution models have successfully been used on viral genome data from well-sampled regions in Asia, Europe, and the United States (U.S.) to infer viral genomic and antigenic evolution, diversification, and transmission patterns [20,21,22,23,24,25,26,27]. Contrary to common belief, influenza surveillance in Africa has improved substantially since the mid-2000s, showing the cocirculation of multiple viral subtypes all year round with distinct peaks in Northern and Southern Africa [28,29,30]. However, data on the genomic characterization of IAVs in Africa remain scanty, limiting our understanding of Africa’s IAVs evolution, contribution to the global influenza ecology, and benefit from viral control strategies. We sought to systematically merge data on the evolution, diversification, drug resistance, zoonosis, phylogenetic clustering and genetic clades, phylodynamics, and phylogeographic patterns of H1N1, H1N1pdm09, and H3N2 virus strains previously sampled in Africa.
2. Materials and Methods
2.1. Definitions and Outcomes
We defined a subtype as a group of viruses with a specific combination of HA and NA genes, for example, H1N1 and H3N2 [7]. A viral lineage is a group of genetically related viruses of the same subtype sharing a common ancestor. A strain is a virus exhibiting minor mutations different from other viruses of the same lineage or subtype.
A phylogeny is a tree-like structure reconstructed using viral gene sequences showing evolutionary relationships between the sampled viruses from a common ancestor. The tree has internal nodes, branches, and tips representing ancestors, genetic distances from the nearest ancestors, and sampled viruses, respectively.
Phylogenetic clusters and clades are often interchanged [31]. Here, we defined a cluster as a subtree of viruses sharing a common ancestor and may share epidemiological linkage [32]. In contrast, a clade is a cluster of viruses with characteristic amino acid substitutions in their HA proteins subunits (HA1 or HA2) [27]. Phylodynamics is the study of evolutionary processes that shape the phylogenies, such as changes in viral effective population sizes [Ne(t)], evolutionary rate, basic reproductive number (R0), selection pressures, and reassortment [25,33]. Additional definitions are provided in Table S1.
2.2. Search Strategy and Selection Criteria
We searched 7 databases: African Journals Online, Embase, Global Health, Google Scholar, PubMed, Scopus, and Web of Science on 30 July 2021 for published articles on sequencing and genomic characterization of IAVs sampled in Africa, according to the PRISMA guidelines [34]. The search keywords included (“influenza”) AND (“sequencing” OR “genomic analysis” OR “phylogenetic analysis”) and their respective medical subject heading (MeSH) terms, AND (each African country, region, and the Indian Ocean) NOT (non-human influenza or other pathogens causing influenza-like-illnesses), as provided in Table S2. We did not filter by language or publication date.
All searched study articles were exported to and deduplicated using Zotero citation manager (https://www.zotero.org/, last accessed on 30 July 2021). Non-English articles were translated using google translate (https://translate.google.com, last accessed on 30 July 2021). Unique articles were subjected to a primary screening for eligibility based on the title, abstract, and method using a predefined eligibility criterion (Table S3). We excluded studies that did not confirm influenza infection by culture or/and polymerase chain reaction (PCR), analyzed only non-human or other human influenza viruses (type B, C, and D), case reports, conference abstracts and presentations, health meetings, and sampled only non-African populations.
Eligible articles identified through the primary screening were subjected to a secondary search by screening their bibliographies for additional relevant articles. Authors were contacted for details on studies with sufficient information to decide on their inclusion. Eligible studies were agreed upon by G.N., R.G., J.M.K., and D.P.K.
2.3. Data Extraction and Synthesis
G.N. and R.G. extracted study data using a predefined data collection form. Data extracted for each study included: country, sampling period and setting, population demographics, selection criteria of samples for sequencing or pre-collected sequences, sample size, sequencing technology, number of viruses sequenced, genes sequenced or/and analyzed, analysis type, epidemiological variables linked to sequence data, and main findings.
Extracted data were stratified according to viral subtype and analysis type. We systematically report results from the earliest to the latest sampling year to highlight differences in viral evolution and transmission patterns between seasons.
2.4. Study Quality Assessment
Eligible studies were assessed for quality and risk-of-bias using the Newcastle-Ottawa Quality Assessment Scale-NOS (adapted for cross-sectional studies) [35] with additional items from the Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases (STROME-ID) checklist [36]. Studies were evaluated based on the sampling, reporting missing data, outcome assessment, and result selection and reporting (Table S4).
The review was reported according to the preferred reporting items for systematic review (PRISMA) guidelines, and its protocol was registered on PROSPERO (https://www.crd.york.ac.uk/PROSPERO/, last accessed on 23 March 2022) under ID CRD42020187011.
3. Results
3.1. Search Outcomes and Study Characteristics
The primary search identified 1337 study articles, 60 of which were eligible (Figure 1). The secondary search identified 11 additional eligible articles, bringing the total to 71 articles included in the final analysis.
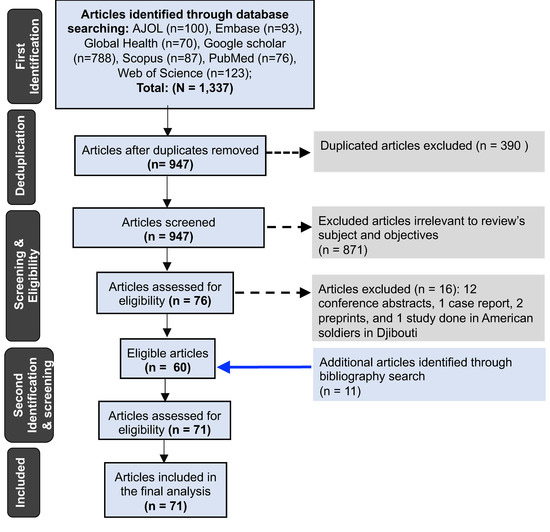
Figure 1.
Flowchart showing the exclusion and inclusion criteria of studies analyzed.
A total of 70% (50/71) of the included studies were nested within surveillance programs, and 30% (21/71) were independent (Table 1). The included studies analyzed IAVs sampled from 34 (63%) of the 54 African countries (Table S5), with most studies characterizing viruses from South Africa (n = 42), Kenya (n = 25), and Ghana (n = 24) (Figure 2). A total of 14, 41, and 42 studies characterized H1N1, H1N1pdm09, and H3N2 viruses sampled for 14 (1996–2009), 10 (2009–2018), and 26 (1993–2018) years, respectively.

Table 1.
Characteristics of eligible studies analyzed.
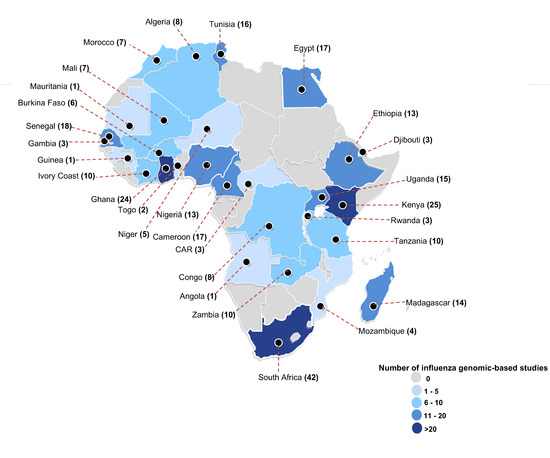
Figure 2.
The geographical distribution of studies that did genomic characterization of influenza type-A viruses (H1N1, H1N1pdm09, and H3N2) sampled in Africa. Each country is highlighted based on the absolute number of studies that analyzed sequences of influenza viruses collected from that country. For each country, the study count also includes any study that included at least one sequence from that country in their virus clade classification using the European Center for Disease Control (ECDC) guidelines [27]. Abbreviations: CAR = Central African Republic. Countries not shown: Cape Verde (n = 1), Reunion (n = 3), Seychelles (n = 4), Mauritania (n = 1), Mauritius (n = 10), and Mayotte (n = 1).
A total of 87% (62/71) of the studies generated and analyzed new viral sequences, and 13% (9/71) analyzed online sequences. Of the studies that generated new sequences, 53% (33/62) and 37% (23/62) sequenced IAVs locally and abroad, especially in the United Kingdom (U.K.) and the United States (U.S.), respectively. Two studies sequenced IAVs both locally and abroad, and four studies did not report where sequencing was performed.
A total of 56% (35/62) of the studies used Sanger sequencing technology and were published between 1996 and 2020. Only 8% (5/62) of the studies used next-generation sequencing (NGS) and were published between 2011 and 2021 [75,76,81,97,98]. Two studies used both Sanger and NGS [26,73]. Another study did not clearly report whether Sanger or NGS was used [94]. Three studies used pyrosequencing or Sanger and pyrosequencing or Sanger and targeted HA analysis. Sixteen studies did not describe the technology used.
A total of 82% (58/71) of included studies sequenced and/or analyzed partial genomes, especially the HA and/or NA and/or MP. A total of 16% (11/71) studies sequenced and/or analyzed whole genomes [26,44,47,54,55,57,63,73,75,95,98]. One study sequenced whole genomes but analyzed only the HA genes [81]. Two studies analyzed only MP or NS genes. Viral phylogenetic patterns were the most characterized by 77% (55/71) of included studies followed by 68% (48/71) studies reported on virus-vaccine genomic variation. Viral drug sensitivity and phylodynamics were described in 39% (28/71) and 17% (12/71) studies, respectively. One study [100] had data on viral recombination.
3.2. Quality of Studies Analyzed
Our quality and risk-of-bias assessment scheme scored each study out of 23 points (Table S4). Given the additional assessment terms, the analyzed studies had a mean quality score of 54.9% (30.4–73.9) (Table S6), lower than the 90% (70–100) reported by Modesti [35]. The detailed assessment of the sampling bias for all included studies is provided in Table S7.
3.3. Molecular Epidemiology of Seasonal H1N1 Viruses in Africa
3.3.1. Genomic Diversity of H1N1 Viruses and Their Relatedness to Vaccines
Africa H1N1 viruses evolved every season. Virus strains sampled before 2007 exhibited minor genomic variation from their corresponding season vaccine strains. For example, five Morocco H1N1 strains sampled between 1996 and 1997 had a single substitution V57I in their HA1 proteins relative to the 1997–1998 season A/Bayern/7/1995(H1N1) vaccine strain and were recognized by antisera against the previous 1987–1996 northern hemisphere (NH) and 1988–1997 southern hemisphere (SH) season A/Singapore/6/1986(H1N1) vaccine strain [83]. Strains sampled later in Dakar (Senegal) during the 1997–1999 and 2000–2003 seasons evolved and were genetically and antigenically similar to the A/Beijing/262/1995(H1N1) (1998–2000 NH and 1999 SH season) and A/New Caledonia/20/1999(H1N1) (2000–2007 NSH season) vaccine strains, respectively [89]. New H1N1 strains similar to the 2008–2010 NH and 2009 SH season A/Brisbane/59/2007(H1N1) vaccine strain emerged in Dakar [89], Kenya [38], and South Africa [37] between 2007 and 2008. While Kenya H1N1 strains sampled in 2007 had three mutated antigenic sites Sb (D190N/G) and Ca2 (D225G), more substitutions at site Sb (N187S, G189N/A, K192R, A193T) and receptor-binding sites (N187S, G189N, A193T) were observed in 2008 relative to A/Brisbane/59/2007(H1N1) strain [38].
3.3.2. Antiviral Drug Sensitivity among H1N1 Viruses
All H1N1 strains that circulated in Egypt, Kenya, and Senegal between 2004 and 2009 lacked the adamantane-resistant marker S31N in their M2 proteins [41,87,105]. The adamantane-susceptible strains sampled in Kenya between 2008 and 2009 had HA1 substitutions R192H, K140E, K145R, and N183S absent in global adamantane-resistant H1N1 strains (with E140K) [105].
Oseltamivir-resistant H1N1 strains with N1 substitution H275Y (H274Y, N2 numbering) emerged at different times in 2008 and circulated at high frequencies (70–100%) in several African countries. The first H1N1-H275Y variant was sampled in Cameroon in late February, Ivory Coast and Seychelles in April, South Africa in May, and Senegal in July 2008 [37,39,40,41,88,104,105]. The H1N1-H275Y variants had additional N1 substitutions unique to the country of sampling: South Africa (M23L, N73K) [37], Senegal (S141N, G185A) [41], and Cameroon, Ivory Coast, and Seychelles (D354G) [40,88]. Neuraminidase inhibition (NAI) assays confirmed that Africa H1N1-H275Y variants were resistant to oseltamivir and peramivir but susceptible to zanamivir [37,39,40,104].
3.3.3. Phylogenetic Clustering Patterns and Circulating Clades among H1N1 Viruses
Phylogenies of Africa H1N1 virus strains showed the cocirculation of multiple viral lineages, clusters, and clades from 1996 through 2009. South Africa H1N1 strains sampled in 1996 were highly clustered (bootstrap > 98) with European-origin A/Bayern/7/1995(H1N1)-like strains [85]. An Asian-origin A/Wuhan/371/1995(H1N1)-like lineage emerged in 1997 and co-circulated with the A/Bayern/7/1995 (H1N1)-like strains in South Africa up to 1999 [85]. Similarly, Asian-origin A/Beijing/262/1995(H1N1)-like strains emerged in Dakar in 1997 and circulated through 1999 [89]. In 2001, novel Oceania-origin A/New Caledonia/20/1999(H1N1)-like strains belonging to clade 1 emerged in Egypt and were also sampled later in Senegal and Reunion (2003), Morocco (2004), South Africa (2005), Egypt (2005 and 2006), and Madagascar (2006) [89] (Figure 3). During the same season (2001), novel reassortant H1N2 viruses with HA1 substitutions V175I, A190T, and A215T emerged in Egypt and co-circulated with H1N1 viruses [89]. H1N2 strains were later sampled in South Africa (2002) and Senegal (2003) [89] but disappeared shortly after their emergence.

Figure 3.
Temporal and geographical distribution of genetic clades of seasonal H1N1 virus strains that circulated in Africa between 2001 and 2009. Details on the characteristic genomic markers (amino acid substitutions in the HA1 proteins) for each clade are described in Table S8.
A South Africa strain, A/Johannesburg/141/2007(H1N1), sampled in 2007, shared a similar H1 gene as an American-origin A/St.Petersburg/10/2007-like(clade 2C) strain with HA1 substitutions T82K, Y94H, and K141E [89]. In the same year (2007), new Australian-origin A/Brisbane/59/2007(H1N1)-like strains (clade 2B, mostly oseltamivir-resistant) with HA1 substitutions (D35N, R159K, E274K) emerged in Egypt, Madagascar, Mauritius, and Senegal [41,89]. Clade 2B was widely spread in 2008 in Northern (Algeria), Central (Cameroon), Eastern (Madagascar, Mauritius, Seychelles, Kenya), Western (Ivory Coast, Ghana, Senegal), and Southern Africa (South Africa), clustering according to country of sampling [41,84,88,89,105]. Clade 2B H1N1 strains sporadically spread in Kenya and Madagascar in 2009 [88,105] (Figure 3, Table S8).
3.4. Molecular Epidemiology of Pandemic H1N1pdm09 Viruses in Africa
We refer to H1N1pdm09 virus strains sampled from 2009 to 2010 and from 2011 onwards as pandemic H1N1pdm09 (pH1N1pdm09) and seasonal H1N1pdm09 (sH1N1pdm09), respectively.
3.4.1. Genomic Diversity of H1N1pdm09 Viruses and Their Relatedness to Vaccines
Africa pH1N1pdm09 virus strains showed early evolution from the A/California/07/2009(H1N1) vaccine strain recommended for global use during the 2010–2016 seasons. The complete H1 gene and protein sequences of the pH1N1pdm09 strains were 97–99.5% and 98.4–99.5% similar to the A/California/07/2009(H1N1) vaccine strain [46,47,48,52,53,54,55,62,65,93,104]. Partial H1 gene sequences of H1N1pdm09 strains sampled from the Central African Republic (CAR) in 2010 [59] and South Africa (Cape Town) in April–July 2011 [65] were 99–100% and 96.6–98.5% similar to the A/California/07/2009(H1N1) strain, respectively.
H1N1pdm09 strains sampled between 2009 and 2011 in Kenya [54,55,93], Tunisia [50], and Morocco [52] had several substitutions at four major antigenic sites: Sa (N125D, S162I, S183P, K164F), Sb (S185T, S190G, S202T), Ca1 (H138Q, G170R, S203T, R205K, S220T, D238E), and Ca2 (D222G/E, D239E) relative to the A/California/07/2009(H1N1) strain. Despite the rapid genetic diversification, pH1N1pdm09 and early post-pandemic H1N1pdm09 strains (sampled in 2011) remained antigenically similar to A/California/07/2009(H1N1) vaccine strain. Except for two South Africa strains sampled in 2010, with HA1 substitutions N125D and V272F, that reacted low to A/California/07/2009(H1N1) antisera [66].
Africa sH1N1pdm09 strains progressively drifted from the A/California/07/2009(H1N1) vaccine strain, with new substitutions observed every year. H1 proteins of sH1N1pdm09 strains sampled in the Democratic Republic of Congo (Congo), Ethiopia, Egypt, Ghana, Mozambique, Cameroon, and Kenya between 2013 and 2016 had all five major antigenic sites: Sa (T120A, A141V), Sb (S203T, E374K, S451N, S162R/N, S185T), Ca1 (D97N, K283E), Ca2 (S203T, K163Q), and Cb (A48S, P83S, S84N, K163Q) mutated relative to the A/California/07/2009(H1N1) vaccine strain [58,61,97,102]. sH1N1pdm09 strains sampled between 2017 and 2018 evolved and were 97.9–99.4% and 97.5–99% similar to the new 2017–2018 season A/Michigan/45/2015(H1N1) vaccine strain at the nucleotide and amino acid level, respectively [91]. Kenya H1N1pdm09 viruses sampled during the 2017–2018 season had several mutated antigenic sites: Sa (T120A, P137S, A141E, I149L), Ca1 (H273Y, I295V), and Ca2 (S164T), Cb (S74R, S162N) relative to A/Michigan/45/2015(H1N1) strain [61]. In addition, Kenya strains sampled in 2018 had mutated antigenic sites, Ca1 (G45R, V298I) and Ca2 (R223Q), relative to the new 2019 NH season A/Brisbane/02/2018(H1N1) vaccine strain [61]. Both antigenic drift and variation in the dominant antigenic sites between seasons, 2009–2010 (Ca1) to 2014–2016 (Cb, Sa, Sb), and 2017–2018 (Ca1, Cb) resulted in low efficacy of the A/California/07/2009(H1N1), A/Michigan/45/2015(H1N1), and A/Brisbane/02/2018(H1N1) vaccine strains of 24.55–35.77%, 39.6–41.8%, and 32.4–42.1% against the 2014–2016 and 2017–2018, and 2018 sH1N1pdm09 viruses, respectively [58,61].
All six classical N-linked glycosylation sites present in the A/California/07/2009(H1N1) strain HA1 protein were conserved among 2009–2011 and 2014–2016 H1N1pdm09 strains sampled in Tunisia [50] and Cameroon [58], respectively. Except for A/Tunisia/15656/2009(H1N1) and A/Tunisia/2144/2011(H1N1) strains with substitutions N276H and T288N, respectively. Kenya strains sampled during the 2015–2016 season retained four (11, 23, 87, and 287) of the six classical N-linked glycosylation sites. While all 2015–2017 and 2017–2018 sH1N1pdm09 strains sampled in Egypt [101] and Kenya [61], respectively, gained a new N-linked glycosylation site (S162N) present in the A/Michigan/45/2015(H1N1) strain H1 protein, except for one A/Kenya/066/2018(H1N1) strain.
Africa H1N1pdm09 strains also evolved from vaccine strains in the remaining seven genes (PB2, PB1, PA, NP, N1, MP, and NS). For example, Kenya H1N1pdm09 strains sampled in 2009 had PB2, PB1, and PA proteins with 99–100%, 98.4–100%, and 99.4–100% similarity to A/California/07/2009(H1N1) strain [55]. All pH1N1pdm09 strains sampled in Uganda, Senegal, Mauritania, Kenya, and South Africa had V106I and N248D in the catalytic site (CS) of their N1 proteins relative to the A/California/07/2009(H1N1) strain [47,48,54,55,56,104]. Quiliano et al. observed 26 unique substitutions among N1 proteins of 59 Africa H1N1pdm09 strains sampled between 2009 and April 2011, with the highest frequency of substitutions in the transmembrane (TM) and L21 domains [64]. Kenya pH1N1pdm09 strain nucleoproteins (NP) and matrix protein (M1 and M2) sequences sampled in 2009 were 98.8–99.6% and 98–100%, similar to the A/California/07/2009(H1N1) strain, respectively. M1 (92–100%) was more conserved than M2 (77–100%) proteins among Kenya pH1N1pdm09 strains [54]. Virus strains sampled from Kenya, Nigeria, Ethiopia, and Mali in 2009 and Senegal in 2010 had NS proteins with a mean amino acid similarity of 98% to A/California/07/2009(H1N1) strain [53]. NS1 was more conserved than NS2 proteins among Kenya pH1N1pdm09 strains [53,54,55].
3.4.2. Genomic Markers of Disease Severity among H1N1pdm09 Viruses
H1N1pdm09 strains with antigenic site Ca2 substitution, D222G, associated with disease severity [106], circulated at lower frequencies in Tunisia (8%, 4/50) and Egypt (9%, 1/17) during the 2009–2011 and 2009 season, respectively [49,50,101]. Another D222G variant was sampled in Egypt later in 2016 [101]. Several HA1 substitutions, S185T, Q293H, K374E, 312I, H138Q, S203T, R205K, D222E, and T288N, were also sporadically observed among patients with severe acute respiratory illnesses (SARI) [49,50,51,66]. The D222E variants widely circulated in Tunisia, South Africa, Morocco, Senegal, and Egypt during the pandemic [48,49,50,51,52,66,101].
3.4.3. Antiviral Drugs Sensitivity among H1N1pdm09 Viruses
All Africa H1N1pdm09 virus strains sampled between 2009 and 2016 had the M2 adamantane-resistant marker, S31N [47,54,55,58,105], but lacked the N1 oseltamivir-resistance marker, H275Y [47,48,51,54,55,58,62,63,66,90,101,104]. Except for a strain sampled from a South African patient after treatment with a standard dose of oseltamivir during the pandemic [43] and an A/Egypt/1424/2016(H1N1) strain had H275Y in their N1 proteins. No H1N1pdm09-H275Y variant was sampled in 2017. All N1 sites, 117, 119, 136, 151, 152, 199, 223, 247, 293, and 295, associated with reduced NAIs inhibition in vitro were conserved among Cameroon 2014–2016 sH1N1pdm09 strains [58].
3.4.4. Phylogenetic Clustering and Circulating Clades among H1N1pdm09 Viruses
Phylogenies showed that Africa H1N1pdm09 virus strains were variants of the A/California/07/2009(H1N1) strain that diverged into multiple clades that co-circulated every season. Analysis of concatenated whole viral genomes (concat-8) formed similar but deeper phylogenies (with a higher number of nodes between root and furthest tip) than those of individual genes [54,55]. Pandemic H1N1pdm09 viruses sampled from different African countries mixed. While sH1N1pdm09 strains clustered distinctly according to time and location (household or province or country) of sampling [47,53,55,60,62,102,103].
The 2009–2011 H1N1pdm09 outbreaks observed in different African countries resulted from the emergence of strains belonging to genetic clades 2, 3, 5, 6, and 7 (Figure 4). Clade 2 pH1N1pdm09 strains, similar to earlier Mexican- and England-origin strains sampled in April–June 2009, were first introduced in Kenya on 19 April 2009 (95% CI: 27 December 2008–6 July 2009) [54,55]. Similar clade 2-like strains with HA1 substitutions N31D and S162N similar to A/Mexico/2466/07/2009(H1N1), A/Wisconsin/02/01/2011(H1N1), and A/Czech Republic/32/2011(H1N1) strains were sampled in South Africa later in 2010 [50,51,52,66]. Novel clade 3 strains with HA1 substitutions S183P and A134T, similar to the A/England/195/2009(H1N1) and A/New York/3177/2009(H1N1) strains, were first sampled in Cameroon in 2009 and circulated through 2010 [48]. Similar clade 3 strains were also observed in Ghana, Senegal, Ivory Coast, and Ethiopia in 2010 and last reported in Tunisia in 2011 [48,50,51,52,54,55,91]. Pandemic clade 5 strains, with HA1 substitutions D97N, R205K, I216V, and V249L, first emerged in 2010 in Morocco [52], then continued to circulate in Morocco, Tunisia, South Africa, and Uganda in 2011 [50,51,52,95,101]. Shockingly, clade 5 remerged in Tunisia later in 2017 [91].
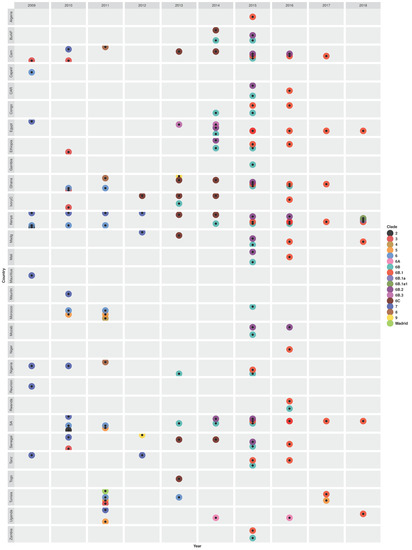
Figure 4.
Temporal and geographical distribution of genetic clades of pandemic H1N1pdm09 (2009–2010) and seasonal H1N1pdm09 (2011 onwards) virus strains that circulated in Africa between 2009 and 2018. Abbreviations: Burkina Faso (BurkF), Cameroon (Cam), Cape Verde (CapeV), Central African Republic (CAR), Ivory Coast (IvoryC), Madagascar (Madg), Mauritania (Mauritn), Mozambique (Mozab), South Africa (SA), Tanzania (Tanz), and A/Madrid/SO8171/2010(H1N1)-like clade (Madrid). Details on the characteristic genomic markers (amino acid substitutions in the HA1 proteins) for each clade are described in Table S9.
Novel clade 7 with HA1 substitutions S185T, S143G, and A197T were first sampled in 2009 in Egypt, Nigeria, Reunion, Mauritius, Tanzania, and Kenya [48,54,55,63,98] (Figure 4). Molecular dating of the H1 phylogeny estimated that clade 7 strains, similar to earlier strains sampled from England, California, and China, were introduced in Kenya (KENB-GC7) on 4 June 2009 (95% CI: 16 April–29 June 2009), 2 months before laboratory detection [54,55] and in the Reunion (RUN) on 19 May 2009 [63]. Contrastingly, whole-genome (concat-8) analysis estimated the most recent common ancestor (TMRCA) of Kenya pH1N1pdm09 strains at an earlier date of 28 February 2009 (95% CI: October 2008–May 2009) [54,55]. Notably, clade 7 diversified in Kenya through independent introductions of multiple subclades: KENC-GC7, KEND-GC7, KENE-GC7, and KENF-GC7 introduced on 8 August 2009 (19 May–2 October 2009), 7 August 2009 (10 June–17 September 2009), 4 August 2009 (10 June–2 October 2009), and 2 October 2009 (20 June–10 December 2009), respectively [54,55]. Similarly, two subclades of clade 7 (RUNA and RUNB), similar to a Japan-origin A/Sapporo/1/2009(H1N1) strain, were introduced in Reunion on the 26th June 2009 and 8th July 2010, respectively [63]. Clade 7 strains sampled in Reunion, Tanzania, and Mauritius were more similar than those sampled in Seychelles and Madagascar. Clade 7 continued to circulate in 2010 (Senegal, Kenya, Mauritania, South Africa, Cameroon, and Nigeria), 2011 (Kenya and Uganda), and 2012 (Tanzania, Kenya, and Madagascar) [48,54,55,84,96].
Another group of the earliest pH1N1pdm09 strains sampled in Africa belonged to clade 6, with HA1 substitutions D97N and S185T, similar to A/New York/3324/2009(H1N1) and A/Shanghai/143T/2009(H1N1), which later became dominant and the most diverse. Clade 6 strains were first sampled in Cape Verde and Kenya in 2009 [48,98]. Clade 6 continued to circulate in Kenya, Morocco, Ghana, South Africa during the 2010–2011 season and in Tunisia (2011 and 2013) [51,52,92,93,98,105]. After the 2009–2010 pandemic, several clade 6 variants: 6A, 6B, 6B.1, 6B.1a, 6B.2, 6B.3, and 6C emerged in different African countries. Specifically, novel clade 6C strains, with HA1 substitutions D97N, S185T, K283E, E499K, and V234I, were first sampled in Ivory Coast in 2012 [99], then circulated in different Western and Eastern African countries between 2013 and 2014 [58,94,96,97,98,99,101,102] (Figure 4 and Table S9). Novel sH1N1pdm09 viruses belonging to clade 6B, with HA1 substitutions K163Q and A256T, emerged in 2013 in Ivory Coast, Nigeria, and South Africa [58,94,97,99,102], before it widely spread in all African regions from 2014 through 2016 [58,94,96,97,98,99,102]. Among the clade 6B variants, 6B.3 strains were sampled only in Egypt during the 2013–2014 season [101]. In contrast, 6B.2 with HA1 substitutions E499K, S84N, and R45K and 6B.1 with S84N, S162N, and I216T circulated in the 2014–2016 and 2015–2018 seasons, respectively, in different countries [58,61,91,96,98,99,101,102]. Clade 6B.1 dominated and further diverged into 6B.1a and 6B.1a1 subclades, which were observed in 2018 in Kenya [98]. A study by Opanda et al. reported the emergence of novel A/Brisbane/02/2018(H1N1)-like clade 6B.1a1 strains in Kenya in 2018 [61]. Strains with a clade 6A-like H1 genes were sampled in 2014 and 2016 only in Uganda [58].
Three novel clades, A/Madrid/SO8171/2010(H1N1)-like with E172K, K308E, and V47I, A/Christchurch/16/2010(H1N1)-like (clade 4) with N125D, and A/Cameroon/LEID/01/11/1450/2011(H1N1)-like (clade 8, unique to Africa) with A186T and V272A substitutions in their HA1 proteins emerged in the early post-pandemic year (2011) circulating in Tunisia, Morocco, and three countries (Ghana, Nigeria, and Cameroon), respectively [50,51,52,95]. Like clade 8, novel clade 9 with HA1 substitutions L32I, D86E, S128T, R259K, S263A, I460V, and V520A was unique to Africa (Figure S10), observed in Senegal and Ghana in 2012 and 2013, respectively [97,102].
Despite the multiple introductions and early diversification of H1N1pdm09 strains, two highly supported viral clusters (I and II, bootstrap > 80%) persisted for >1.8 years in Western Africa between 2010 and 2012 [60]. Cluster I contained strains with H1 substitutions S145T and R276K that circulated in Senegal, Ghana, and Ivory Coast between March 2011 and January 2013, similar to a France-origin strain sampled earlier in 2010. The largest cluster II contained both Western Africa (n = 26, including the A/Ghana/763/2011(H1N1)-clade 8 strain) and non-Western Africa (Ethiopia, France, Italy, Norway, Sweden, and Minnesota, n = 11) strains with H1 substitutions A15T, N490D, T491K, and V537, sampled during the November 2010–October 2012 and November 2012–January 2013 seasons, respectively. All cluster II non-Western Africa strains clustered monophyletically (bootstrap = 100%) [60].
Phylogeographic analysis by Owuor using the Bayesian Ancestral state reconstruction (BSSVS) confirmed the introduction of H1N1pdm09 strains into Africa between 2009 and 2018 from outside Africa, with the highest viral migration rate of 1.48 (Bayes factor, BF ≥ 3) from East and Southeast Asia [98]. Introduction events were inferred as clusters with bootstrap ≥ 80%, size of n ≥ 2 strains, and >80% of strains sampled from Africa. The BSSVS model estimated strongly supported H1N1pdm09 viral migration from Northern to Southern Africa (BF ≥ 1000), between countries (100 ≤ BF < 1000), and within the country (Kenya) (rate = 0.81–0.93, BF ≥ 1000) [98]. Overall, H1N1pdm09 strains sampled in Africa belonged to similar globally recognized clades. However, most clades first circulated elsewhere before their introduction in Africa, except clades 8 and 9 were unique to Africa (Figure S10).
3.4.5. Population Dynamics, Evolutionary Rates, Selection, and Reassortment among H1N1pdm09 Viruses
Upon viral introduction in Kenya, the population sizes (genomic diversity) of H1N1pdm09 strains fluctuated continuously, with the highest peaks observed during the 2009–2010 and 2018 seasons [98].
Using a relaxed molecular clock, concatenated whole genomes of Kenya pH1N1pdm09 strains were estimated to evolve at a mean rate of 4.9 × 10−3 (2.6–7.2) substitutions per site per year (subs/site/year). While their individual genes evolved at different mean rates such as MP at 9.88 × 10−3 (5.58–14.5), H1 at 5.58 × 10−3 (2.75–9.28), NS at 5.22 × 10−3 (1.64–9.17), N1 at 4.07 × 10−3 (1.47–7.73), PB2 at 4.01 × 10−3 (1.47–6.45), PB1 at 3.89 × 10−3 (1.29–7.22), PA at 1.86 × 10−3 (3.03–6.19), and NP at 0.80 × 10−3 (0.4–2.04) subs/site/year [54,55]. Venter et al. applied a strict clock on the HA1 genes of South Africa pH1N1pdm09 strains and estimated a lower mean rate of 0.9 × 10−4 subs/site/year [66]. All South Africa and Kenya H1N1pdm09 strains sampled during the 2009–2010 and 2015–2018 seasons, respectively, exhibited lower rates of nonsynonymous (dN) than synonymous substitutions (dS) with dN/dS ratio of 0.6–0.8 in their HA1 genes, confirming purifying (negative) selection [61,66]. Single likelihood ancestor counting (SLAC) and fixed-effects likelihood (FEL) predicted no codon site under selection at a p-value <0.05 [61].
Gachara et al. [55] did not predict any reassortant among Kenya pH1N1pdm09 strains using the FluGenome web tool [107]. However, H1N1pdm09 strains sampled from 2009 to 2016 across Africa shared similar H1 but different N1 genes, suggesting a different origin of N1 genes [58,60]. Furthermore, two Uganda strains (A/Uganda/856/2014(H1N1) and A/Uganda/1744/2016(H1N1)) had clade 6A-like H1 genes, but clade 6C- and 6B-like N1 genes, respectively, suggesting reassortment [58].
3.5. Molecular Epidemiology of Seasonal H3N2 Viruses in Africa
3.5.1. Genomic Diversity of H3N2 Viruses and Their Relatedness to Vaccines
Africa seasonal H3N2 strains showed an early and continuous evolution from vaccine strains every year. Three South Africa H3N2 strains sampled in 1993 had eight to nine substitutions at three major antigenic sites A, B, and C relative to the season’s A/Beijing/353/1989(H3N2) vaccine strain [68]. South Africa strains sampled in 1994 evolved at four major antigenic sites, A, B, C, and D, and were genetically and antigenically similar to a new A/Guangdong/25/1993(H3N2) strain [68]. Besselaar et al. sampled an A/South Africa/1147/1996(H3N2) strain with an HA1 protein similar to the new A/Nanchang/933/1995(H3N2) vaccine strain [69]. A/Nanchang/933/1995(H3N2)-like strains widely circulated later in 1997 in South Africa, Senegal, and Morocco [69,83,89]. H3N2 strains sampled in February 1998 (early) and later in 1998 had HA1 proteins similar to the different vaccine strains, A/Nanchang/933/1995(H3N2) and new A/Sydney/5/1997(H3N2), respectively [69,83,85]. Hemagglutinin-inhibition confirmed that all and the majority of H3N2 strains sampled in 1997 and 1998, respectively, reacted to antisera raised against A/Nanchang/933/1995(H3N2) and A/Sydney/5/1997(H3N2) strains, respectively, except for A/Jhb/1/1998(H3N2) and A/Jhb/2/1998(H3N2) strains [69,83,85]. Two new viral lineages of Europe-origin (A/Finland/620/1999(H3N2)) and Central America-origin (A/Panama/2007/1999(H3N2)) emerged in South Africa in 1999 [85]. A/Panama/2007/1999-like strains circulated in Senegal and South Africa from 1999 to 2002 [70,89] and caused the 2002 outbreak in Bosobolo (Congo) [82].
The following year (2003), novel A/Fujian/411/2002(H3N2)-like strains with HA1 substitutions A131T and H155T emerged in Senegal and caused an outbreak in a police residential college in Pretoria, South Africa [70,89]. The Pretoria outbreak strains lacked an HA1 Q156H present in strains sampled the same year from Johannesburg and Middleburg (South Africa). Dakar (Senegal) H3N2 strains sampled in 2004 were genetically similar to the 2005 SH season vaccine strain, A/Wellington/1/2004(H3N2) [89]. Four strains sampled in Nairobi (Kenya) between July and September 2006 were antigenically similar to the 2006 SH season vaccine variants, A/California/07/2004(H3N2) and A/New York/55/2004(H3N2) [71]. Kenya H3N2 strains sampled in 2007 had mutated major antigenic sites A (G50E, D122N, S138A, K140I), B (V186G), and I223V relative to the 2007 SH season A/Wisconsin/67/2005(H3N2) vaccine strain but remained antigenically similar to the vaccine [71,80]. A total of 50 Uganda H3N2 strains sampled between October and December 2008 had all 8 genes similar to the 2007–2008 NH A/Wisconsin/67/2005(H3N2) and 2008–2009 SH A/Brisbane/10/2007(H3N2) vaccine strains [75].
New A/Perth/16/2009(H3N2)-like strains emerged and co-circulated with pH1N1pdm09 strains in Uganda and Senegal in 2009 [73,89]. The Uganda H3N2 strains sampled in 2009 differed from the 2010–2012 SH season A/Perth/16/2009(H3N2) vaccine strain at antigenic sites B (N189K, N144S, K158N) and D (T212A) [73]. More mutated antigenic sites E (K62E), C (S45N), B (L157S), A (K144N), and E (G78D) were observed among Kenya strains sampled in the 2010–2011 season relative to the A/Perth/16/2009(H3N2) strain [80,93]. Kenya H3N2 strains sampled in 2013 differed from the 2013 SH season A/Victoria/361/2011(H3N2) vaccine strain, at mutated antigenic sites A (I140R, I140G, N145S), B (T128A, R156H, V186G), C (N278K), D (G78D), Q33R, R142G, E190D, and Y219S [80]. The dominant antigenic sites varied among Kenya strains in 2007 (A: D122N, S138A, K140I), 2008 (A: N144S/K), 2009 (B: K158N and N189K), 2010 (A: N144K), and 2013 (A: T128A, R156H, and V186G), resulting in a vaccine efficacy of 17%, 72.36%, 49.98%, 72.36%, and 17%, respectively.
In 2014, new strains similar to the 2014 SH season A/Texas/50/2012(H3N2) vaccine strain were sampled in Congo [97], South Africa [77], and Southern and Northern Cameroon [78,79]. The A/Texas/50/2012(H3N2) vaccine showed low efficacy in South Africa (VE: −18.4% (−171.5 to −48.4) [77], and the majority of the sampled Congo and South Cameroon strains were also antigenically similar to the A/Switzerland/9715293/2013(H3N2) vaccine strain used later in 2015 [78,97]. A total of 50% (6/12) of Egypt H3N2 strains sampled in October–December 2014 had substitution N225D in H3 and N151D in N2 associated with non-agglutination of avian red blood cells (RBCs) [67]. H3N2 strains sampled in Egypt strains between 2015 and 2017 had several mutated antigenic sites A (T135K, R142G/K/A, N144K/S, N145S), B (L157S, F159Y, K160T), C (D53N/G, N278K), and D (N121D/K/S, N225D), and all had the non-agglutinating substitution N225D [101]. Strains sampled in the same period (2015–2017) from Northern and Southern Cameroon were similar to the 2016–2017 season SNH A/Hong Kong/4801/2014(H3N2) vaccine strain but had different dominant antigenic sites resulting in different efficacy of the A/Switzerland/9715293/2013(H3N2) vaccine in Northern (80.55%) and Southern Cameroon (17%) in 2015 [78,79].
Africa H3N2 strains also evolved from vaccine strains by altering potential N-glycosylation sites (pNGS) in their H3 proteins. Specifically, 100% and 89.5% of Kenya H3N2 strains sampled in the 2010 and 2010–2011 season gained new pNGS K144N and S45N, respectively, absent in the A/Perth/16/2009(H3N2) vaccine strain [93]. Another substitution N144D resulted in a loss of a pNGS among strains sampled in Ghana, Nigeria, and South Africa in 2011, and one A/Johannesburg/27/2011(H3N2) strain gained a pNGS due to S45N [95]. A total of 5 Tunisia H3N2 strains sampled in January–February 2013 had 1 mutated receptor-binding site (S145N) and 10 substitutions at 5 pNGS (45, 124, 128, 144, and 145) relative to the A/Perth/16/2009(H3N2) strain [74]. A total of 60% (6/10) of the pNGS substitutions were among severe and fatal cases [74]. Two strains, A/Tunisia/2494/2013(H3N2)-fatal and A/Tunisia/1987/2013(H3N2)-severe, had an additional mutated receptor-binding site (RBS) A198S [74]. Contrastingly, Nyang’au et al. predicted six HA1 pNGS (8, 22, 63, 133, 165, and 285) as conserved (threshold > 0.5) among 115 Kenya strains sampled from 2007 to 2013 [80]. South Africa and Cameroon H3N2 strains sampled during the 2014–2016 seasons had varying numbers of pNGS (8–11) [77,78,79]. However, the number of pNGS did not affect viral reaction to the A/Texas/50/2012(H3N2) vaccine antisera among South Africa strains sampled in 2014 (p-value = 0.6236) [77]. Cameroon strains sampled in the 2014–2016 seasons had pNGS at H3 positions (8, 22, 38, 45, 63, 128, 133, 160, 165, 246, and 285). Substitutions A128T and K160T created new pNGSs [78,79]. Virus strains sampled in 2015 from Mozambique lost two (L3I, N144S) and gained three (F159Y, K160T, N225D) pNGSs [102], and the Gambia lost one (N144S) and gained one (K160T) pNGSs [96] in their H3 proteins. A total of 94.7% (71/75) of strains sampled in Kilifi (coastal Kenya) between December 2015 and March 2017 gained a new pNGS K160T, but their RBS (98, 136, 153, 183, 190, 194, 195, and 228) remained conserved [81].
All Uganda H3N2 strains sampled during the 2008–2009 season had the classical receptor-binding site motif with 19Y, 136S, 153W, 183H, 195Y, and 225–228 NIPS conserved [73].
South Africa and Cameroon H3N2 strains sampled between 2009 and 2016 also continuously evolved from vaccine strains in their N2 and MP genes [78,104].
3.5.2. Antiviral Drugs Sensitivity among H3N2 Viruses
Six H3N2 strains sampled from Egypt and South Africa during the 2004–2006 seasons lacked the primary adamantane-resistance marker S31N in their M2 proteins [87]. Adamantane-resistance variants (with S31N) emerged in 2008 and continued to circulate in Uganda, Kenya, and Cameroon during the 2008–2009, 2008–2011, and 2014–2016 seasons, respectively [72,73,78,79,105]. H3N2-S31N variants sampled in Kenya, Malaysia, and Singapore between 2008 and 2011 shared HA1 substitutions F193S and I140K [105]. A/KEN/164/2011(H3N2) [105] and A/Cameroon/16v-6529/2016(H3N2) strains [79] had four (L26I, V27S, A30C, and S31D) and one (V27A) secondary M2 adamantane-resistant markers, respectively.
All H3N2 strains sampled in Africa between 2007 and 2016 lacked the oseltamivir-resistance marker H275Y in their N2 proteins [73,78,79,90,104]. Neuraminidase inhibition (NAI) assays confirmed that all strains sampled in Cameroon, Ivory Coast, Madagascar, Niger, Senegal, Congo, and Mozambique during the 2008–2010 and 2014–2015 seasons were sensitive to oseltamivir and zanamivir drugs [88,102].
3.5.3. Phylogenetic Clustering and Circulating Clades among H3N2 Viruses
Phylogenetic trees of Africa H3N2 strains showed the cocirculation of multiple viral lineages, temporal evolution, and mixture with global strains [26,73,83,85,89]. The earliest strains sampled from South Africa in 1994 (A/Johannesburg/33/1994(H3N2), A/Johannesburg/47/1994(H3N2)) and 1997 (A/Johannesburg/10/1997(H3N2)) clustered with Asian-origin A/Beijing/32/1992(H3N2) and A/Wuhan/359/1995(H3N2)-like strains, respectively [26]. Using an automated Graph-incompatibility-based Reassortment Finder (GiRaF) tool, Westgeest et al. predicted the 3 Johannesburg strains had reassorted between H3 and PB1, NP, N2, and MP genes [26]. South Africa strains sampled in late 1997, 1998, 1999, and December 1999–2002 seasons clustered with earlier strains sampled from Asia and Europe(A/Guangdong/8/1996(H3N2), A/Bratislava/6/1997(H3N2)), Australia (A/Sydney/5/1997(H3N2)), Europe (A/Finland/620/1999), and Central America (A/Panama/2007/1999), respectively [70,85].
During the 2003 season, novel strains with HA1 substitution Q156H belonging to the A/Fujian/411/2002(H3N2) clade emerged in South Africa, Senegal, and Madagascar [70,71,89] (Figure 5). The following year (2014), A/Fujian/411/2002(H3N2) clade was replaced by novel A/Wellington/1/2004(H3N2)-like strains in Senegal and South Africa [71,89]. No study reported clades for H3N2 strains sampled in 2005. However, Deyde et al. sampled an A/Egypt/4864/2005(H3N2) strain that had HA1 substitution P227S, similar to an earlier A/Indonesia/4027/2004 strain [87].
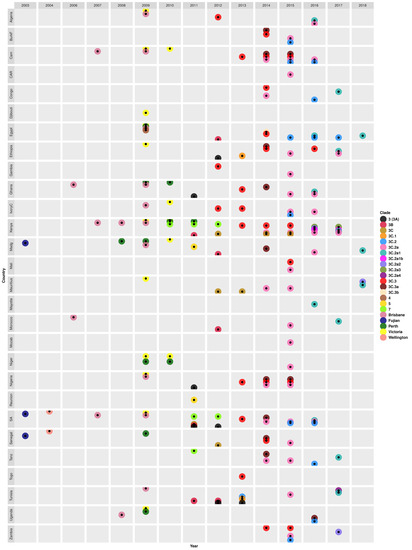
Figure 5.
Temporal and geographical distribution of genetic clades of seasonal H3N2 virus strains that circulated in Africa between 2003 and 2018. Abbreviations: Burkina Faso (BurkF), Cameroon (Cam), Cape Verde (CapeV), Central African Republic (CAR), Ivory Coast (IvoryC), Madagascar (Madg), Mozambique (Mozab), South Africa (SA), Tanzania (Tanz). A/Brisbane/10/2007(H3N2)-like clade (Brisbane), A/Fujian/411/2002(H3N2)-like clade (Fujian), A/Perth/16/2009(H3N2)-like clade (Perth), A/Victoria/208/2009(H3N2-like clade (Victoria), and A/Wellington/1/2004(H3N2)-like clade (Wellington). Details on the characteristic genomic markers (amino acid substitutions in the HA1 proteins) for each clade are described in Table S11.
The H3N2 epidemics in 2006 (Ghana and Morocco) and 2007 (Cameroon, Kenya, and South Africa) were caused by strains belonging to a novel A/Brisbane/10/2007(H3N2)-like clade with HA1 substitution K140I [70,71,80]. A/Brisbane/10/2007(H3N2)-like strains sampled in Kenya and Uganda later in 2008 acquired new HA1 substitutions I140K, V112I, and F193S [73,80,105]. H3N2 strains with HA1 substitution V112I and an additional K158R were also observed in Cameroon and Senegal in May–August 2009 [88]. Novel A/Perth/16/2009(H3N2) clade strains, with HA1 substitution E62K, emerged in Madagascar in 2008 [88].
During the H1N1pdm09 pandemic year (2009), H3N2 strains belonging to three clades A/Brisbane/10/2007(H3N2), A/Perth/16/2009(H3N2), and A/Victoria/208/2009(H3N2) co-circulated in different Africa countries (Figure 5 and Table S11). Specifically, A/Brisbane/10/2007(H3N2)-like strains, with K173Q, were sampled in Ghana, Egypt, Kenya, South Africa, Algeria, Madagascar, Tunisia, Ivory Coast, Nigeria, and Cameroon [73,80]. Furthermore, A/Perth/16/2009(H3N2)-like strains, with more characteristic substitutions (E62K, N144K, K158N, N189K, and I230V) circulated in Madagascar, Niger, Senegal, Uganda, Ghana, and Egypt in 2009 [73,80,88,89,105], Niger, Kenya, and Ghana in 2010 [88,95,105], and in Kenya in 2011 [105]. The novel A/Victoria/208/2009(H3N2)-like strains with HA1 substitutions K158N, N189K, T212A, and N312S, emerged and circulated in different countries across all regions from 2009 through 2010 [73,80,81,88]. The earliest variants of the A/Victoria/208/2009(H3N2) clade to be observed belonged to clades 4 and 7, which emerged in Egypt in 2009 [101] and in Kenya in 2010, respectively [81]. Clade 7 strains with HA1 substitutions S45N were later sampled in South Africa, Tanzania, and Kenya in 2011 and in South Africa and Kenya in 2012 [81,95].
During the early post-pandemic year (2011), new A/Victoria/208/2009(H3N2) clade variants, clades 5, 3 or 3A, 3B, and 3C, emerged in Africa. Novel clade 5 strains with HA1 substitutions D53N, Y94H, I230V, and E280A, similar to the A/Alabama/05/2010(H3N2) strain, were only sampled in 2011 in Madagascar and Reunion [95]. Novel European-origin (A/Stockholm/18/2011(H3N2)-like) clade 3 (same as 3A) with HA1 substitutions N145S, V223I, and N144D, were also first sampled in 2011 in Ghana, Nigeria, and South Africa [95], and later sampled South Africa and Ethiopia in 2012 [92,101] and in Tunisia during the 2012–2013 season [91]. Novel clade 3B strains with HA1 substitutions N312S and A198S were first sampled in 2011 in Kenya, Tunisia, and South Africa, then in Tunisia, Egypt, Morocco, Madagascar in 2012 [80,81,92]. The novel Asian-origin A/Hong Kong/3969/2011(H3N2)-like clade 3C strains with HA1 substitutions S45N, T48I, A198S, and V223I were first sampled in 2011 in South Africa [80,95].
H3N2 virus epidemics that occurred in Africa in 2012 onwards were dominated by clade 3C and its variants (3C.1, 3C.2, 3C.2a, 3C.2a1, 3C.2a1b, 3C.2a2, 3C.2a3, 3C.2a4, 3C.3, 3C.3a, and 3C.3b) (Figure 5). The classical clade 3C strains continued to be sampled during 2012 (in Senegal and Kenya), 2012–2013 (in Mauritius), and 2014–2015 (in Kenya) seasons [80,81,92]. The earliest clade 3C variants belonged to clade 3C.3 and were sampled in Algeria, the Gambia, and Ivory Coast in 2012 [99,101]. Notably, multiple clade 3C variants co-circulated each year from 2013 through 2018. For example, novel clade 3C.1 strains with HA1 substitutions Q33R, N145S, and N278K emerged in Tunisia and Ethiopia in 2013 [80]. In the same year (2013), novel A/Hong Kong/146/2013(H3N2)-like clade 3C.2 strains with N145S and A/Samara/73/2013(H3N2)-like clade 3C.3 strains with T128A and R142G were sampled in Tunisia [91] and different countries (Nigeria, Ivory Coast, South Africa, Ghana, Kenya, Cameroon, and Togo) [80,99,101], respectively. Clades 3C.3 and 3C.2 continued to circulate in different countries during the 2014–2016 [78,97,99] and 2015–2017 [78,101] seasons, respectively.
During the 2014 season, clade 3C.3 diverged into 3C.3a and 3C.3b. Clade 3C.3b strains had HA1 substitutions K83R and R261Q and were sampled in 2014 only in Egypt [97]. While clade 3C.3a strains with HA1 substitutions N225D (some with A138S, F159S, Y94H, or K326R) widely spread in Burkina Faso, Ghana, Senegal, South Africa, Ethiopia, Tanzania, Madagascar, Cameroon, and Nigeria [77,78,79,96,97,99]. Clade 3C.3a strains continued to circulate in Nigeria and Cameroon the following year (2015) and were last observed in 2016 in Uganda [79,96,99].
Novel clade 3C.2a, with HA1 substitutions L3I, K160T, N225D, F159Y, and N128K, dominated the 2014–2017 seasons, circulating in 65% (22/34) of the countries sampled [76,77,79,81,96,97,98,99,102]. However, there were notable differences in the patterns of clade circulation within countries. For example, both clade 3C.2a and 3C.3a strains co-circulated in Northern and Southern Cameroon during the 2014–2015 seasons, but 3C.3a strains emerged in Northern in 2015, later than in Southern Cameroon [78,79]. In addition, subclade 3C.3a circulated in Burkina Faso from May to mid-September 2014, but 3C.2a emerged later in August–October 2015 [99]. Furthermore, H3N2 strains sampled in Cameroon, Tanzania, Ethiopia, and Nigeria between 2014 and 2016 switched between clades 3C.3, 3C.2a, and 3C.3a in their N2 and MP phylogenies suggestive of reassortment [78,79].
Phylogenetic analysis revealed a highly supported (bootstrap = 99%) transmission of novel subclade 3C.2a1 viruses with N171K between two South African provinces. Specifically, a strain sampled in the Western Cape Province A/South Africa/3743/2016(H3N2) was introduced and resulted in an outbreak in a boarding school in the Eastern Cape Province between 13 and 29 July 2016 [76]. In 2016, clade 3C.2a diverged into novel subclades 3C.2a1, 3C.2a1b, 3C.2a2, and 3C.2a3 and all four variants co-circulated in 2017 in different countries. Specifically, clade 3C.2a2 strains with T131K, R142K, and R261Q were only sampled in Mauritius (2018) and Zambia (2017) [79]. A study by Owuor et al. observed both 3C.2a1b and 3C.2a3 strains in Kenya in 2017 [81]. Another novel subclade 3C.2a4 with HA1 substitutions N31S, D53N, N162K, and I192T emerged in Tunisia later in 2017 [79]. Subclades 3C.2a1 strains with HA1 substitutions dominated the 2017 season in Ethiopia, Morocco, Tunisia, Congo, Tanzania, and Kenya, and the 2018 season in Egypt, Madagascar, and Mauritius [79] (Figure 5 and Table S11).
Similar to H1N1pdm09, we observed differences in circulating H3N2 clades between Africa and elsewhere in the world since 2009 (Figure S12).
3.5.4. Viral Population Dynamics, Evolutionary Rates, Selection, and Phylogeographic Patterns among H3N2 Viruses
Bayesian phylogenetics and molecular dating confirmed multiple introductions of new H3N2 strains in Africa from elsewhere in the world. Nyang’au et al. applied a relaxed clock on the HA1 phylogeny of Kenya H3N2 strains sampled between 2007 and 2013 and dated their most recent common ancestor (TMRCA) on September 2001 (95% high probability density (HPD) = September 1998–October 2003) [80]. Molecular dating of concatenated whole genomes of Kenya strains sampled during the 2015–2016 season estimated different TMRCA ages for clades 3C.2a, 3C.2a1b, and 3C.2a2 on 12 January 2013, 26 June 2014, and 2 May 2014, respectively [98]. The HA1 genes of the 2007–2013 season Kenya H3N2 strains evolved at a mean rate of 4.17 × 10−3 (3.09–5.31) subs/site/year and underwent negative selection (dN/dS = 0.56). None of their codons was under selection [80].
Lemey et al. did Bayesian analysis on 1529 global H3N2 virus sequences (including Africa, n = 31) sampled between 2002 and 2007, human morbidity, and air transportation data. Lemey et al. identified South Africa as the only and largest air community connecting Eastern, Western, and Southern Africa [21]. Trunk analysis showed that mainland China and Southeast China contributed 60% and 15% of the strains in the global H3 phylogeny trunk, respectively. Africa did not contribute to the tree trunk [21]. Bayesian ancestral state reconstruction (BSSVs) analysis using concatenated H3N2 whole genomes confirmed highly supported viral migration rates at 0.62–3.34 from East and Southeast Asia to Africa and other continents between January 2010 and December 2013 [98]. Furthermore, H3N2 viral migration within Africa was also highly supported, from Northern to Southern hemispheres (BF ≥ 1000) and within and between geographical regions (10 ≤ BF < 100) of the continent. Dense populations and close locations favored viral migration in Kenya [98].
3.6. Reassortment between H1N1pdm09 and H3N2 Viruses and Zoonotic Exchanges
Four Kenya H3N2 strains, A/Kenya/201/2010(H3N2), A/Kenya/206/2010(H3N2), A/Kenya/209/2010(H3N2), and A/Kenya/214/2010(H3N2), sampled between October and December 2010 were identified as reassortants with A/Perth/16/2009(H3N2)-like H3 and A/California/7/2009(H1N1)-like MP genes [72]. Another study later confirmed three of these strains, A/Kenya/201/2010(H3N2), A/Kenya/206/2010(H3N2), and A/Kenya/209/2010(H3N2), to have reassorted M2 proteins with substitutions I11T, N13S, G16E, V28I, and I54R similar to H1N1pdm09 strains sampled in the 2009–2011 season [105].
There was evidence of swine infection with human IAVs and phylogenetic mixing of avian, swine, and human IAVs in Africa. Phylogenetic analysis 102 virus sequences sampled in Tunisia from humans (H1, H3, N1, and N2) and avian (H9, PB2, NP, and MP) between 2009 and 2013 showed clustering of human (H1, H3) and an avian A/chicken/Tunisia/848/2011(H9N2) strain [100]. Soli et al. used the Recombination Detection Program (RDP) software v.4.beta79 [108] and predicated 12 recombination events (p-value < 10−5) among H3, H1, and H9, with H1 and H9 sequences as major parents in 9 and 2 events, respectively. A total of 67% (8/12) of the events resulted in H3 sequences with recombined sites ≥ 100 base pairs [100].
A total of 14% (31/227) of swine sampled in Nigeria between July 2010 and June 2012 were infected with human H1N1pdm09 strains, and all 31 infections were observed in 2011. The Nigeria swine H1N1pdm09 strains clustered with human H1N1pdm09 virus strains sampled earlier during the 2010–2011 season in Western Africa and globally [57]. A total of 46 human and 4 swine H1N1pdm09 strains sampled during the 2009–2013 and 2010–2013 seasons, respectively, mixed regardless of their country of sampling in their partial H1 (positions 1–138 and 1069–1752 deleted) and N1 (positions 1–598 and 1318–1410 deleted) phylogenies [56,57]. A syndromic survey detected human H1N1pdm09 and H3N2 strains among 9.8% (13/132) and 2.3% (3/132) of Ghanaian swine sampled between 2014 and 2015, respectively. Phylogenetic analysis of the MP genes showed clustering of a human (A/Ghana/KM001/2015(H1N1)) and swine H1N1pdm09 strains (A/swine/Nigeria/IBDVR004/2015(H1N1) and A/swine/Nigeria/IBDVR005/2015) (bootstrap = 68%) [42]. The following season (2016–2017), H1N1pdm09 strains were detected in 1.4% (17/1200) of Ghanaian swine [44]. All swine H1N1pdm09 strains sampled in Nigeria, Ghana, Cameroon, Kenya, Togo, and Egypt between 2014 and 2017 evolved from the human A/California/04/2009(H1N1) vaccine strain and clustered according to the year of sampling in the H1 and MP phylogenies [42,44]. Antigenic analysis found antibodies against human H3N2 and H1N1pdm09 strains in swine and farmers sera, confirming bidirectional viral transfer [44].
4. Discussion
We successfully merged data on the genomic evolution, diversity, transmission, and migration patterns of influenza type-A virus (IAV) strains that previously circulated among African human populations between 1993 and 2018. We found that sequencing and genomic characterization of Africa IAVs have increased over the years. However, the implementation of viral whole-genome sequencing using next-generation sequencing (NGS) technology remains low in Africa.
Sequence comparisons showed that IAV strains sampled in Africa evolved continuously, acquiring unique amino acid substitutions in their surface, transmembrane, and internal proteins relative to vaccine strains recommended for use every year. The substitutions affected all the five major antigenic sites (Sa, Sb, Ca1, Ca2, and Cb) and (A, B, C, D, and E) of the H1 and H3 proteins, respectively. Seasonal H1N1 viruses went extinct in 2009 as a result of host antibodies elicited by the novel pH1N1pdm09 viruses [109]. Both Africa [52,54,55,66] and global pH1N1pdm09 viruses [110,111,112] underwent rapid diversification driven by stochastic substitutions and increased transmission reflected as comb-like phylogenies. Seasonal H1N1pdm09 formed ladder-like phylogenies resulting from immunological pressures due to natural infection or vaccination [98,112,113,114]. Substitutions at antigenic site B (positions 155, 156, 158, 159, 189, and 193) responsible for the antigenic drift and fitness among global 1968–2003 season H3N2 viruses [12,115,116,117,118] were also observed among Africa H3N2 strains [70,73,80,88,89,97,101,102,105]. Both the antigenic drift and annual variation in dominant antigenic sites among Africa IAVs resulted in sub-optimal vaccine protectiveness against circulating strains [78,79,80] hence the need to update vaccine strain compositions every year.
IAVs also evolve from vaccines by altering potential N-glycosylation sites (pNGSs). For example, the earliest Hong Kong H3N2 strain, A/HK/1968(H3N2), had two pNGSs (81 and 165), while strains sampled later gained 10–11 and 8 pNGSs in their H3 and N2 proteins, respectively [15,16,119]. We observed several substitutions resulting in gain (S45N, S124N, A128T, K144N, F159Y, K160T, N225D) and loss (L3I, N144S) of pNGS among Africa H3N2 strains [67,78,79,80,81,93,95,96,97,101], similar to global strains [15]. The HA receptor-binding sites among African IAV strains mostly remained conserved [73,74,81]. Africa pH1N1pdm09 strains had one mutated N1 antigenic site (N248D) [47,48,54,55,56,104] also observed in the USA and reported to alter the antibody recognition site hence affecting the A/California/07/2009(H1N1) vaccine efficacy [120].
Despite the availability of antiviral drugs in 65% (19/31) of African countries as of January 2013, drug use remains very low [121]; hence, drug-resistant variants may have been largely imported into Africa. There was a drastic increase in the global frequency of adamantane-resistant H1N1 (0.3% to 15.6%) and H3N2 (15% to 90.6%) viruses during the 2000–2006 and 2004–2006 seasons, respectively [122]. However, all Africa H1N1 and H3N2 strains sampled during the 2004–2009 and 2004–2006 season, respectively, remained sensitive to adamantanes [41,87,105]. Adamantane-resistant H3N2 strains were introduced in Africa in 2008 [72,73,78,79,105]. The majority of H1N1pm09 and H3N2 strains sampled in Africa and globally between 2009 and 2019 were adamantane-resistant [110,123,124,125]. A new population of adamantane-resistant variants with mutated M2 positions 26, 27, 30, 34, and 38 emerged and circulated in Africa and globally from 2011 through 2016 [122,126,127].
Neuraminidase inhibitors (NAIs), oseltamivir, and zanamivir started to be used between 1999 and 2002 [122]. The first oseltamivir-resistant H1N1-H274Y variants were sampled in France in 2006 (A/Lyon/381/2006(H1N1) [128] and Australia between October 2007 and April 2008 before their introduction in South Africa in 2008 [39,104]. A total of 90% of global H1N1 strains sampled between 2008 and 2009 were oseltamivir-resistant but sensitive to zanamivir [122]. Oseltamivir-resistant H1N1pdm09 strains were sporadically circulated in South Africa [43], globally between 2009 and 2011 [64,95,122], the Middle East and North Africa in 2015 [91], and Myanmar in 2017 [129]. However, there is no sufficient evidence that the resistant H1N1pdm09 variants resulted from oseltamivir treatment. Our analysis shows that the majority of African and global H1N1pdm09 and H3N2 strains sampled during the 2009–2018 season remained sensitive to NAI drugs, making oseltamivir suitable for pre- or post-exposure prophylaxis treatment.
Time-resolved phylogenies showed the cocirculation of multiple IAV lineages and clades each year in Africa as observed globally [130,131]. Africa IAVs phylogenetically mixed with earlier global strains, suggestive of viral introductions into Africa from the rest of the world. Circulating strains in a given year replaced strains in the previous year. However, some H1N1pdm09 lineages persisted through consecutive seasons in the United Kingdom [132], China [113], and Western Africa [60]. Phylogeographical and tree trunk analysis confirmed significant viral migration from South Asia and Europe to Africa (Bayes Factor > 1000) but low migration rates from Africa to elsewhere [21,98,112,133]. Another study by Chan et al. fitted a probabilistic model on the global H1N1 and H3N2 virus HA1 sequences and confirmed Europe and North America as the main importers of IAVs to Africa [86]. Together, the studies confirm Africa as a sink for new IAVs. However, this pattern could be an artifact of inconsistent and insufficient viral sampling in Africa [134].
Molecular dating estimated the ages of TMRCA for Africa and global pH1N1pdm09 strains earlier than their first laboratory detection [55,56,112,135,136]. Following their introduction, IAVs continued to evolve and spread within and between African households, countries, and regions, showing a temporal and geographical evolution structure [47,53,55,60,62,102,103] consistent with previous studies [137]. Global H3N2 and H1N1pdm09 viruses sampled during the 2000–2012 and 2009–2019 season, respectively, had their H3, H1, and N1 genes evolve at mean rate ranges of 4.8–5.2 × 10−3, 4.4–5.34 × 10−3, and 3.8–5.21 × 10−3, respectively [112,113,124,133]. Kenya pH1N1pdm09 virus H1 and N1 and 2007–2013 season H3 genes evolved at comparable mean rates at 5.58 × 10−3 (2.75–9.28) and 4.07 × 10−3 (1.47–7.73) and 4.17 × 10−3 (3.09–5.31) subs/site/year, respectively [54,55,80], confirming a relatively constant viral evolution rate [138]. The surface genes of IAVs evolve more than internal genes because they are subjected to strong immunological selection pressures. All Africa and global 2007–2013 season H3N2 [80] and 2009–2019 season H1N1pdm09 strains [61,66,112,113,139] had their HA genes under negative selection with dN/dS < 1. Except Venter et al. reported dN/dS of 1.6 among HA1 genes of global H1N1pdm09 strains sampled in 2011 [66]. Skyride analysis showed strong oscillations of population sizes of both H1N1pdm09 and H3N2 strains sampled in Africa [54] and globally [113,140,141,142] between 2007 and 2019, indicating that viruses underwent bottlenecks during their evolution and transmission.
Despite the similarity in circulating viral clades, evolutionary rates, and population dynamics, we observed some differences in viral evolution and transmission patterns between Africa and elsewhere. First, the seasonal H1N1 strains went extinct in Africa upon the emergence of pH1N1pdm09 in 2009 but were sporadically sampled elsewhere as of May 2010 [3]. Second, our results show only H1N1 strains belonging to clades 1, 2B, and 2C circulated in Africa between 2001 and 2008. However, additional clades 1, 2A, 2C.1, 2B.1, 2C.2, and 2B.2 circulated in Taiwan between 2005 and 2008 [143]. Third, novel reassortant H1N2 viruses circulated in 2000 in Thailand before they were detected in the USA, Europe, and Africa during the 2001–2003 seasons [89,144,145,146]. Fourth, out of the seven global pandemic H1NIpdm09 genetic clades(1–7) [114], only clades 2, 3, 5, 6, and 7 viruses were pandemic in Africa. Clades 4 and A/Madrid/SO8171/2010-clade strains emerged later in Africa in 2011 [50,51,147]. Fifth, novel H1N1pdm09 clades 8 and 9 circulated only in Africa [50,51,95,97,102,148]. Sixth, H3N2 virus clades 3C.2a and 3C.3a circulated in Switzerland in 2013 before their emergence in Africa in 2014 [77]. Although clades 3C.3a and 3C.2a continued to co-circulate globally through 2018 [15], no 3C.3a-like strain was sampled in Africa after 2016. These differences may be due to the inconsistent and under-sampling of IAVs in Africa. Most studies sampled specific geographical locations, especially urban or cities, and over short periods. In contrast, some countries were un-sampled, creating geographical and temporal sampling biases in our results [134].
Various automated methods based on phylogenetic incongruence, TMRCAs, genetic distances, and coalescent modeling have detected intra- and inter-subtype reassortment among global IAVs [26,113,138,139,149,150,151,152,153]. Global H3N2 strains reassorted more at a rate of 0.3–0.55 events per lineage per year than the H1N1pdm09 (0.1–0.45) [151]. However, reassortment among Africa IAVs has largely been inferred manually based on viral sequences occupying different positions between phylogenies of two or more gene segments [58,60,73,78]. Owuor used the automated reassortment detection tool (GiRaF) and identified no reassortant H3N2 strains in Kilifi (coastal Kenya) between December 2015 and December 2016 [98]. Contrastingly, our ongoing GIRAF analysis of ~1500 Africa virus whole genomes sampled between 1994 and 2020 identified ~573 reassortants, some of which circulated in Kilifi [154]. Such differences could be due to different sampling periods, given that the number of reassortants observed depends on the number of genomes sampled (genomic diversity) [150,154]. Inter-subtype reassortment events observed in Africa between 2009 and 2013 involved human-avian and human-human viral gene exchanges [72,100,105]. However, resulting reassortants could have been less fit to circulate [155]. Africa and global H1N1pdm09 strains sampled in humans and swine mixed phylogenetically indicative of a bidirectional zoonosis and continuous evolution of H1N1pdm09 strains among swine worldwide [42].
5. Conclusions
Phylogenetic and phylogeographic analyses showed that multiple introductions of new strains from outside Africa and extensive local viral transmission sustain the high genomic diversity, continuous antigenic drift, and persistence of influenza viruses among African human populations. Circulating strains had several new amino acid substitutions affecting major antigenic and N-linked glycosylation sites in their hemagglutinin (HA) proteins, which could dramatically affect antibody recognition among vaccinated and exposed unvaccinated populations. The role of reassortment and zoonosis in the evolution and diversification of IAVs in Africa needs to be determined. We observed substitutions and clades and persistent viral lineages unique to Africa. Therefore, Africa’s contribution to the global influenza ecology should not be understated. Thus, there is a need to expand influenza surveillance across Africa and prioritize routine whole-genome sequencing using modern NGS technology and genomic analysis to monitor circulating strains and early detection of emerging ones. Such knowledge could inform public health policies and appropriate vaccine development and selection.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms10050900/s1, Table S1: Terms and definitions; Table S2: Keywords and their corresponding mesh terms included in the search strategy; Table S3: Study article inclusion and exclusion criteria; Table S4: Study quality and risk-of-bias assessment scheme; Table S5: List of all African countries with or without viral sequence data analysed in the included studies; Table S6: Study quality and risk of bias assessment; Table S7: Detailed study sampling bias assessment; Table S8: Circulating genetic clades among Africa H1N1 viruses sampled between 2001 and 2009, Table S9: Circulating genetic clades among Africa H1N1pdm09 viruses between 2009 and 2018; Figure S10: Viral diversification and distribution of genetic clades among H1N1pdm09 viruses that circulated in Africa versus elsewhere during the 2009–2020 seasons; Table S11: Circulating genetic clades among Africa H3N2 viruses between 2004 and 2018; and Figure S12: Viral diversification and distribution of genetic clades among H3N2 viruses that circulated in Africa versus elsewhere during the 2009–2020 seasons.
Author Contributions
Conceptualization, G.N.; methodology, G.N., R.G., J.M.K. and D.P.K.; validation, R.G., J.M.K. and D.P.K.; formal analysis, G.N. and R.G.; data curation, G.N. and R.G.; writing—original draft preparation, G.N.; writing—review and editing, G.N., R.G., J.M.K., D.P.K. and S.D.W.F.; visualization, G.N.; supervision, J.M.K., D.P.K. and S.D.W.F.; funding acquisition, G.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Makerere University-Uganda Virus Research Institute Centre of Excellence for Infection and Immunity Research and Training (MUII). MUII is supported through the DELTAS Africa Initiative (Grant no. 107743). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS), Alliance for Accelerating Excellence in Science in Africa (AESA), and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (Grant no. 107743) and the UK Government.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Special thanks to all African researchers and surveillance programs for availing their data used in the review. The authors also thank the Science for Africa Foundation for paying the article processing charges to ensure this paper is openly accessible.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Taubenberger, J.K.; Morens, D.M. Influenza: The once and future pandemic. Public Health Rep. 2010, 125 (Suppl. 3), 16–26. [Google Scholar] [PubMed]
- McDonald, S.M.; Nelson, M.I.; Turner, P.E.; Patton, J.T. Reassortment in Segmented RNA Viruses: Mechanisms and Outcomes. Nat. Rev. Microbiol. 2016, 14, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [PubMed]
- Zimmer, S.M.; Burke, D.S. Historical Perspective—Emergence of Influenza A (H1N1) Viruses. N. Engl. J. Med. 2009, 361, 279–285. [Google Scholar] [CrossRef]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of Global Seasonal Influenza-Associated Respiratory Mortality: A Modelling Study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53. [Google Scholar]
- Centers for Disease Control and Prevention Types of Influenza Viruses. Available online: https://www.cdc.gov/flu/about/viruses/types.htm (accessed on 28 July 2021).
- Chen, R.; Holmes, E.C. Avian influenza virus exhibits rapid evolutionary dynamics. Mol. Biol. Evol. 2006, 23, 2336–2341. [Google Scholar]
- Boni, M.F.; Zhou, Y.; Taubenberger, J.K.; Holmes, E.C. Homologous Recombination Is Very Rare or Absent in Human Influenza A Virus. J. Virol. 2008, 82, 4807–4811. [Google Scholar] [CrossRef][Green Version]
- Caton, A.J.; Brownlee, G.G.; Yewdell, J.W.; Gerhard, W. The antigenic structure of the influenza virus A/PR/8/ 34 hemagglutinin (H1 subtype). Cell 1982, 31, 417–427. [Google Scholar]
- Lee, M.S.; Chen, J.S. Predicting Antigenic Variants of Influenza A/H3N2 Viruses. Emerg. Infect. Dis. 2004, 10, 1385. [Google Scholar] [CrossRef]
- Wiley, D.C.; Wilson, I.A.; Skehel, J.J. Structural Identification of the Antibody-Binding Sites of Hong Kong Influenza Haemagglutinin and Their Involvement in Antigenic Variation. Nature 1981, 289, 6162101. [Google Scholar]
- Air, G.M.; Laver, W.G. The Neuraminidase of Influenza Virus. Proteins 1989, 6, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Colman, P.M.; Varghese, J.N.; Laver, W.G. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 1983, 303, 41–44. [Google Scholar] [PubMed]
- Allen, J.D.; Ross, T.M. H3N2 Influenza Viruses in Humans: Viral Mechanisms, Evolution, and Evaluation. Hum. Vaccines Immunother. 2018, 14, 1840–1847. [Google Scholar] [CrossRef]
- Skehel, J.J.; Wiley, D.C. Receptor Binding and Membrane Fusion in Virus Entry: The Influenza Hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef]
- Belser, J.A.; Maines, T.R.; Tumpey, T.M.; Katz, J.M. Influenza A Virus Transmission: Contributing Factors and Clinical Implications. Expert Rev. Mol. Med. 2010, 12, e39. [Google Scholar] [CrossRef]
- Lyons, D.M.; Lauring, A.S. Mutation and Epistasis in Influenza Virus Evolution. Viruses 2018, 10, 407. [Google Scholar] [CrossRef]
- Tscherne, D.M.; Garcia-Sastre, A. Virulence determinants of pandemic influenza viruses. J. Clin. Investig. 2011, 121, 6–13. [Google Scholar]
- Lemey, P.; Suchard, M.; Rambaut, A. Reconstructing the Initial Global Spread of a Human Influenza Pandemic: A Bayesian Spatial-Temporal Model for the Global Spread of H1N1pdm. PLoS Curr. 2009, 1, RRN1031. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.; Bielejec, F.; Baele, G. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Pathog. 2014, 10, e1002947. [Google Scholar] [CrossRef]
- Pond, S.; Frost, S.; Muse, S. HyPhy: Hypothesis Testing Using Phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Bromham, L. Estimating Divergence Dates from Molecular Sequences. Mol. Biol. Evol. 1998, 15, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J. Mapping the Antigenic and Genetic Evolution of Influenza Virus. Science 2004, 305, 371–376. [Google Scholar] [PubMed]
- Volz, E.M.; Koelle, K.; Bedford, T. Viral Phylodynamics. PLoS Comput. Biol. 2013, 9, e1002947. [Google Scholar] [CrossRef]
- Westgeest, K.B.; Russell, C.A.; Lin, X.; Spronken, M.I.J.; Bestebroer, T.M.; Bahl, J.; van Beek, R.; Skepner, E.; Halpin, R.A.; de Jong, J.C.; et al. Genomewide Analysis of Reassortment and Evolution of Human Influenza A(H3N2) Viruses Circulating between 1968 and 2011. J. Virol. 2014, 88, 2844–2857. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control Influenza Virus Characterisation Reports, Summary Europe. Available online: https://www.ecdc.europa.eu/en/seasonal-influenza/surveillance-and-disease-data/influenza-virus-characterisation (accessed on 23 March 2022).
- Green, A. Progress in Influenza Surveillance in Africa. Lancet 2018, 391, 1345–1346. [Google Scholar] [CrossRef]
- Igboh, L.S.; McMorrow, M.; Tempia, S.; Emukule, G.O.; Talla Nzussouo, N.; McCarron, M.; Williams, T.; Weatherspoon, V.; Moen, A.; Fawzi, D.; et al. Influenza Surveillance Capacity Improvements in Africa during 2011–2017. Influenza Other Respir. Viruses 2021, 15, 495–505. [Google Scholar] [CrossRef]
- Radin, J.M.; Katz, M.A.; Tempia, S.; Talla Nzussouo, N.; Davis, R.; Duque, J.; Adedeji, A.; Adjabeng, M.J.; Ampofo, W.K.; Ayele, W.; et al. Influenza Surveillance in 15 Countries in Africa, 2006–2010. J. Infect. Dis. 2012, 206 (Suppl. 1), S14–S21. [Google Scholar] [CrossRef]
- Han, A.X.; Parker, E.; Scholer, F.; Maurer-Stroh, S.; Russell, C.A. Phylogenetic Clustering by Linear Integer Programming (PhyCLIP). Mol. Biol. Evol. 2019, 36, 1580–1595. [Google Scholar] [CrossRef]
- Hassan, A.S.; Pybus, O.G.; Sanders, E.J.; Albert, J.; Esbjörnsson, J. Defining HIV-1 Transmission Clusters Based on Sequence Data. AIDS 2017, 31, 1211–1222. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA Statement for Reporting Systematic Reviews and Meta-Analyses of Studies That Evaluate Health Care Interventions: Explanation and Elaboration. PLoS Med. 2009, 6, e1000100. [Google Scholar] [CrossRef]
- Modesti, P.A.; Reboldi, G.; Cappuccio, F.P.; Agyemang, C.; Remuzzi, G.; Rapi, S.; Perruolo, E.; Parati, G. ESH Working Group on CV Risk in Low Resource Settings Panethnic Differences in Blood Pressure in Europe: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0147601. [Google Scholar] [CrossRef]
- Field, N.; Cohen, T.; Struelens, M.J.; Palm, D.; Cookson, B.; Glynn, J.R.; Gallo, V.; Ramsay, M.; Sonnenberg, P.; Maccannell, D.; et al. Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases (STROME-ID): An Extension of the STROBE Statement. Lancet Infect. Dis. 2014, 14, 341–352. [Google Scholar] [CrossRef]
- Besselaar, T.G.; Naidoo, D.; Buys, A.; Gregory, V.; McAnerney, J.; Manamela, J.M.; Blumberg, L.; Schoub, B.D. Widespread Oseltamivir Resistance in Influenza A Viruses (H1N1), South Africa. Emerg. Infect. Dis. 2008, 14, 1809–1810. [Google Scholar]
- Bulimo, W.D.; Achilla, R.A.; Majanja, J.; Mukunzi, S.; Wadegu, M.; Osunna, F.; Mwangi, J.; Njiri, J.; Wangui, J.; Nyambura, J.; et al. Molecular Characterization and Phylogenetic Analysis of the Hemagglutinin 1 Protein of Human Influenza A Virus Subtype H1N1 Circulating in Kenya During 2007–2008. J. Infect. Dis. 2012, 206, S46–S52. [Google Scholar] [CrossRef][Green Version]
- Hurt, A.C.; Ernest, J.; Deng, Y.M.; Iannello, P.; Besselaar, T.G.; Birch, C.; Buchy, P.; Chittaganpitch, M.; Chiu, S.C.; Dwyer, D.; et al. Emergence and spread of oseltamivir-resistant A(H1N1) influenza viruses in Oceania, South East Asia and South Africa. Antivir. Res. 2009, 83, 90–93. [Google Scholar]
- Njouom, R.; Mba, S.A.; Noah, D.N.; Gregory, V.; Collins, P.; Cappy, P. Circulation of human influenza viruses and emergence of Oseltamivir-resistant A(H1N1) viruses in Cameroon, Central Africa. BMC Infect. Dis. 2010, 10, 56. [Google Scholar] [CrossRef]
- Dia, N.; Niang, M.N.; Diadhiou, S.A.; Goudiaby, D.G.; Faye, A.; Kiori, D.; Bâ, M.; Michel, R.; Diop, O.M. Spread of Influenza A(H1N1) Oseltamivir-Resistant Viruses in Africa in 2008 Confirmed by Multiple Introductions in Senegal. BMC Infect. Dis. 2013, 13, 106. [Google Scholar] [CrossRef]
- Adeola, O.A.; Olugasa, B.O.; Emikpe, B.O.; Folitse, R.D. Syndromic Survey and Molecular Analysis of Influenza Viruses at the Human–Swine Interface in Two West African Cosmopolitan Cities Suggest the Possibility of Bidirectional Interspecies Transmission. Zoonoses Public Health 2019, 66, 232–247. [Google Scholar] [CrossRef]
- Aspinall, S.; Pretorius, B.; Vally, T.; Sebopa, B.; Turner-Babb, C. An Observational Study of Infuenza A(H1N1)Pdm09 Viral Shedding and Resistance under Standard-Duration Oseltamivir Treatment. South. Afr. J. Epidemiol. Infect. 2013, 28, 122–125. [Google Scholar] [CrossRef]
- Ayim-Akonor, M.; Mertens, E.; May, J.; Harder, T. Exposure of Domestic Swine to Influenza A Viruses in Ghana Suggests Unidirectional, Reverse Zoonotic Transmission at the Human–Animal Interface. Zoonoses Public Health 2020, 67, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Ben Hamed, S.; Elargoubi, A.; Harrabi, M.; Srihi, H.; Souiai, O.; Mastouri, M.; Almalki, M.A.; Gharbi, J.; Ben M’hadheb, M. Phylogenetic Analysis of the Neuraminidase Segment Gene of Influenza A/H1N1 Strains Isolated from Monastir Region (Tunisia) during the 2017–2018 Outbreak. Biologia 2021, 76, 1797–1806. [Google Scholar] [CrossRef]
- Bonney, J.H.; Kronmann, K.C.; Lindan, C.P. Virological Surveillance of Influenza-like Illness among Children in Ghana, 2008–2010. J. Infect. Dis. 2012, 206, S108–S113. [Google Scholar]
- Byarugaba, D.K.; Erima, B.; Millard, M.; Kibuuka, H.; Lkwago, L.; Bwogi, J.; Mimbe, D.; Kiconco, J.B.; Tugume, T.; Mworozi, E.A.; et al. Whole-Genome Analysis of Influenza A(H1N1)Pdm09 Viruses Isolated in Uganda from 2009 to 2011. Influenza Other Respir. Viruses 2016, 10, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Dia, N.; Ndiaye, M.N.; de Lourdes Monteiro, M.; Koivogui, L.; Bara, M.O.; Diop, O.M. A Subregional Analysis of Epidemiologic and Genetic Characteristics of Influenza A(H1N1)Pdm09 in Africa: Senegal, Cape Verde, Mauritania, and Guinea, 2009–2010. Am. J. Trop. Med. Hyg. 2013, 88, 946. [Google Scholar] [CrossRef]
- El Moussi, A.; Kacem, M.A.B.H.; Pozo, F.; Ledesma, J.; Cuevas, M.T.; Casas, I.; Slim, A. Frequency of D222G Haemagglutinin Mutant of Pandemic (H1N1) Pdm09 Influenza Virus in Tunisia between 2009 and 2011. Diagn. Pathol. 2013, 8, 124. [Google Scholar]
- El Moussi, A.; Ben Hadj Kacem, M.A.; Pozo, F.; Ledesma, J.; Cuevas, M.T.; Casas, I.; Slim, A. Genetic Diversity of HA1 Domain of Heammaglutinin Gene of Influenza A(H1N1)Pdm09 in Tunisia. Virol. J. 2013, 10, 150. [Google Scholar] [CrossRef]
- El Moussi, A.; Pozo, F.; Kacem, M.A.B.H.; Ledesma, J.; Cuevas, M.T.; Casas, I.; Slim, A. Virological Surveillance of Influenza Viruses during the 2008–09, 2009–2010 and 2010–11 Seasons in Tunisia. PLoS ONE 2013, 8, e74064. [Google Scholar]
- El Rhaffouli, H.; Fahime, E.; Laraqui, A.; Bajjou, T.; Melloul, M.; Obeid, S.; Fathallah, L.; Lahlou Amine, I. Evolution of the Hemagglutinin Gene of the Influenza A(H1N1)Pdm09 Virus in Morocco during Two Influenza Seasons 2009–2011. Curr. Microbiol. 2013, 68, 372–380. [Google Scholar] [CrossRef]
- Gachara, G.; Symekher, S.; Mbithi, J.N.; Simwa, J.; Ng’ayo, M.; Magana, J.; Bulimo, W.D. Amino Acid Sequence Analysis and Identification of Mutations in the NS Gene of 2009 Influenza A (H1N1) Isolates from Kenya. Virus Genes 2011, 43, 27–32. [Google Scholar]
- Gachara, G.M. Molecular Epidemiology and Evolution of Influenza A(H1N1) Pdm09 Virus in Kenya. Master’s Thesis, Kenyatta University, Nairobi, Kenya, 2014. Available online: https://ir-library.ku.ac.ke (accessed on 21 March 2022).
- Gachara, G.; Symekher, S.; Otieno, M.; Magana, J.; Opot, B.; Bulimo, W. Whole Genome Characterization of Human Influenza A(H1N1)Pdm09 Viruses Isolated from Kenya during the 2009 Pandemic. Infect. Genet. Evol. 2016, 40, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Meseko, C.A.; Odurinde, O.O.; Olaniran, B.O.; Heidari, A.; Oluwayelu, D.O. Pandemic Influenza A/H1N1 Virus Incursion into Africa: Countries, Hosts and Phylogenetic Analysis. Niger. Vet. J. 2015, 36, 1251–1261. [Google Scholar]
- Meseko, C.A.; Heidari, A.; Odaibo, G.N.; Olaleye, D.O. Complete Genome Sequencing of H1N1pdm09 Swine Influenza Isolates from Nigeria Reveals Likely Reverse Zoonotic Transmission at the Human-Animal Interface in Intensive Piggery. Infect. Ecol. Epidemiol. 2019, 9, 1696632. [Google Scholar] [CrossRef] [PubMed]
- Monamele, C.G.; Njifon, H.L.M.; Vernet, M.-A.; Njankouo, M.R.; Kenmoe, S.; Yahaya, A.A. Molecular Characterization of Influenza A (H1N1) Pdm09 in Cameroon during the 2014–2016 Influenza Seasons. PLoS ONE 2019, 14, e0210119. [Google Scholar] [CrossRef]
- Nakouné, E.; Tricou, V.; Manirakiza, A.; Komoyo, F.; Selekon, B.; Gody, J.C.; Victoir, K.; Buchy, P.; Kazanji, M. First Introduction of Pandemic Influenza A/H1N1 and Detection of Respiratory Viruses in Pediatric Patients in Central African Republic. Virol. J. 2013, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Njouom, R.; Viboud, C.; Niang, M.N.; Kadjo, H.; Ampofo, W. Multiyear Persistence of 2 Pandemic A/H1N1 Influenza Virus Lineages in West Africa. J. Infect. Dis. 2014, 210, 121–125. [Google Scholar] [CrossRef][Green Version]
- Opanda, S.; Bulimo, W.; Gachara, G.; Ekuttan, C.; Amukoye, E. Assessing Antigenic Drift and Phylogeny of Influenza A (H1N1) Pdm09 Virus in Kenya Using HA1 Sub-Unit of the Hemagglutinin Gene. PLoS ONE 2020, 15, e0228029. [Google Scholar] [CrossRef]
- Orelle, A.; Razanajatovo, N.H.; Rajatonirina, S.; Hoffmann, J.; Randrianasolo, L.; Razafitrimo, G.M.; Naidoo, D.; Richard, V.; Heraud, J.-M. Epidemiological and Virological Characterization of 2009 Pandemic Influenza A Virus Subtype H1N1 in Madagascar. J. Infect. Dis. 2012, 206, S140–S147. [Google Scholar] [CrossRef]
- Pascalis, H.; Temmam, S.; DA, W. Molecular Evolutionary Analysis of PH1N1 2009 Influenza Virus in Reunion Island, South West Indian Ocean Region: A Cohort Study. PLoS ONE 2012, 7, e43742. [Google Scholar]
- Quiliano, M.; Valdivia-Olarte, H.; Olivares, C.; Requena, D.; Gutiérrez, A.H.; Reyes-Loyola, P.; Tolentino-Lopez, L.E.; Sheen, P.; Briz, V.; Muñoz-Fernández, M.A.; et al. Molecular Distribution of Amino Acid Substitutions on Neuraminidase from the 2009 (H1N1) Human Influenza Pandemic Virus. Bioinformation 2013, 9, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Valley-Omar, Z.; Nindo, F.; Mudau, M.; Hsiao, M.; Martin, D.P. Phylogenetic Exploration of Nosocomial Transmission Chains of 2009 Influenza A/H1N1 among Children Admitted at Red Cross War Memorial Children’s Hospital, Cape Town, South Africa in 2011. PLoS ONE 2015, 10, e0141744. [Google Scholar] [CrossRef]
- Venter, M.; Naidoo, D.; Pretorius, M.; Buys, A.; McAnerney, J.; Blumberg, L.; Madhi, S.A.; Cohen, C.; Schoub, B. Evolutionary Dynamics of 2009 Pandemic Influenza A Virus Subtype H1N1 in South Africa during 2009–2010. J. Infect. Dis. 2012, 206 (Suppl. 1), 166–172. [Google Scholar]
- Aboualy, M.; Mohareb, E.; Fahim, M.; Roshdy, W.; Younan, M.; Said, M.; Imam, M.; Abdelsalam, E.T. Identification of Non-Haemagglutinating Influenza A/H3 Virus and Characterization of Haemagglutinin (HA) and Neuraminidase (NA) Strain Mutations in Influenza-Like-Illness Cases in Egypt on MDCK Cell Line. Egypt. Acad. J. Biol. Sci. G. Microbiol. 2018, 10, 1–8. [Google Scholar] [CrossRef]
- Besselaar, T.G.; Blackburn, N.K.; Schoub, B.D. The Molecular Characterization of Influenza Virus Strains Isolated in South Africa during 1993 and 1994. Res. Virol. 1996, 147, 239–245. [Google Scholar]
- Besselaar, T.G.; Schoub, B.D.; Blackburn, N.K. Impact of the Introduction of A/Sydney/5/97 H3N2 Influenza Virus into South Africa. J. Med. Virol. 1999, 59, 561–568. [Google Scholar] [CrossRef]
- Besselaar, T.G.; Botha, L.; McAnerney, J.M.; Schoub, B.D. Antigenic and Molecular Analysis of Influenza A (H3N2) Virus Strains Isolated from a Localised Influenza Outbreak in South Africa in 2003. J. Med. Virol. 2004, 73, 71–78. [Google Scholar]
- Bulimo, W.D.; Garner, J.L.; Schnabel, D.C.; Bedno, S.A.; Njenga, M.K.; Ochieng, W.O.; Amukoye, E.; Magana, J.M.; Simwa, J.M.; Ofula, V.O.; et al. Genetic Analysis of H3N2 Influenza A Viruses Isolated in 2006–2007 in Nairobi, Kenya. Influenza Other Respir. Viruses 2008, 2, 107–113. [Google Scholar]
- Bulimo, W.D.; Gachara, G.; Opot, B.H.; MW, M.; Wurapa, E.K. Evidence in Kenya of Reassortment Between Seasonal Influenza A(H3N2) and Influenza A(H1N1)Pdm09 to Yield A(H3N2) Variants With the Matrix Gene Segment of A(H1N1)Pdm09. Afr. J. Pharmacol. Ther 2012, 1, 1–7. [Google Scholar]
- Byarugaba, D.K.; Ducatez, M.F.; Erima, B.; Mworozi, E.A.; Millard, M.; Kibuuka, H.; Lukwago, L.; Bwogi, J.; Kaira, B.B.; Mimbe, D.; et al. Molecular Epidemiology of Influenza A/H3N2 Viruses Circulating in Uganda. PLoS ONE 2011, 6, e27803. [Google Scholar] [CrossRef]
- El Moussi, A.; Kacem, M.A.B.H.; Slim, A. Loss and Gain of N-Linked Glycosylation Sites in Globular Head and Stem of HA Found in A/H3N2 Flu Fatal and Severe Cases during 2013 Tunisia Flu Seasonal Survey. Virus Genes 2014, 48, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Kaira, B.B. Detection and Characterization of Human Influenza a Virus Isolates from Patients Attending Kayunga and Mulago Hospitals in Uganda. Master’s Thesis, Makerere University, Kampala, Uganda, 2011. Available online: http://makir.mak.ac.ug (accessed on 21 March 2022).
- Kleynhans, J.; Treurnicht, F.K.; Cohen, C.; Vedan, T.; Seleka, M.; Maki, L.; von Gottberg, A.; McCarthy, K.; Ramkrishna, W.; McMorrow, M. Outbreak of Influenza A in a Boarding School in South Africa, 2016. Pan Afr. Med. J. 2019, 33. [Google Scholar]
- McAnerney, J.M.; Treurnicht, F.; Walaza, S.; Cohen, A.L.; Tempia, S.; Mtshali, S. Evaluation of Influenza Vaccine Effectiveness and Description of Circulating Strains in Outpatient Settings in South Africa, 2014. Influenza Other Respir. Viruses 2015, 9. [Google Scholar] [CrossRef]
- Monamele, G.C.; Torre, J.C.; Vernet, M.-A. Genetic and Antigenic Characterization of Influenza A(H3N2) in Cameroon during the 2014–2016 Influenza Seasons. PLoS ONE 2017, 12, 0184411. [Google Scholar]
- Njifon, H.L.M.; Monamele, C.G.; Vernet, M.-A.; Njankouo, M.R.; Deweerdt, L.; Nono, R.; Kenmoe, S.; Mbacham, W.; Njouom, R. Genetic Diversity of Influenza A(H3N2) Viruses in Northern Cameroon during the 2014–2016 Influenza Seasons. J. Med. Virol. 2019, 91, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Nyang’au, E.M.; Bulimo, W.D.; Mobegi, V.; Opanda, S.; Magiri, E. Genetic Analysis of HA1 Domain of Influenza A/H3N2 Viruses Isolated in Kenya During the 2007 to 2013 Seasons Reveal Significant Divergence from WHO-Recommended Vaccine Strains. Int. J. Infect. Dis. 2020, 95, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Owuor, D.C.; Ngoi, J.M.; Otieno, J.R.; Otieno, G.P.; Nyasimi, F.M.; Nyiro, J.U.; Agoti, C.N.; Chaves, S.S.; Nokes, D.J. Genetic Characterization of Influenza A(H3N2) Viruses Circulating in Coastal Kenya, 2009–2017. Influenza Other Respir. Viruses 2020. [Google Scholar] [CrossRef]
- WHO. Influenza Outbreak in the District of Bosobolo, Democratic Republic of the Congo, November—December 2002: Epidemiological Report = Flambée de Grippe Dans Le District de Bosobolo, République Démocratique Du Congo, Novembre-Décembre 2002: Epidémiologie. Wkly. Epidemiol. Rec. 2003, 78, 94–96. [Google Scholar]
- Barakat, A.; Benjouad, A.; Manuguerra, J.C.; El Aouad, R.; Werf, S. Virological Surveillance in Africa Can Contribute to Early Detection of New Genetic and Antigenic Lineages of Influenza Viruses. J. Infect. Dev. Ctries. 2011, 5, 270–277. [Google Scholar]
- Barr, I.G.; McCauley, J.; Cox, N.; Daniels, R.; Engelhardt, O.G.; Fukuda, K.; Grohmann, G.; Hay, A.; Kelso, A.; Klimov, A.; et al. Epidemiological, Antigenic and Genetic Characteristics of Seasonal Influenza A(H1N1), A(H3N2) and B Influenza Viruses: Basis for the WHO Recommendation on the Composition of Influenza Vaccines for Use in the 2009–2010 Northern Hemisphere Season. Vaccine 2010, 28, 1156–1167. [Google Scholar]
- Besselaar, T.G.; Schoub, B.D.; McAnerney, J.M. Phylogenetic Studies of South African Influenza A Viruses: 1997–1999. In Options for the Control of Influenza IV; Osterhaus, A., Cox, N., Hampson, A., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2001; pp. 139–145. [Google Scholar]
- Chan, J.; Holmes, A.; Rabadan, R. Network Analysis of Global Influenza Spread. PLoS Comput. Biol. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Deyde, V.M.; Xu, X.; Bright, R.A.; Shaw, M.; Smith, C.B.; Zhang, Y.; Shu, Y.; Gubareva, L.V.; NJ, C.; Klimov, A.I. Surveillance of Resistance to Adamantanes among Influenza A (H3N2) and A (H1N1) Viruses Isolated Worldwide. Am. J. Infect. Dis 2007, 196, 249–257. [Google Scholar]
- Heraud, J.M.; Njouom, R.; Rousset, D. Spatiotemporal Circulation of Influenza Viruses in 5 African Countries during 2008–2009: A Collaborative Study of the Institut Pasteur International Network. J. Infect. Dis 2012, 206, S5–S13. [Google Scholar] [PubMed]
- Niang, M.N.; Dosseh, A.; Ndiaye, K.; Sagna, M.; Gregory, V.; Goudiaby, D.; Hay, A.; Diop, O.M. Sentinel Surveillance for Influenza in Senegal, 1996–2009. J. Infect. Dis. 2012, 206 (Suppl. 1), S129–S135. [Google Scholar] [CrossRef] [PubMed]
- Ait-Aissa, A.; Derrar, F.; Hanoun, D.; Gradi, E.-A.; Scaravelli, D.; Bouslama, Z. Surveillance for Antiviral Resistance among Influenza Viruses Circulating in Algeria during Five Consecutive Influenza Seasons (2009 to 2014). J. Med. Virol. 2018, 90, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Al Khatib, H.A.; Al Thani, A.A.; Gallouzi, I.; Yassine, H.M. Epidemiological and Genetic Characterization of PH1N1 and H3N2 Influenza Viruses Circulated in MENA Region during 2009–2017. BMC Infect. Dis. 2019, 19, 314. [Google Scholar] [CrossRef]
- Barr, I.G.; Russell, C.; Besselaar, T.G.; Cox, N.J.; Daniels, R.S.; Donis, R.; Engelhardt, O.G.; Grohmann, G.; Itamura, S.; Kelso, A.; et al. WHO Recommendations for the Viruses Used in the 2013–2014 Northern Hemisphere Influenza Vaccine: Epidemiology, Antigenic and Genetic Characteristics of Influenza A(H1N1)Pdm09, A(H3N2) and B Influenza Viruses Collected from October 2012 to January 2013. Vaccine 2014, 32, 4713–4725. [Google Scholar] [CrossRef]
- Bulimo, W.D.; Mukunzi, S.; Achilla, R.; Opot, B.; Osuna, F.; Majanja, J. Were the WHO-Recommended Human Influenza Vaccine Formulations Appropriate for Kenya During the 2010–2011 Season? Inferences from the HA1 Gene Analysis. Afr. J. Pharmacol. Ther. 2012, 1, 46–54. [Google Scholar]
- Kavunga-Membo, H.; Nkwembe, E.; Simulundu, E.; Karhemere, S.; Babakazo, P.; Manya, L.; Kabamba, J.; Okitolonda, E.; Ahuka-Mundeke, S.; Muyembe, J.J. Epidemiology of Circulating Human Influenza Viruses from the Democratic Republic of Congo, 2015. PLoS ONE 2018, 13, e0203995. [Google Scholar] [CrossRef]
- Klimov, A.I.; Garten, R.; Russell, C.; Barr, I.G.; Besselaar, T.G.; Daniels, R.; Engelhardt, O.G.; Grohmann, G.; Itamura, S.; Kelso, A.; et al. WHO Recommendations for the Viruses to Be Used in the 2012 Southern Hemisphere Influenza Vaccine: Epidemiology, Antigenic and Genetic Characteristics of Influenza A(H1N1)Pdm09, A(H3N2) and B Influenza Viruses Collected from February to September 2011. Vaccine 2012, 30, 6461–6471. [Google Scholar] [CrossRef]
- Mackenzie, G.A.; Vilane, A.; Salaudeen, R.; Hogerwerf, L.; van den Brink, S.; Wijsman, L.A.; Overduin, P.; Janssens, T.K.; de Silva, T.I.; van der Sande, M.A. Respiratory Syncytial, Parainfluenza and Influenza Virus Infection in Young Children with Acute Lower Respiratory Infection in Rural Gambia. Sci. Rep. 2019, 9, 17965. [Google Scholar] [PubMed]
- Nkwembe, E.; Cintron, R.; Sessions, W.; Kavunga, H.; Babakazo, P.; Manya, L.; Muyembe, J.J. Molecular Analysis of Influenza A(H3N2) and A(H1N1)Pdm09 Viruses Circulating in the Democratic Republic of Congo, 2014. J. Harmon. Res. Med. Health Sci. 2016, 3, 247–264. [Google Scholar] [PubMed]
- Owuor, D.C. Sequence Diversity, Evolution and Transmission of Influenza A(H1N1)Pdm09 and A(H3N2) Viruses in Kenya, 2009–2018. Ph.D. Thesis, The Open University, Milton Keynes, UK, 2021. Available online: https://doi.org/10.21954/ou.ro.000126d0 (accessed on 21 March 2022).
- Sanou, A.M.; Wandaogo, S.C.M.; Poda, A.; Tamini, L.; Kyere, A.E.; Sagna, T. Epidemiology and Molecular Characterization of Influenza Viruses in Burkina Faso, Sub-Saharan Africa. Influenza Other Respir. Viruses 2018, 12, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Soli, R.; Kaabi, B.; Barhoumi, M.; Maktouf, C.; Ahmed, S.B.-H. Bayesian Phylogenetic Analysis of the Influenza-A Virus Genomes Isolated in Tunisia, and Determination of Potential Recombination Events. Mol. Phylogenetics Evol. 2019, 134, 253–268. [Google Scholar] [CrossRef]
- Soliman, M.S.; El-Kholy, A.A.; Kamel, M.M.; Alorabi, J.A.; Mohamed, N.M.; Abdel-Moneim, A.S. Characterization and Mutational Analysis of Haemagglutinin and Neuraminidase of H3N2 and H1N1pdm09 Human Influenza A Viruses in Egypt. Virus Dis. 2020, 31, 262–269. [Google Scholar] [CrossRef]
- Tivane, A.; Daniels, R.; Nguenha, N. Antigenic and Genetic Characterization of Influenza Viruses Isolated in Mozambique during the 2015 Season. PLoS ONE 2018, 13, e0201248. [Google Scholar]
- Valley-Omar, Z.; Iyengar, P.; von Mollendorf, C.; Tempia, S.; Moerdyk, A.; Hellferscee, O.; Martinson, N.; McMorrow, M.; Variava, E.; Masonoke, K.; et al. Intra-Host and Intra-Household Diversity of Influenza A Viruses during Household Transmissions in the 2013 Season in 2 Peri-Urban Communities of South Africa. PLoS ONE 2018, 13, e0198101. [Google Scholar] [CrossRef]
- Treurnicht, F.K.; Buys, A.; Tempia, S.; Seleka, M.; Cohen, A.L.; Walaza, S.; Glass, A.J.; Rossouw, I.; McAnerney, J.; Blumberg, L.; et al. Replacement of Neuraminidase Inhibitor-Susceptible Influenza A(H1N1) with Resistant Phenotype in 2008 and Circulation of Susceptible Influenza A and B Viruses during 2009–2013, South Africa. Influenza Other Respir. Viruses 2019, 13, 54–63. [Google Scholar] [CrossRef]
- Wadegu, M.; Wamunyokoli, F.; Osanjo, G.; Opanda, S.; Majanja, J.; Coldren, R.; Bulimo, W. Molecular Surveillance of Adamantane Resistance among Human Influenza A Viruses Isolated in Four Epidemic Seasons in Kenya. Afr. J. Pharmacol. Ther. 2016, 5, 181–192. [Google Scholar]
- World Health Organization. World Health Organization: Preliminary review of D222G amino acid substitution in the haemagglutinin of pandemic influenza A(H1N1) 2009 viruses. Wkly. Epidemiol. Rec. 2010, 16, 23. [Google Scholar]
- Lu, G.; Rowley, T.; Garten, R.; Donis, R.O. FluGenome: A Web Tool for Genotyping Influenza A Virus. Nucleic Acids Res. 2007, 35, W275–W279. [Google Scholar] [CrossRef] [PubMed]
- Heath, L.; Walt, E.; Varsani, A.; Martin, D.P. Recombination Patterns in Aphthoviruses Mirror Those Found in Other Picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [PubMed]
- Pica, N.; Hai, R.; Krammer, F.; Wang, T.T.; Maamary, J.; Eggink, D.; Tan, G.S.; Krause, J.C.; Moran, T.; Stein, C.R.; et al. Hemagglutinin Stalk Antibodies Elicited by the 2009 Pandemic Influenza Virus as a Mechanism for the Extinction of Seasonal H1N1 Viruses. Proc. Natl. Acad. Sci. USA 2012, 109, 2573–2578. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, N.; Haanpaa, M.; Ronkko, E.; Lyytikainen, O.; Kuusi, M.; Ruutu, P.; KallioKokko, H.; Mannonen, L.; Lappalainen, M.; Ziegler, T.; et al. Genetic Diversity of the 2009 Pandemic Influenza A(H1N1) Viruses in Finland. PLoS ONE 2010, 5, e13329. [Google Scholar]
- Strengell, M.; Ikonen, N.; Ziegler, T.; Julkunen, I. Minor Changes in the Hemagglutinin of Influenza A(H1N1)2009 Virus Alter Its Antigenic Properties. PLoS ONE 2011, 6, e25848. [Google Scholar] [CrossRef]
- Su, Y.C.F.; Bahl, J.; Joseph, U.; Butt, K.M.; Peck, H.A.; Koay, E.S.C.; Oon, L.L.E.; Barr, I.G.; Vijaykrishna, D.; Smith, G.J.D. Phylodynamics of H1N1/2009 Influenza Reveals the Transition from Host Adaptation to Immune-Driven Selection. Nat. Commun. 2015, 6, 7952. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, K.; Yin, Y.; Qin, J.; Zhou, Y.-H.; Yang, J.; Li, S.; Poon, L.L.M.; Zhang, C. The Phylodynamics of Seasonal Influenza A/H1N1pdm Virus in China Between 2009 and 2019. Front. Microbiol. 2020, 11, 735. [Google Scholar] [CrossRef]
- Nelson, M.; Spiro, D.; Wentworth, D.; Fan, J.; Beck, E.; George, K.S. The Early Diversification of Influenza A/H1N1pdm. PLoS Curr. 2009, 1, RRN1126. [Google Scholar] [CrossRef]
- Koel, B.F.; Burke, D.F.; Bestebroer, T.M.; van der Vliet, S.; Zondag, G.C.M.; Vervaet, G.; Skepner, E.; Lewis, N.S.; Spronken, M.I.J.; Russell, C.A.; et al. Substitutions near the Receptor Binding Site Determine Major Antigenic Change during Influenza Virus Evolution. Science 2013, 342, 976–979. [Google Scholar] [CrossRef]
- Koelle, K.; Rasmussen, D.A. The Effects of a Deleterious Mutation Load on Patterns of Influenza A/H3N2′s Antigenic Evolution in Humans. eLife 2015, 4, e07361. [Google Scholar] [CrossRef]
- Petrova, V.N.; Russell, C.A. The Evolution of Seasonal Influenza Viruses. Nat. Rev. Microbiol. 2018, 16, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Otwinowski, J.; Thompson, A.J.; Nycholat, C.M.; Nourmohammad, A.; Wilson, I.A. Major Antigenic Site B of Human Influenza H3N2 Viruses Has an Evolving Local Fitness Landscape. Nat. Commun. 2020, 11, 1233. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Stevens, D.J.; Daniels, R.S.; Douglas, A.R.; Knossow, M.; Wilson, I.A.; Wiley, D.C. A Carbohydrate Side Chain on Hemagglutinins of Hong Kong Influenza Viruses Inhibits Recognition by a Monoclonal Antibody. Proc. Natl. Acad. Sci. USA 1984, 81, 1779–1783. [Google Scholar] [PubMed]
- Maurer-Stroh, S.; Ma, J.; Lee, R.T.; Sirota, F.L.; Eisenhaber, F. Mapping the Sequence Mutations of the 2009 H1N1 Influenza A Virus Neuraminidase Relative to Drug and Antibody Binding Sites. Biol. Direct. 2009, 4, 18. [Google Scholar]
- Duque, J.; McMorrow, M.L.; Cohen, A.L. Influenza Vaccines and Influenza Antiviral Drugs in Africa: Are They Available and Do Guidelines for Their Use Exist? BMC Public Health 2014, 14, 41. [Google Scholar]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug Resistance in Influenza A Virus: The Epidemiology and Management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef]
- Badar, N.; Salman, M.; Aamir, U.B.; Ansari, J.; Ranjha, M.A.; Khan, M.A.; Ikram, A.; Nisar, N.; Mushtaq, N.; Mirza, H.A. Evolutionary Analysis of Influenza A(H1N1)Pdm09 during the Pandemic and Post-Pandemic Period in Pakistan. J. Infect. Public Health 2020, 13, 407–413. [Google Scholar] [CrossRef]
- Jones, S.; Nelson-Sathi, S.; Wang, Y.; Prasad, R.; Rayen, S.; Nandel, V.; Hu, Y.; Zhang, W.; Nair, R.; Dharmaseelan, S.; et al. Evolutionary, Genetic, Structural Characterization and Its Functional Implications for the Influenza A (H1N1) Infection Outbreak in India from 2009 to 2017. Sci. Rep. 2019, 9, 14690. [Google Scholar] [CrossRef]
- Saha, P.; Biswas, M.; Gupta, R.; Majumdar, A.; Mitra, S.; Banerjee, A.; Mukherjee, A.; Dutta, S.; Chawla-Sarkar, M. Molecular Characterization of Influenza A Pandemic H1N1 Viruses Circulating in Eastern India during 2017–19: Antigenic Diversity in Comparison to the Vaccine Strains. Infect. Genet. Evol. 2020, 81, 104270. [Google Scholar] [CrossRef]
- Dong, G.; Peng, C.; Luo, J.; Wang, C.; Han, L.; Wu, B.; Ji, G.; He, H. Adamantane-Resistant Influenza a Viruses in the World (1902–2013): Frequency and Distribution of M2 Gene Mutations. PLoS ONE 2015, 10, e0119115. [Google Scholar] [CrossRef]
- He, W.; Zhang, W.; Yan, H.; Xu, H.; Xie, Y.; Wu, Q.; Wang, C.; Dong, G. Distribution and Evolution of H1N1 Influenza A Viruses with Adamantanes-Resistant Mutations Worldwide from 1918 to 2019. J. Med. Virol. 2021, 93, 3473–3483. [Google Scholar] [CrossRef] [PubMed]
- Escuret, V.; Frobert, E.; Bouscambert-Duchamp, M. Detection of Human Influenza A (H1N1) and B Strains with Reduced Sensitivity to Neuraminidase Inhibitors. J. Clin. Virol. 2008, 41, 25–28. [Google Scholar] [PubMed]
- Kyaw Win, S.M.; Saito, R.; Win, N.C.; Lasham, D.J.; Kyaw, Y.; Lin, N.; Thein, K.N.; Chon, I.; Odagiri, T.; Thein, W.; et al. Epidemic of Influenza A(H1N1)Pdm09 Analyzed by Full Genome Sequences and the First Case of Oseltamivir-Resistant Strain in Myanmar 2017. PLoS ONE 2020, 15, e0229601. [Google Scholar] [CrossRef]
- Shen, J.; Ma, J.; Wang, Q. Evolutionary trends of A(H1N1) influenza virus hemagglutinin since 1918. PLoS ONE 2009, 4, 7789. [Google Scholar]
- Nelson, M.N.; Edelman, L.; Spiro, D.J.; Boyne, A.R.; Bera, J.; Halpin, R.; Ghedin, E.; Miller, M.A.; Simonsen, L.; Viboud, C.; et al. Molecular Epidemiology of A/H3N2 and A/H1N1 Influenza Virus during a Single Epidemic Season in the United States. PLoS Pathog. 2008, 4, e1000133. [Google Scholar]
- Baillie, G.J.; Galiano, M.; Agapow, P.M.; Myers, R.; Chiam, R.; Gall, A.; Palser, A.L.; Watson, S.J.; Hedge, J.; Underwood, A.; et al. Evolutionary Dynamics of Local Pandemic H1N1/2009 Influenza Virus Lineages Revealed by Whole-Genome Analysis. J. Virol. 2012, 86, 11–18. [Google Scholar]
- Bedford, T.; Riley, S.; Barr, I.G.; Broor, S.; Chadha, M.; Cox, N.J. Global Circulation Patterns of Seasonal Influenza Viruses Vary with Antigenic Drift. Nature 2015, 523, 26053121. [Google Scholar] [CrossRef]
- Viboud, C.; Nelson, M.I.; Tan, Y.; Holmes, E.C. Contrasting the epidemiological and evolutionary dynamics of influenza spatial transmission. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120199. [Google Scholar]
- CDC. Outbreak of Swine-Origin Influenza A (H1N1) Virus Infection-Mexico, March-April 2009. MMWR Morb. Mortal. Wkly. Rep. 2009, 58, 19444150. [Google Scholar]
- CDC. 2009 H1N1 Flu Pandemic Timeline. Available online: https://www.cdc.gov/flu/pandemic-resources/2009-pandemic-timeline.html (accessed on 21 July 2021).
- Bedford, T.; Cobey, S.; Pascual, M. Strength and Tempo of Selection Revealed in Viral Gene Genealogies. BMC Evol. Biol. 2011, 11, 220. [Google Scholar] [CrossRef]
- Nelson, M.I.; Holmes, E.C. The Evolution of Epidemic Influenza. Nat. Rev. Genet. 2007, 26, 32. [Google Scholar]
- Christman, M.C.; Kedwaii, A.; Xu, J.; Donis, R.O.; Lu, G. Pandemic (H1N1) 2009 Virus Revisited: An Evolutionary Retrospective. Infect. Genet. Evol. 2011, 11, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Bahl, J.; Nelson, M.I.; Chan, K.H. Temporally structured metapopulation dynamics and persistence of influenza A H3N2 virus in humans. Proc. Natl. Acad. Sci. USA 2011, 108, 19359–19364. [Google Scholar] [PubMed]
- Ebranati, E.; Pariani, E.; Piralla, A.; Gozalo-Margüello, M.; Veo, C.; Bubba, L.; Amendola, A.; Ciccozzi, M.; Galli, M.; Zanetti, A.R.; et al. Reconstruction of the Evolutionary Dynamics of A(H3N2) Influenza Viruses Circulating in Italy from 2004 to 2012. PLoS ONE 2015, 10, e0137099. [Google Scholar] [CrossRef]
- Gong, Y.-N.; Tsao, K.-C.; Chen, G.-W. Inferring the Global Phylodynamics of Influenza A/H3N2 Viruses in Taiwan. J. Formos. Med. Assoc. 2019, 118, 116–124. [Google Scholar] [CrossRef]
- Yang, J.R.; Lin, Y.C.; Huang, Y.P.; Su, C.H.; Lo, J.; Ho, Y.L.; Yao, C.Y.; Hsu, L.C.; Wu, H.S.; Liu, M.T. Reassortment and Mutations Associated with Emergence and Spread of Oseltamivirresistant Seasonal Influenza A/H1N1 Viruses in 2005–2009. PLoS ONE 2011, 6, e18177. [Google Scholar]
- Bragstad, K.; Nielsen, L.P.; Fomsgaard, A. The Evolution of Human Influenza A Viruses from 1999 to 2006: A Complete Genome Study. Virol. J. 2008, 5, 40. [Google Scholar] [CrossRef]
- Komadina, N.; McVernon, J.; Hall, R.; Leder, K. A Historical Perspective of Influenza A(H1N2) Virus. Emerg. Infect. Dis. 2014, 20, 6–12. [Google Scholar] [CrossRef]
- World Health Organization. Recommended Composition of Influenza Virus Vaccines for Use in the 2002–2003 Season = Composition Recommandée Des Vaccins Antigrippaux Pour La Saison 2002–2003. Wkly. Epidemiol. Rec. 2002, 77, 62–66. [Google Scholar]
- Ledesma, J.; Pozo, F.; Reina, G.; Blasco, M.; Rodríguez, G.; Montes, M.; López-Miragaya, I.; Salvador, C.; Reina, J.; Ortíz de Lejarazu, R.; et al. Genetic Diversity of Influenza A(H1N1)2009 Virus Circulating during the Season 2010–2011 in Spain. J. Clin. Virol. 2012, 53, 16–21. [Google Scholar] [CrossRef]
- Kyncl, J.; Havlickova, M.; Nagy, A.; Jirincova, H.; Piskova, I. Early and Unexpectedly Severe Start of Influenza Epidemic in the Czech Republic during Influenza Season 2012–13. Eurosurveillance 2013, 18, 20396. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Ghedin, E.; Miller, N.; Taylor, J.; Bao, Y. Whole-genome analysis of human influenza A virus reveals multiple persistent lineages and reassortment among recent H3N2 viruses. PLoS Biol. 2005, 3, 300. [Google Scholar]
- Maljkovic Berry, I.; Melendrez, M.C.; Li, T.; Hawksworth, A.W.; Brice, G.T.; Blair, P.J.; Halsey, E.S.; Williams, M.; Fernandez, S.; Yoon, I.-K.; et al. Frequency of Influenza H3N2 Intra-Subtype Reassortment: Attributes and Implications of Reassortant Spread. BMC Biol. 2016, 14, 117. [Google Scholar] [CrossRef]
- Müller, N.F.; Stolz, U.; Dudas, G.; Stadler, T.; Vaughan, T.G. Bayesian Inference of Reassortment Networks Reveals Fitness Benefits of Reassortment in Human Influenza Viruses. Proc. Natl. Acad. Sci. USA 2020, 117, 17104–17111. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453, 615–619. [Google Scholar]
- Simonsen, L.; Viboud, C.; Grenfell, B.; Dushoff, J.; Jennings, L.; Smit, M.; Hata, M.; Macken, C.; Gog, J.; Miller, M.A.; et al. The genesis and spread of reassortment human influenza A/H3N2 viruses conferring adamantane resistance. Mol. Biol. Evol. 2007, 24, 24. [Google Scholar]
- Nabakooza, G.; Pastusiak, A.; Kateete, D.P.; Lutwama, J.J.; Kitayimbwa, J.M.; Frost, S.D.W. Whole-Genome Analysis to Determine the Rate and Patterns of Intra-Subtype Reassortment among Influenza Type-A Viruses in Africa. Virus Evol. 2022, 8, veac005. [Google Scholar] [CrossRef]
- Phipps, K.L.; Marshall, N.; Tao, H.; Danzy, S.; Onuoha, N.; Steel, J.; Lowen, A.C. Seasonal H3N2 and 2009 Pandemic H1N1 Influenza A Viruses Reassort Efficiently but Produce Attenuated Progeny. J. Virol. 2017, 91, e00830-17. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).