
eGFP Gene Integration in HO: A Metabolomic Impact?

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Microbial Strains
2.2. Constructions of Yeast Strains Labeled with EGFP Using CRISPR-Cas9 Tool
2.3. Fermentation Process
2.4. Flow Cytometry Analysis
2.4.1. Yeast Viability
2.4.2. Flow Cytometry Settings
2.5. Two-Photon Microscopy
2.6. Analytical Method
2.6.1. Enological Analysis
2.6.2. UHPLC-Q-ToF-MS Analysis
3. Results and Discussion
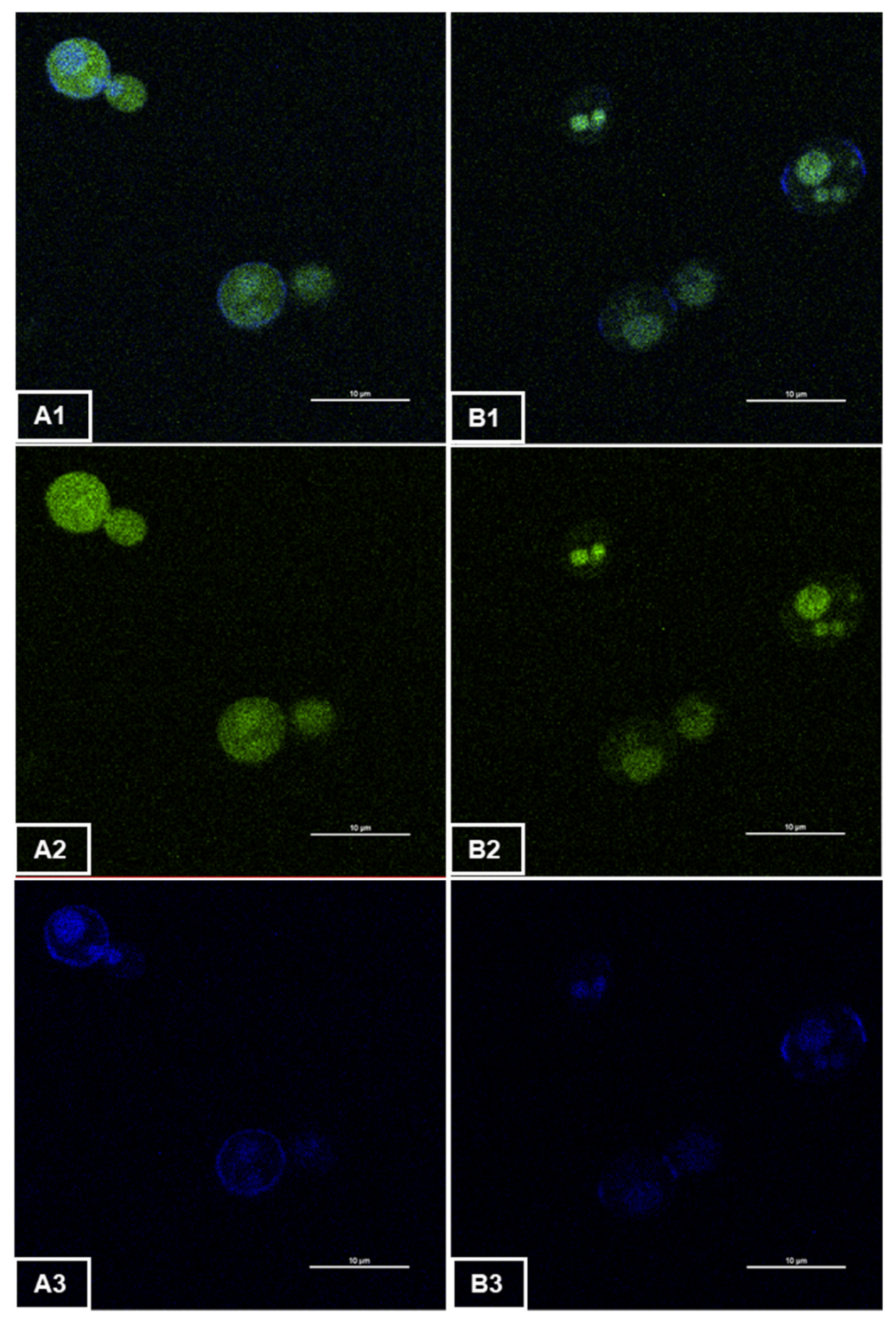
3.1. Fluorescence Emission of Recombinant Yeast GFP
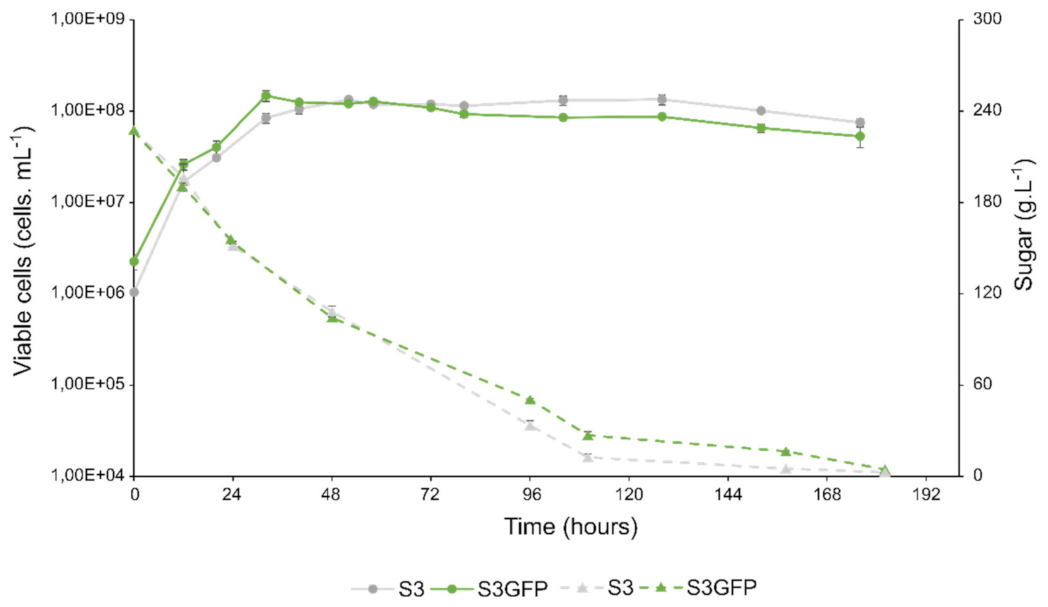
3.2. Growth and Fermentation Kinetics
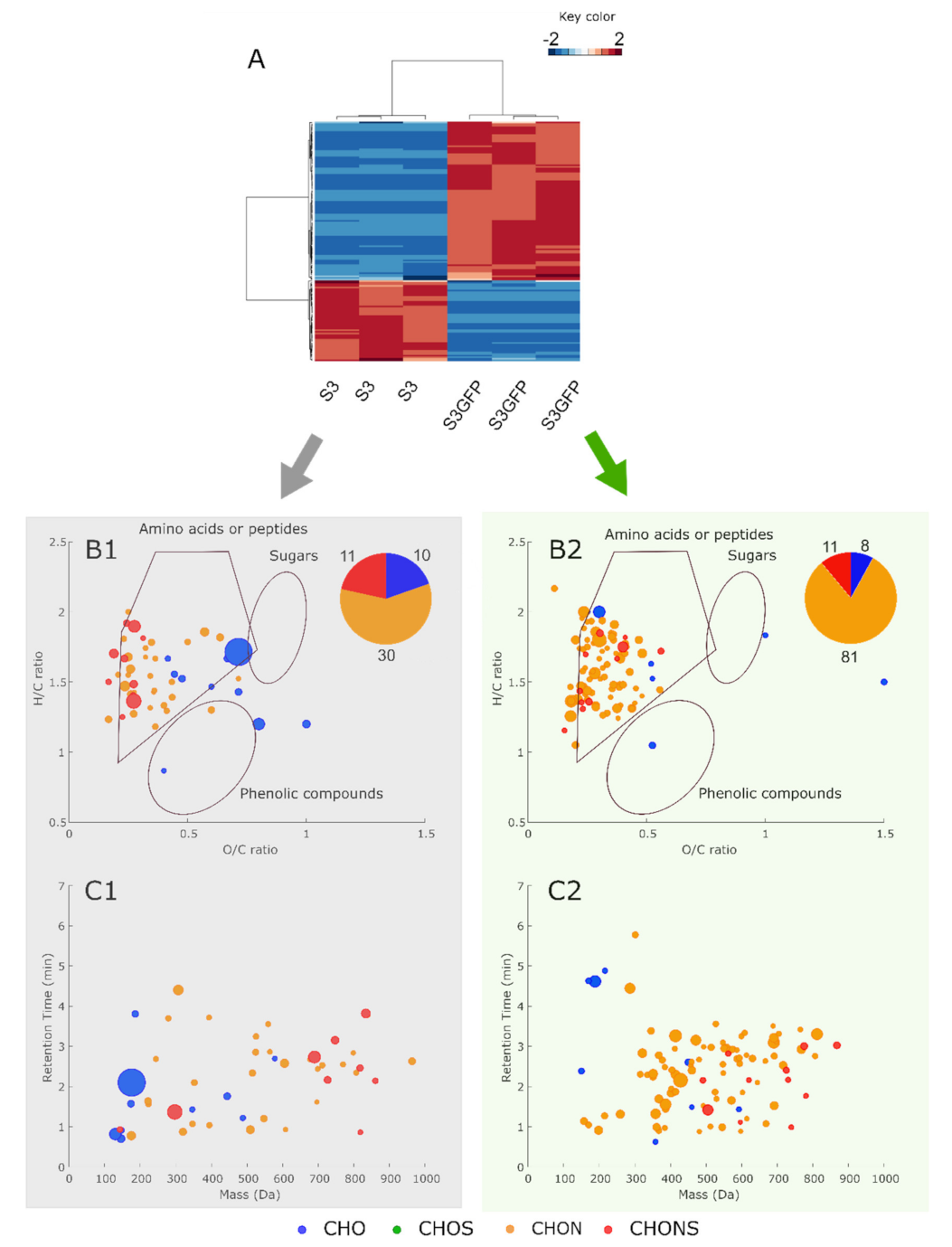
3.3. Metabolomic Pattern
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lorang, J.M.; Tuori, R.P.; Martinez, J.P.; Sawyer, T.L.; Redman, R.S.; Rollins, J.A.; Wolpert, T.J.; Johnson, K.B.; Rodriguez, R.J.; Dickman, M.B.; et al. Green fluorescent protein is lighting up fungal biology. Appl. Environ. Microbiol. 2001, 67, 1987–1994. [Google Scholar] [CrossRef]
- Tatchell, K.; Robinson, L.C. Use of green fluorescent protein in living yeast cells. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2002; Volume 351, pp. 661–683. [Google Scholar]
- Momeni, B.; Brileya, K.A.; Fields, M.W.; Shou, W. Strong inter-population cooperation leads to partner intermixing in microbial communities. eLife 2013, 2, e00230. [Google Scholar] [CrossRef]
- Petitgonnet, C.; Klein, G.L.; Roullier-Gall, C.; Schmitt-Kopplin, P.; Quintanilla-Casas, B.; Vichi, S.; Julien-David, D.; Alexandre, H. Influence of cell-cell contact between L. thermotolerans and S. cerevisiae on yeast interactions and the exo-metabolome. Food Microbiol. 2019, 83, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Conacher, C.G.; Naidoo-Blassoples, R.K.; Rossouw, D.; Bauer, F.F. Real-time monitoring of population dynamics and physical interactions in a synthetic yeast ecosystem by use of multicolour flow cytometry. Appl. Microbiol. Biotechnol. 2020, 104, 5547–5562. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell. Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef]
- Heim, R.; Prasher, D.C.; Tsien, R.Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 1994, 91, 12501–12504. [Google Scholar] [CrossRef] [PubMed]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the Green Fluorescent Protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Kostriken, R.; Strathern, J.N.; Klar, A.J.S.; Hicks, J.B.; Heffron, F. A site-specific endonuclease essential for mating-type switching in Saccharomyces cerevisiae. Cell 1983, 35, 167–174. [Google Scholar] [CrossRef]
- Russell, D.W.; Smith, M.; Herskowitz, I. Structure of the Saccharomyces cerevisiae HO gene and analysis of its upstream regulatory region. Mol. Cell. Biol. 1986, 6, 14. [Google Scholar]
- Herskowitz, I. Control of cell type in Saccharomyces cerevisiae: Mating type and mating-type interconversion. Mol. Biol. Yeast Saccharomyces Life Cycle Inherit. 1981, 181–209. [Google Scholar]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; William, W.W.; Douglas, C.P. Green fluorescent protein as a marker for gene expression 1994, 263, 802–804. Science 1994, 263, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Weill, U.; Yofe, I.; Sass, E.; Stynen, B.; Davidi, D.; Natarajan, J.; Ben-Menachem, R.; Avihou, Z.; Goldman, O.; Harpaz, N.; et al. Genome-wide SWAp-tag yeast libraries for proteome exploration. Nat. Methods 2018, 15, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Weill, U.; Krieger, G.; Avihou, Z.; Milo, R.; Schuldiner, M.; Davidi, D. Assessment of GFP tag position on protein localization and growth fitness in yeast. J. Mol. Biol. 2019, 431, 636–641. [Google Scholar] [CrossRef]
- Generoso, W.C.; Gottardi, M.; Oreb, M.; Boles, E. Simplified CRISPR-cas genome editing for Saccharomyces cerevisiae. J. Microbiol. Methods 2016, 127, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K.; Cameron, D.M.; Collins, S.R.; Schuldiner, M.; Stewart-Ornstein, J.; Newman, H.W.; Braun, S.; Madhani, H.D.; Krogan, N.J.; Weissman, J.S. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods 2008, 5, 711–718. [Google Scholar] [CrossRef]
- KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.genome.jp/kegg/ (accessed on 27 January 2020).
- Metlin. Available online: https://metlin.scripps.edu/landing_page.php?pgcontent=mainPage (accessed on 27 January 2020).
- Liu, Y.; Forcisi, S.; Lucio, M.; Harir, M.; Bahut, F.; Deleris-Bou, M.; Krieger-Weber, S.; Gougeon, R.D.; Alexandre, H.; Schmitt-Kopplin, P. Digging into the low molecular weight peptidome with the OligoNet web server. Sci. Rep. 2017, 7, 11692. [Google Scholar] [CrossRef] [PubMed]
- Brachat, A.; Liebundguth, N.; Rebischung, C.; Lemire, S.; Schärer, F.; Hoepfner, D.; Demchyshyn, V.; Howald, I.; Düsterhöft, A.; Möstl, D.; et al. Analysis of deletion phenotypes and GFP fusions of 21 novel Saccharomyces cerevisiae open reading frames. Yeast 2000, 16, 241–253. [Google Scholar] [CrossRef]
- Styles, E.B.; Friesen, H.; Boone, C.; Andrews, B.J. High-throughput microscopy-based screening in Saccharomyces cerevisiae. Cold Spring Harb Protoc. 2016, 2016, pdbtop087593. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Namiki, Y.; Okada, H.; Mori, Y.; Furukawa, H.; Wang, J.; Ohkusu, M.; Kawamoto, S. Structome of Saccharomyces cerevisiae determined by freeze-substitution and serial ultrathin-sectioning electron microscopy. Microscopy 2011, 60, 321–335. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gui, Q.; Deng, S.; Zhou, Z.; Cao, W.; Zhang, X.; Shi, W.; Cai, X.; Jiang, W.; Cui, Z.; Hu, Z.; et al. Transcriptome analysis in yeast reveals the externality of position effects. Mol. Biol. Evol. 2021, 38, 3294–3307. [Google Scholar] [CrossRef] [PubMed]
- Baganz, F.; Hayes, A.; Marren, D.; Gardner, D.C.J.; Oliver, S.G. Suitability of replacement markers for functional analysis studies Saccharomyces Cerevisiae. Yeast 1997, 13, 1563–1573. [Google Scholar] [CrossRef]
- Kafri, M.; Metzl-Raz, E.; Jona, G.; Barkai, N. The cost of protein production. Cell Rep. 2016, 14, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Baichwal, V.R.; Cunningham, T.S.; Gatzek, P.R.; Kohlhaw, G.B. Leucine biosynthesis in yeast: Identification of two genes (LEU4, LEU5) that affect α-Isopropylmalate synthase activity and evidence that LEU1 and LEU2 gene expression is controlled by α-Isopropylmalate and the product of a regulatory gene. Curr. Genet. 1983, 7, 369–377. [Google Scholar] [CrossRef]
- Ganini, D.; Leinisch, F.; Kumar, A.; Jiang, J.; Tokar, E.J.; Malone, C.C.; Petrovich, R.M.; Mason, R.P. Fluorescent proteins such as EGFP lead to catalytic oxidative stress in cells. Redox Biol. 2017, 12, 462–468. [Google Scholar] [CrossRef]
- Lupo, S.; Aranda, C.; Olivera, H.; Riego, L.; González, A.; Miranda-Ham, L.; Servin, L. Tyrosine is involved in protection from oxidative stress in Saccharomyces cerevisiae. Can. J. Microbiol. 1997, 43, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Samaranayaka, A.G.P.; Li-Chan, E.C.Y. Food-derived peptidic antioxidants: A review of their production, assessment, and potential applications. J. Funct. Foods 2011, 3, 229–254. [Google Scholar] [CrossRef]
- Pedelacq, J.-D.; Cabantous, S. Development and applications of superfolder and split fluorescent protein detection systems in biology. Int. J. Mol. Sci. 2019, 20, 3479. [Google Scholar] [CrossRef]
- Eguchi, Y.; Makanae, K.; Hasunuma, T.; Ishibashi, Y.; Kito, K.; Moriya, H. Estimating the protein burden limit of yeast cells by measuring the expression limits of glycolytic proteins. eLife 2018, 7, e34595. [Google Scholar] [CrossRef] [PubMed]
- Moriya, H. Quantitative nature of overexpression experiments. MBoC 2015, 26, 3932–3939. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) * |
|---|---|
| gRNA-oligotarget1-HO | CTCCGCAGTGAAAGATAAATGATCAAATTGGGCATTACGCCCGA GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGG |
| V1-AS-F | GTAGTGCCCTCTTGGGCTA |
| Fluo-F-HO | AAAAAGGCAAAAGACAAAGGCGAAAAATTGGGCATTACGCCGGTCGACGGATCCCCGGGTT |
| Fluo-R-HO | CACATTTTATACACTCTGGTCCTTTAACTGGCAAACCTTCGTCGATGAATTCGAGCTCGTT |
| VerifGFP-F | ACGGCGTCGTACAAGAGAAC |
| VerifGFP-R | TACATAACCTTCGGGCATGG |
| V1HO-F | CATACGACTGTAATGTTGCT |
| V2-HO-R | AAACTGTAAGATTCCGCCAC |
| Strain | Maximum Population Growth Rate: µ Max (h−1) | Generation Number | Maximum Population: K (Viable Cells.mL−1) | Maximum Sugar Consumption Rate: Vmax (gL−1h−1) | Time to T50 of Sugar Degradation (Hours) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| S3 | 1.37 × 10−1 ± 5 × 10−3 | }NS | 4.75 ± 0.18 | }NS | 1.42 × 108 ± 2 × 106 | }NS | 1.80 ± 0.32 | }NS | 45.3 ± 0.9 | }NS |
| S3GFP | 1.39 × 10−1 ± 4 × 10−3 | 4.54 ± 0.16 | 1.50 × 108 ± 1 × 107 | 2.06 ± 0.12 | 43.3 ± 0.4 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bordet, F.; Romanet, R.; Eicher, C.; Grandvalet, C.; Klein, G.; Gougeon, R.; Julien-Ortiz, A.; Roullier-Gall, C.; Alexandre, H. eGFP Gene Integration in HO: A Metabolomic Impact? Microorganisms 2022, 10, 781. https://doi.org/10.3390/microorganisms10040781
Bordet F, Romanet R, Eicher C, Grandvalet C, Klein G, Gougeon R, Julien-Ortiz A, Roullier-Gall C, Alexandre H. eGFP Gene Integration in HO: A Metabolomic Impact? Microorganisms. 2022; 10(4):781. https://doi.org/10.3390/microorganisms10040781
Chicago/Turabian StyleBordet, Fanny, Rémy Romanet, Camille Eicher, Cosette Grandvalet, Géraldine Klein, Régis Gougeon, Anne Julien-Ortiz, Chloé Roullier-Gall, and Hervé Alexandre. 2022. "eGFP Gene Integration in HO: A Metabolomic Impact?" Microorganisms 10, no. 4: 781. https://doi.org/10.3390/microorganisms10040781
APA StyleBordet, F., Romanet, R., Eicher, C., Grandvalet, C., Klein, G., Gougeon, R., Julien-Ortiz, A., Roullier-Gall, C., & Alexandre, H. (2022). eGFP Gene Integration in HO: A Metabolomic Impact? Microorganisms, 10(4), 781. https://doi.org/10.3390/microorganisms10040781