
Metabolism of the Genus Guyparkeria Revealed by Pangenome Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Microorganisms and Culturing Media
2.2. Genomic DNA Extraction and Sequencing of Strains B1-1 and R3
2.3. Quality Control and Assembly of Sequencing Data
2.4. Gene Annotation of Draft Genome B1-1 and R3
2.5. Pangenome and Core Genome Analyses
2.6. Phylogenetic Analyses
3. Results and Discussion
3.1. Basic Summary of Guyparkeria Genomes
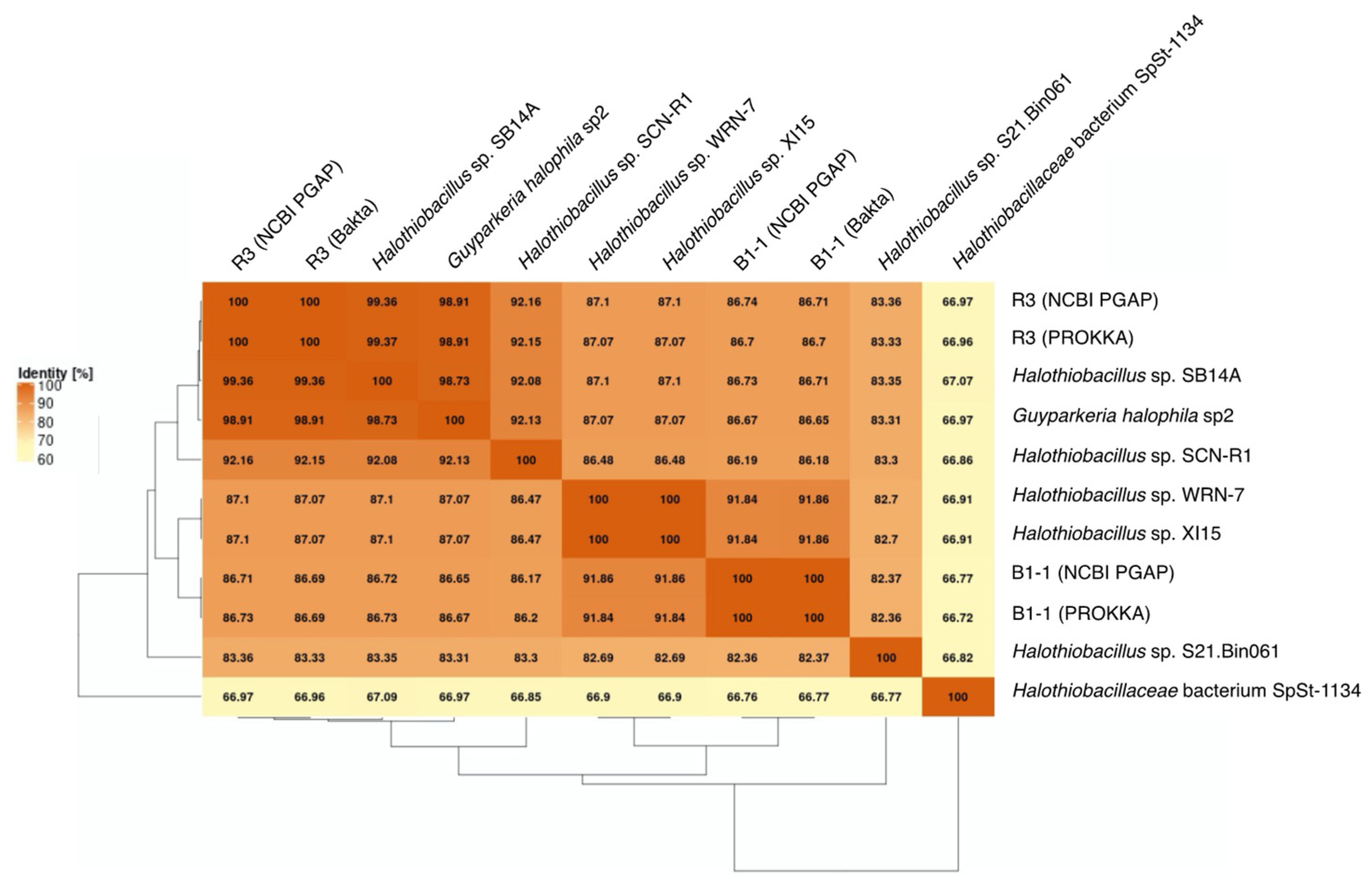
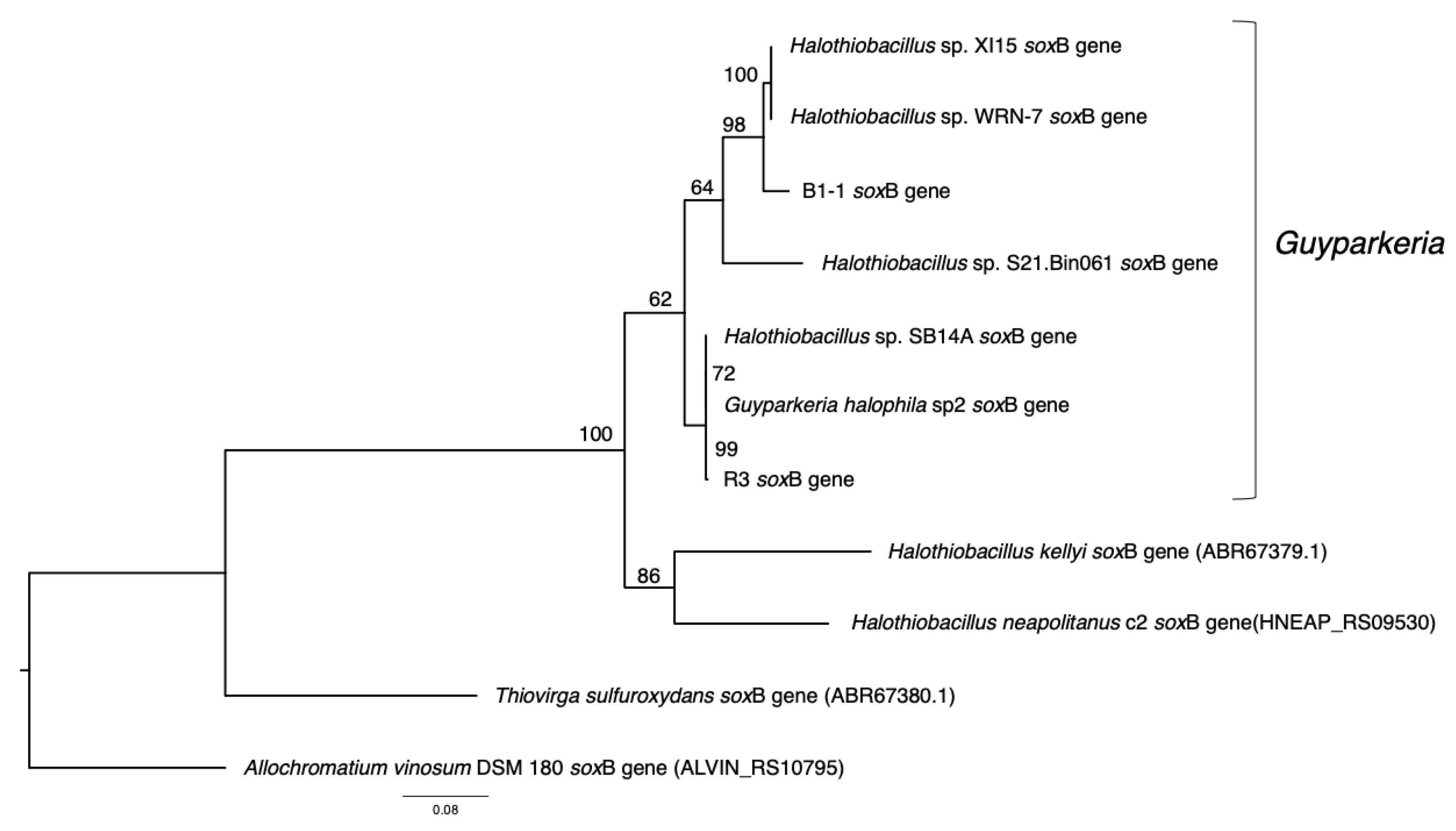
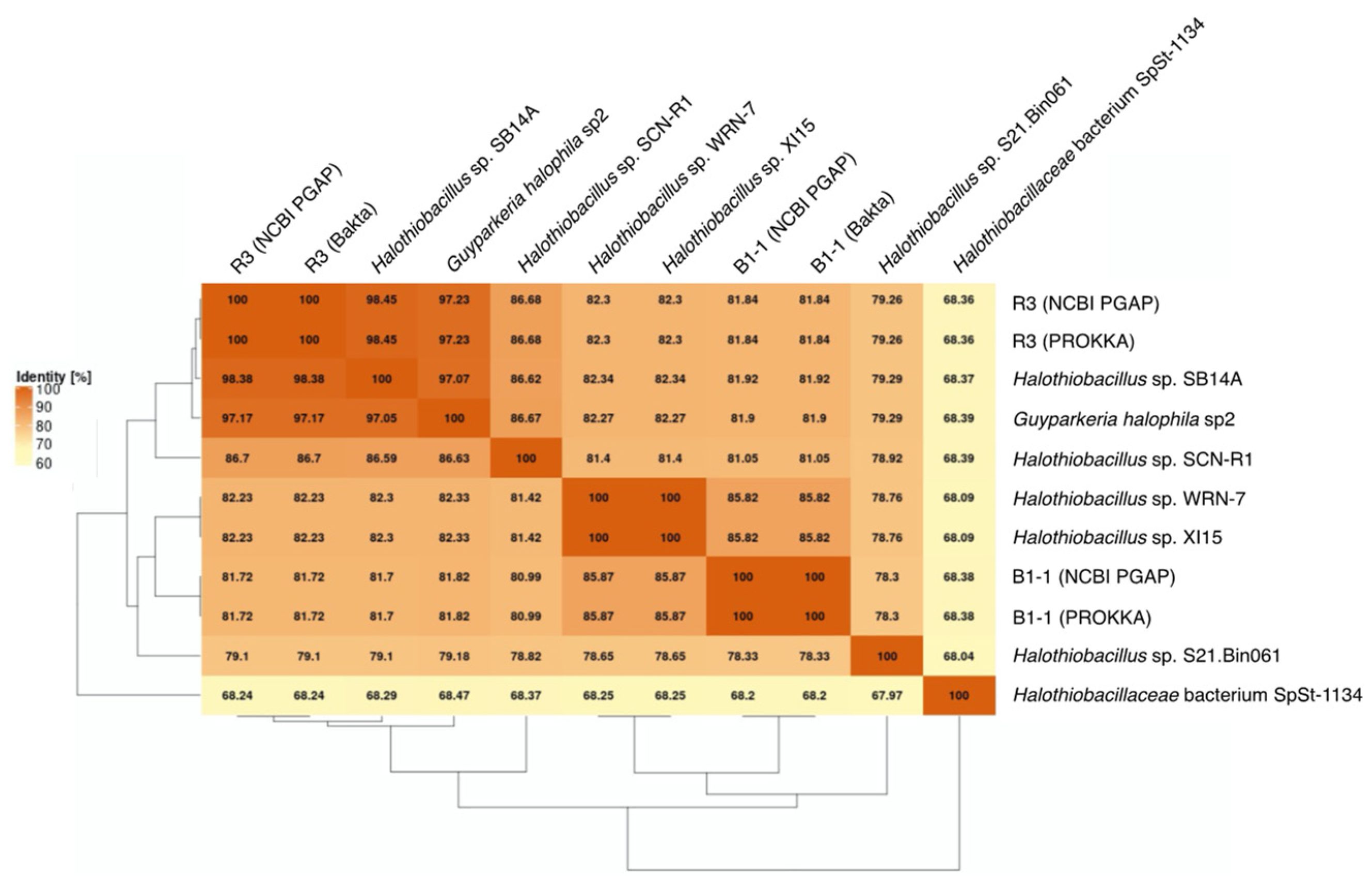
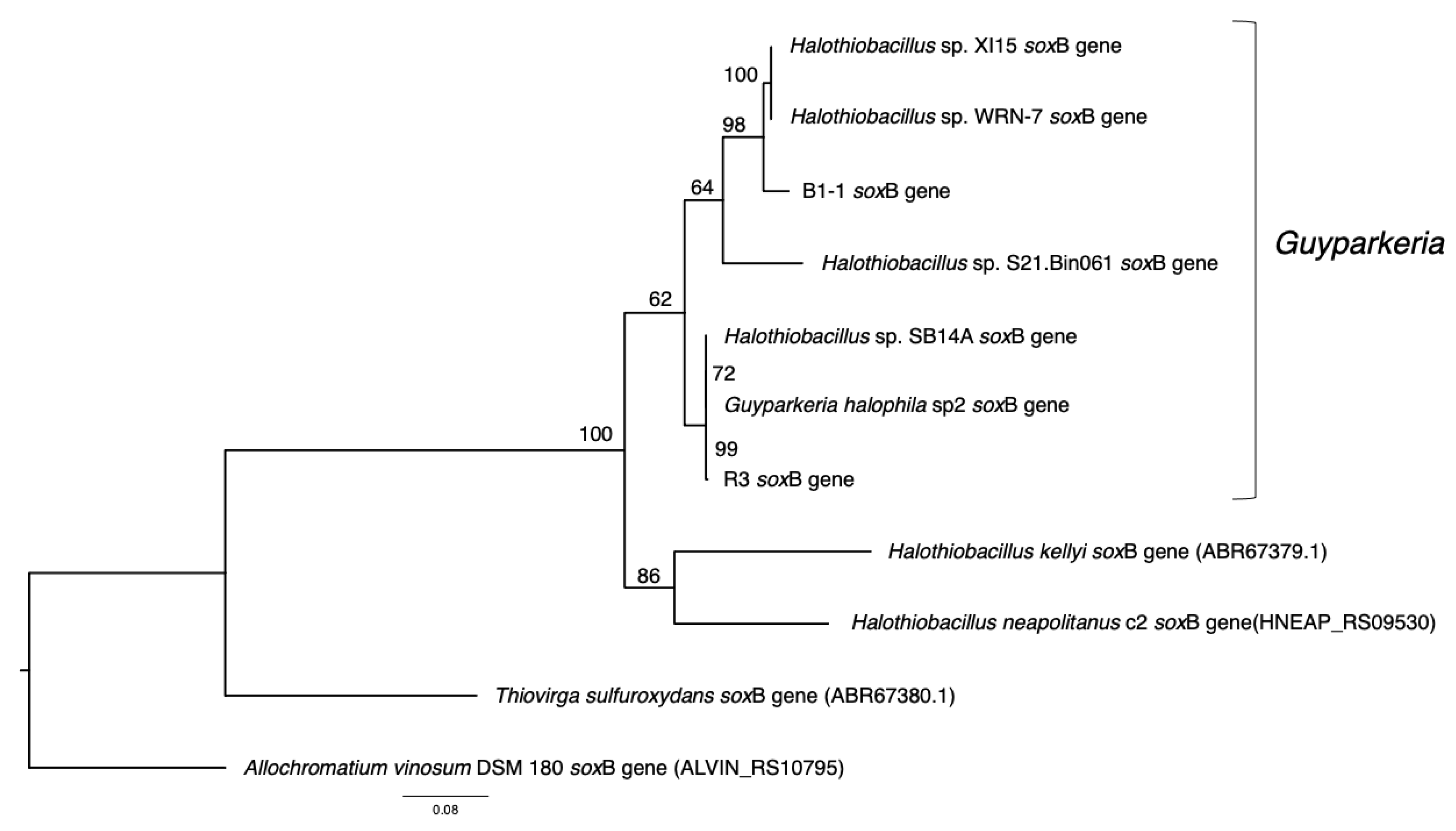
3.2. Phylogenetic Relatedness of Guyparkeria 16S rRNA Genes, Genomes and soxB Genes
3.3. Pangenome of Guyparkeria
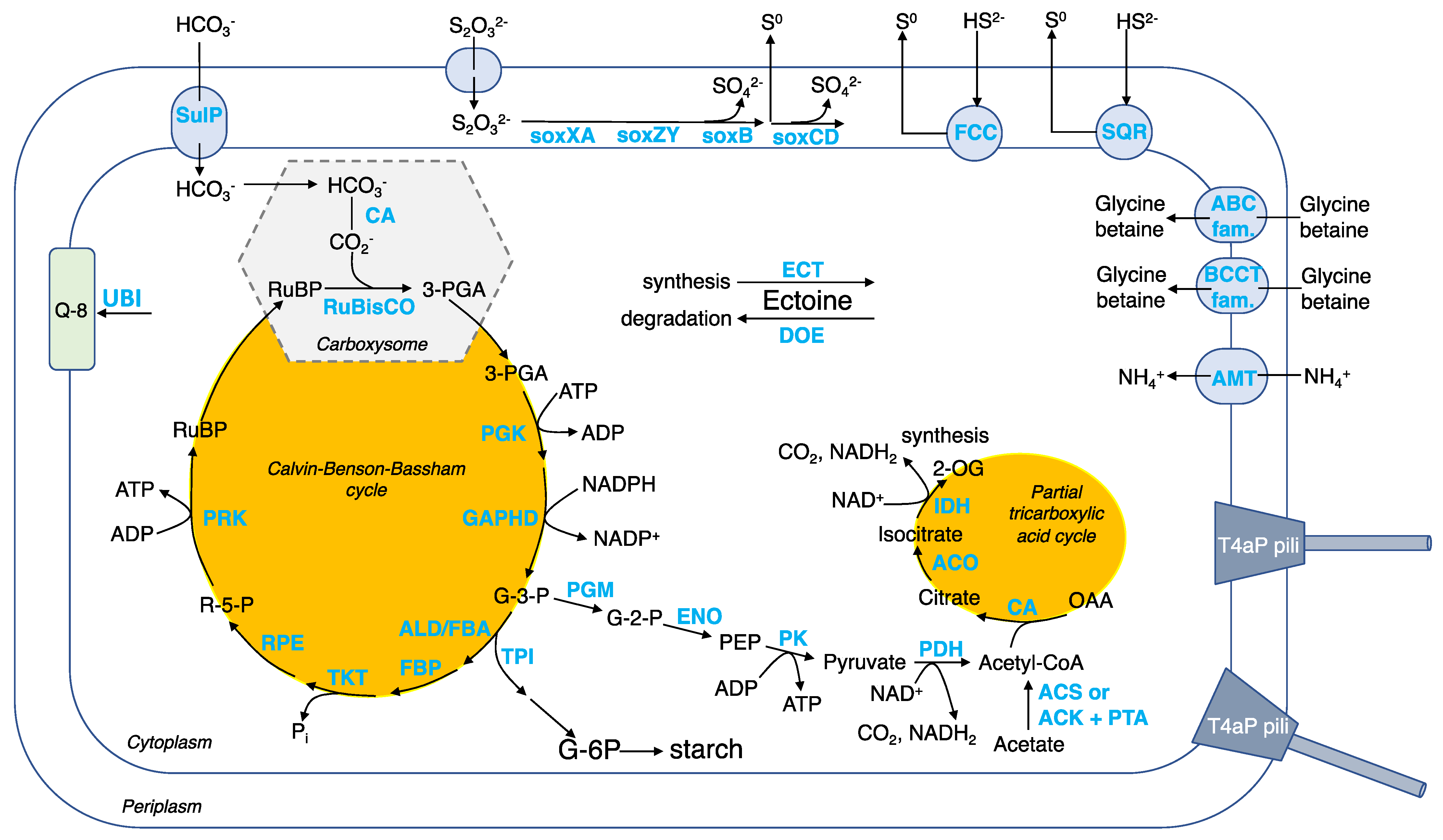
3.4. Genetic Basis of Key Metabolic Functions of Guyparkeria
3.5. Highlights on Some Accessory Genes in B1-1 and R3 Genomes
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boden, R. Reclassification of Halothiobacillus hydrothermalis and Halothiobacillus halophilus to Guyparkeria gen. nov. in the Thioalkalibacteraceae fam. nov., with emended descriptions of the genus Halothiobacillus and family Halothiobacillaceae. Int. J. Syst. Evol. Microbiol. 2017, 67, 3919–3928. [Google Scholar] [CrossRef] [PubMed]
- Durand, P.; Reysenbach, A.; Prieur, D.; Pace, N. Isolation and characterization of Thiobacillus hydrothermalis sp. nov., a mesophilic obligately chemolithotrophic bacterium isolated from a deep-sea hydrothermal vent in Fiji Basin. Arch. Microbiol. 1993, 159, 39–44. [Google Scholar] [CrossRef]
- Kelly, D.P.; Wood, A.P. Reclassification of some species of Thiobacillus to the newly designated genera Acidithiobacillus gen. nov., Halothiobacillus gen. nov. and Thermithiobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 2000, 50, 511–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, A.P.; Kelly, D.P. Isolation and characterisation of Thiobacillus halophilus sp. nov., a sulphur-oxidising autotrophic eubacterium from a Western Australian hypersaline lake. Arch. Microbiol. 1991, 156, 277–280. [Google Scholar] [CrossRef]
- Abashina, T.; Yachkula, A.; Kaparullina, E.; Vainshtein, M. Intensification of nickel bioleaching with neutrophilic bacteria Guyparkeria halophila as an approach to limitation of sulfuric acid pollution. Microorganisms 2021, 9, 2461. [Google Scholar] [CrossRef]
- Tsallagov, S.I.; Sorokin, D.Y.; Tikhonova, T.V.; Popov, V.O.; Muyzer, G. Comparative genomics of Thiohalobacter thiocyanaticus HRh1T and Guyparkeria sp. SCN-R1, halophilic chemolithoautotrophic sulfur-oxidizing gammaproteobacteria capable of using thiocyanate as energy source. Front. Microbiol. 2019, 10, 898. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, D.Y.; Abbas, B.; Van Zessen, E.; Muyzer, G. Isolation and characterization of an obligately chemolithoautotrophic Halothiobacillus strain capable of growth on thiocyanate as an energy source. FEMS Microbiol. Lett. 2014, 354, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Zheng, H.; Chen, Y.; Xu, Y.; Chen, C.; Luo, D. Isolation, identification and characteristics of a sulfide-oxidizing bacterium in sediment of marine cage culture area. J. Fish. Res. 2016, 38, 431–436. (In Chinese) [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Schwengers, O.; Jelonek, L.; Dieckmann, M.A.; Beyvers, S.; Blom, J.; Goesmann, A. Bakta: Rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genom. 2021, 7, 000685. [Google Scholar] [CrossRef]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, 182–185. [Google Scholar] [CrossRef] [Green Version]
- Dieckmann, M.A.; Beyvers, S.; Nkouamedjo-Fankep, R.C.; Hanel, P.H.G.; Jelonek, L.; Blom, J.; Goesmann, A. EDGAR3.0: Comparative genomics and phylogenomics on a scalable infrastructure. Nucleic Acids Res. 2021, 49, W185–W192. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 12, 472–477. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinidis, K.T.; Rosselló-Móra, R.; Amann, R. Uncultivated microbes in need of their own taxonomy. ISME J. 2017, 11, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.S.; Guimarães, L.C.; Silva, A.; Soares, S.C.; Baraúna, R.A. First Steps in the analysis of prokaryotic pan-genomes. Bioinform. Biol. Insights 2020, 14, 1–9. [Google Scholar] [CrossRef]
- Moulana, A.; Anderson, R.E.; Fortunato, C.S.; Huber, J.A. Selection is a significant driver of gene gain and loss in the pangenome of the bacterial genus Sulfurovum in geographically distinct deep-sea hydrothermal vents. mSystems 2020, 5, e00673-19. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Jiang, L.; Hu, Q.; Cui, L.; Zhu, B.; Fu, X.; Lai, Q.; Shao, Z.; Yang, S. Characterization of Sulfurimonas hydrogeniphila sp. nov., a novel bacterium predominant in deep-sea hydrothermal vents and comparative genomic analyses of the genus Sulfurimonas. Front. Microbiol. 2021, 12, 626705. [Google Scholar] [CrossRef]
- Yeates, T.O.; Kerfeld, C.A.; Heinhorst, S.; Cannon, G.C.; Shively, J.M. Protein-based organelles in bacteria: Carboxysomes and related microcompartments. Nat. Rev. Microbiol. 2008, 6, 681–691. [Google Scholar] [CrossRef]
- Price, G.D.; Woodger, F.J.; Badger, M.R.; Howitt, S.M.; Tucker, L. Identification of a SulP-type bicarbonate transporter in marine cyanobacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 18228–18233. [Google Scholar] [CrossRef] [Green Version]
- Dobrinski, K.P.; Longo, D.L.; Scott, K.M. The carbon-concentrating mechanism of the hydrothermal vent chemolithoautotroph Thiomicrospira crunogena. J. Bacteriol. 2005, 187, 5761–5766. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.M.; Sievert, S.M.; Abril, F.N.; Ball, L.A.; Barrett, C.J.; Blake, R.A.; Boller, A.J.; Chain, P.S.G.; Clark, J.A.; Davis, C.R.; et al. The genome of deep-sea vent chemolithoautotroph Thiomicrospira crunogena XCL-2. PLoS Biol. 2006, 4, e383. [Google Scholar] [CrossRef]
- Houghton, J.L.; Foustoukos, D.I.; Flynn, T.M.; Vetriani, C.; Bradley, A.S.; Fike, D.A. Thiosulfate oxidation by Thiomicrospira thermophila: Metabolic flexibility in response to ambient geochemistry. Environ. Microbiol. 2016, 18, 3057–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, C.G.; Bardischewsky, F.; Rother, D.; Quentmeier, A.; Fischer, J. Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 2005, 8, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. The bioenergetic basis for the decrease in metabolic diversity at increasing salt concentrations: Implications for the functioning of salt lake ecosystems. Hydrobiologia 2001, 466, 61–72. [Google Scholar] [CrossRef]
- Chen, X. Identification and characterization of a sulfur-oxidizing bacterium B1-1 isolated from the sediment of marine cage culture area. Microbiol. China 2018, 45, 2082–2090. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Yin, W.; Wang, Y.; Liu, L.; He, J. Biofilms: The microbial “protective clothing” in extreme environments. Int. J. Mol. Sci. 2019, 20, 3423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellison, C.K.; Whitfield, G.B.; Brun, Y.V. Type IV Pili: Dynamic bacterial nanomachines. FEMS Microbiol. Rev. 2021, 46, fuab053. [Google Scholar] [CrossRef]
- Ghosh, W.; Dam, B. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 2009, 33, 999–1043. [Google Scholar] [CrossRef] [Green Version]
- Rameez, M.J.; Pyne, P.; Mandal, S.; Chatterjee, S.; Alam, M.; Bhattacharya, S.; Mondal, N.; Sarkar, J.; Ghosh, W. Two pathways for thiosulfate oxidation in the alphaproteobacterial chemolithotroph Paracoccus thiocyanatus SST. Microbiol. Res. 2020, 230, 126345. [Google Scholar] [CrossRef]
- Ortmann, A.C.; Suttle, C.A. High abundances of viruses in a deep-sea hydrothermal vent system indicates viral mediated microbial mortality. Deep. Res. Part I Oceanogr. Res. Pap. 2005, 52, 1515–1527. [Google Scholar] [CrossRef]
- Nigro, O.D.; Jungbluth, S.P.; Lin, H.-T.; Hsieh, C.-C.; Miranda, J.A.; Schvarcz, C.R.; Rappé, M.S. Viruses in the oceanic basement. MBio 2017, 8, e02129-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Morono, Y.; Inagaki, F.; Takai, K. An improved method for extracting viruses from sediment: Detection of far more viruses in the subseafloor than previously reported. Front. Microbiol. 2019, 10, 878. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCBI Organism Name | Guyparkeria hydrothermalis | Guyparkeria hydrothermalis | Guyparkeria halophila * | Halothiobacillaceae bacterium | Halothiobacillus sp.^ | Halothiobacillus sp. XI15 ^ | Halothiobacillus sp. WRN-7 | Guyparkeria sp. SCN-R1 ^ | Halothiobacillus sp. SB14A ^ |
|---|---|---|---|---|---|---|---|---|---|
| Strain | B1-1 | R3 | sp2 | SpSt-1134 | S21.Bin061 | XI15 | WRN-7 | SCN-R1 | SB14A |
| Habitat | Sediment in marine cage culture area, Fujian, China | Hydrothermal vent chimney, North Fiji Basin, Pacific Ocean | Sediment in cold seep | Hot spring sediment, California, USA | Lacustrine sediment, Tibet, China | Kebrit deep brine-seawater interface, Red Sea, Saudi Arabia | Saline-alkaline soil, Tian Jin City, China | Thiocyanate-degrading bioreactor, Eerbeek, Netherlands | Ocean sediment, Arabian Sea Oxygen Minimum Zone, India |
| Isolate? | Yes | Yes | Yes | No | No | No | No | Yes | Yes |
| NCBI BioSample ID | SAMN23673796 | SAMN23673797 | SAMN13381662 | SAMN09639045 | SAMN13520459 | SAMN04318430 | SAMN04419354 | SAMN10095268 | SAMN11475377 |
| NCBI Assembly ID | GCF_009734265.1 | GCA_011380105.1 | GCA_011389965.1 | GCF_001469965.1 | GCF_001641825.1 | GCF_003932495.1 | GCF_005096345.1 | ||
| GTDB species representative | - | - | yes | yes | yes | - | yes | yes | - |
| Number of scaffolds | 1 | 9 | 1 (complete genome) | 235 | 130 | 14 | 14 | 42 | 62 |
| Number of bases (nt) | 2392942 | 2433989 | 2594469 | 1188407 | 2016313 | 2291306 | 2291306 | 2406866 | 2428842 |
| Completeness (%) | 100 | 100 | 100 | 68.91 | 92.53 | 100 | 100 | 100 | 99.43 |
| Contamination (%) | 0 | 0 | 0 | 1.15 | 0 | 0 | 0 | 0 | 0 |
| GC content (%) | 65.2 | 66.5 | 66.2 | 52.4 | 63.9 | 66.2 | 66.2 | 64.7 | 66.5 |
| Number of rRNAs (5S, 16S, 23S) | 2, 2, 2 | 2, 2, 2 | 2, 2, 2 | 0 | 1, 1, 1 | 1, 1, 1 | 1, 1, 1 | 1, 1, 1 | 2, 1, 1 |
| Number of tRNAs | 47 | 46 | 50 | 14 | 40 | 45 | 45 | 49 | 44 |
| Number of coding sequences CDS (predicted by CheckM) | 2172 | 2142 | 2325 | 1222 | 1943 | 2081 | 2081 | 2180 | 2207 |
| Number of coding sequences CDS (predicted by PROKKA) | 2168 | 2141 | 2320 | 1033 | 1875 | 2067 | 2067 | 2170 | 2172 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau Vetter, M.C.Y.; Huang, B.; Fenske, L.; Blom, J. Metabolism of the Genus Guyparkeria Revealed by Pangenome Analysis. Microorganisms 2022, 10, 724. https://doi.org/10.3390/microorganisms10040724
Lau Vetter MCY, Huang B, Fenske L, Blom J. Metabolism of the Genus Guyparkeria Revealed by Pangenome Analysis. Microorganisms. 2022; 10(4):724. https://doi.org/10.3390/microorganisms10040724
Chicago/Turabian StyleLau Vetter, Maggie C. Y., Baowei Huang, Linda Fenske, and Jochen Blom. 2022. "Metabolism of the Genus Guyparkeria Revealed by Pangenome Analysis" Microorganisms 10, no. 4: 724. https://doi.org/10.3390/microorganisms10040724
APA StyleLau Vetter, M. C. Y., Huang, B., Fenske, L., & Blom, J. (2022). Metabolism of the Genus Guyparkeria Revealed by Pangenome Analysis. Microorganisms, 10(4), 724. https://doi.org/10.3390/microorganisms10040724