Abstract
Extraintestinal pathogenic Escherichia coli (ExPEC) isolates are a main cause of bloodstream infections. The aim of this study was to characterize 256 β-lactam–resistant, bacteremia-causing E. coli isolates collected from 12 healthcare institutions in Lithuania in 2014 and 2018. All isolates were identified as E. coli via MALDI-TOF MS and VITEK ®2. In addition, the isolates were analyzed for the presence of 29 resistance genes and 13 virulence genes, divided into phylogenetic groups (A, B1, B2, C, D, E, and F), and characterized using rep-PCR genotyping methods (BOX-PCR and (GTG)5-PCR). Analyzing the results of this study showed tetA-strB-sul2-TEM-NDM-strA-fosA-AIM-sul3-aadA-CTX-M-9 to be the most common resistance gene combination (67.2% of all isolates). Additionally, the most common virulence genes established were fimH (98.4% of all isolates), fyuA (91.8%), and traT (81.3%) and the most common gene combination was fuyA-fimH-iroN (58.6% of all isolates). Next, the isolates were separated into four phylogenetic groups: A, B1, B2, and F, where group A isolates were detected at a significantly higher frequency (79.3% of all isolates). Finally, a total of 235 genotyping profiles were established using rep-PCR methods, and all profiles were separated into fourteen genotypic clusters, with each cluster containing profiles with a variety of virulence and resistance genes not restricted to any specific cluster. The results of this study elucidate E. coli antimicrobial resistance patterns by highlighting the variability and diversity of resistance and virulence genes and providing phylogenetic classification, genetic profiling, and clustering data. These results may improve clinical control of multidrug-resistant infections in healthcare institutions and contribute to the prevention of potential outbreaks.
1. Introduction
Escherichia coli is an opportunistic bacteria, and the most frequently isolated organism in patients with bacteremia [1]. In some cases, E. coli infections result in death [2]. E. coli strains that induce extraintestinal diseases and survive in the bloodstream are classified as extraintestinal pathogenic E. coli (ExPEC) [3]. The most common extraintestinal site colonized by these bacteria is the urinary tract, which is a common source of bloodstream infections [4].
According to the current phylogenetic classification, E. coli isolates are divided into seven groups (A, B1, B2, C, D, E, and F) [5]. ExPEC strains generally belong to groups B2 and D, and the commensal strains that survive in the intestines generally belong to groups A or B1 [5].
E. coli is resistant to a large group of relevant antimicrobial agents, including aminoglycosides, polymyxins, and broad-spectrum β-lactams. Therefore, all known mechanisms of antibiotic resistance, including enzymatic degradation, altered targets, and active efflux, can be found in E. coli isolates. The genes associated with resistance can be located in chromosomes or plasmids. They can be transmitted between isolates or transferred to another Enterobacteriaceae species [6]. Moreover, ExPEC strains may have several virulence factors, such as host defense subverting mechanisms, iron acquisition systems, toxins and adhesion, all of which may be important factors in host infection. Furthermore, bacteria without virulence factor coding genes in the bacterial chromosome may acquire them by transmission from bacteria with virulence factor coding genes as a result of these genes usually being located on mobile genetic elements, such as pathogenicity island or plasmids [7]. Several virulence factors may facilitate bacterial cells to infect and colonize the host and have been associated with bloodstream infections [1]. However, predictions of initial severity based on the presence of bacterial virulence factors are not fully accurate [8].
E. coli is a major cause of bloodstream infections worldwide. According to previous studies [9], E. coli was classified as the most or second-most common pathogen associated with bloodstream infections in Australia, Canada, Denmark, Finland, Iceland, New Zealand, Sweden, and the USA. However, it is less widespread in Lithuania, as the third-most common cause of bacterial infection at healthcare institutions. Moreover, in Lithuania the number of multidrug-resistant bloodstream infections caused by E. coli is increasing annually [10]. Therefore, knowledge of virulence factors, antimicrobial resistance determinants, and genotype classifications of bacteremia-causing E. coli strains is important for determining the epidemiological profiles of bloodstream infections, which could be beneficial for establishing specific interventions.
The aim of this study was to characterize multidrug-resistant, bacteremia-causing E. coli isolates collected from different healthcare institutions in Lithuania in 2014 and 2018 and compare the mechanisms of spread and pathogenicity observed in 2014 with those observed in 2018.
2. Materials and Methods
2.1. Collection of Clinical E. coli Isolates, Identification, and Genomic DNA Extraction
A total of 256 β-lactam–resistant E. coli isolates were selected from samples collected in 2014 and 2018 from patients with bloodstream infections at 12 different healthcare institutions (I, II, III, IV, V, VI, VII, VIII, IX, X, XI, and XII) in Lithuania (Supplementary Table S1). All isolates were cultivated by hospitals on blood agar for 17–24 h and then sent to National Public Health Surveillance Laboratory for final antibiotic resistance confirmation and pure culture identification using a MALDI-TOF Biotyper (Bruker, Bollerica, MA, USA). Resistance to β-lactam antibiotics was confirmed using the disc diffusion method on Müller–Hinton agar and an automated VITEK ®2 system (bioMérieux, Marcy-l’Étoile, France). Antibiotic resistance data were evaluated using the EUCAST E. coli breakpoints, and commercial disks of the following antibiotics were used: ampicillin (10 µg), amoxicillin/clavulanic acid (20/10 µg), meropenem (10 µg), cefuroxime (30 µg), amikacin (30 µg), ampicillin/sulbactam (10/10 µg), cefotaxime (5 µg), piperacillin/tazobactam (30/6 µg), ceftazidime (10 µg), ciprofloxacin (5 µg), trimethoprim/sulfamethoxazole (1.25/23.75 µg, gentamicin (10 µg), and imipenem (10 µg). The colistin E-test was also used (BioMaxima, Lublin, Poland).
To extract genomic DNA, the isolates were cultivated on tryptic soy agar at 30 °C for 12 h. Genomic DNA was extracted using the GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol.
2.2. Characterization of E. coli Genes Associated with Antibiotic Resistance and Virulence
Genes associated with antibiotic resistance and virulence were detected after the collection and identification of E. coli isolates. A total of 29 antibiotic resistance genes and 13 bacterial virulence genes were identified via PCR, using primers reported in previously published studies (Table 1). The PCR reaction mixture had a total volume of 50 µL, which included DreamTaq Green PCR Master Mix (2X) (Thermo Fisher Scientific, Waltham, MA, USA), 0.25 μM of each primer, and 1 µL of E. coli genomic DNA. The PCR conditions were established according to previously published protocols. After PCR analysis, the amplification products were analyzed by 1% agarose gel electrophoresis.

Table 1.
Characterization of genes associated with antibiotic resistance and virulence.
2.3. Phylogenetic Classification
To assign E. coli isolates to one of the seven main phylogenetic groups, four marker genes (chuA, yjaA, TspE4C2, and arpA) and an additional gene (trpA) were targeted. Quadruplex PCR was performed as previously described [5,22]. Based on the PCR products, the strains were classified as lineage A, B1, B2, C, D, E, or F.
2.4. Genotyping of E. coli Isolates
To analyze the spread of multidrug-resistant E. coli clinical isolates in Lithuania, two genotyping methods were used. First, BOX-PCR genotyping was performed. Next, the results were evaluated to identify isolates belonging to the same strain. These isolates were then analyzed via (GTG)5-PCR. BOX-PCR and (GTG)5-PCR were performed according to Dombek et al. [23] and Versalovic et al. [24], respectively. The reaction mixture used for both PCR methods had a total volume of 50 μL, containing DreamTaq Green PCR Master Mix (2X) (Thermo Fisher Scientific, Waltham, MA, USA), 0.5 μM of primer (BOX-PCR: BOXA1R, 5′-CTA CGG CAA GGC GAC GCT GAC G-3′; (GTG)5: 5′-GTGGTGGTGGTGGTG-3′), and 1 µL of bacterial DNA. PCR conditions were established as described by Versalovic et al. [24]. The amplification products were analyzed by 1% agarose gel electrophoresis.
2.5. Data Analysis
Electrophoresis profiles of the BOX-PCR and (GTG)5-PCR products were determined using BioNumerics 8.0 software (Applied Maths, Sint-Martens-Latem, Belgium) and Tree Of Life (iTOL) v6, an online tool for phylogenetic tree preparation [25]. Next, dendrograms were constructed using the unweighted-pair group method with the arithmetic mean (UPGMA).
The χ2 test or Fisher’s exact test was used to compare categorical variables. Statistical significance was set at p < 0.05. Statistical analyses were performed using SPSS software (version 10.0; SPSS Inc., Chicago, IL, USA).
3. Results
3.1. Identification and Characterization of E. coli Antibiotic Resistance Genes
In 2014 and 2018, 256 clinical isolates of E. coli were collected from 12 different healthcare institutions in Lithuania. All isolates were identified as E. coli. Phenotypic resistance to antibiotics was determined according to EUCAST recommendations. All isolates were resistant to one or more third generation cephalosporins and/or one or more carbapenems. Next, the presence of genetic resistance determinants was confirmed in all isolates. The genetic resistance determinants were then divided into groups according to the six types of antibiotic resistance observed in clinical E. coli cases: β-lactam resistance, sulfonamide resistance, aminoglycoside resistance, tetracycline resistance, fosfomycin resistance, and polymyxin (colistin) resistance (Table 1). Screening of these gene groups showed that the most common resistance genes were tetA (100% of isolates), strB (99.2%), sul2 (99.2%), TEM (98.8%), NDM (98.4%), strA (98.4%), fosA (97.3%), AIM (97.3%), sul3 (92.6%), aadA (92.6%), and the CTX-M-9 group (90.6%). Four genes (TEM, the CTX-M-9 group, AIM, and NDM) cause resistance to β-lactams; three genes (aadA, strA, and strB), to aminoglycosides; two genes (sul2 and sul3), to sulfonamides; one gene (fosA), to fosfomycin; and one gene (tetA), to tetracyclines. Moreover, the most common resistance gene combination was tetA-strB-sul2-TEM-NDM-strA-fosA-AIM-sul3-aadA-CTX-M-9, detected in 67.2% of all isolates collected in 2014 and 2018.
Resistance genes CTX-M-8, CTX-M-25, SIM, DIM, and GIM were not found in any of the isolates. SHV and SPM were found only in three and two isolates, respectively, collected in 2018.
A comparison of the resistance genes detected in isolates from 2014 and 2018 showed that their frequencies changed over the five-year period. Three genes (KPC, VIM and aac3(IV)) were more common in 2018 than in 2014 (Table 2). Conversely, three genes (CTX-M-1, tetC, and mrc-1) were more common in 2014 than in 2018. The frequency of three genes (TEM, strA, and tetA) remained almost constant.
Table 2.
Distribution of different antibiotic resistance genes in 2014 and 2018.
3.2. Identification of Virulence Genes in E. coli
Virulence genes were analyzed in all isolates collected in 2014 and 2018. Thirteen virulence genes from four groups common to bloodstream-associated E. coli infections (toxins, serum resistance factors, iron uptake factors (siderophores), and adhesins) were tested (Table 3). The most common virulence genes were fimH (98.4% of all isolates), fyuA (91.8%), and traT (81.3%), which are responsible for type 1 fimbria adhesin activation, siderophore (yersiniabactin) activation, and bacterial serum resistance, respectively. The least common virulence gene was ibeA, which is responsible for activating adhesion group virulence factors. This gene was detected in only 17 isolates (6.6%, p < 0.05), which were mostly collected from healthcare institution II. Seven of these seventeen isolates were from 2014. The most common virulence gene combination was fuyA-fimH-iroN, found in 150 isolates (58.6%). fuyA and iroN encode siderophores, and fimH is responsible for bacterial adhesion to host cells.
Table 3.
Distribution of virulence genes at different healthcare institutions.
Next, the virulence gene analysis results from 2014 and 2018 were compared to determine how the virulence determinants changed over time. The distribution of the virulence genes changed over the five-year period; some virulence genes increased in frequency, and others decreased (p value < 0.05) (Table 3). The frequency of three virulence genes (hlyA, cnf1 and sfaD/E) increased from 2014 to 2018, whereas that of four genes (cvaC, ibeA, papC and afaB/C) decreased.
3.3. E. coli Phylogenetic Classification
Phylogenetic group determination was performed for all isolates. The isolates belonged to four phylogenetic groups: A, B1, B2, and F. Group A isolates were detected at a significantly higher frequency (79.3% of all isolates, p < 0.05) than isolates of groups B1, B2, and F (Table 4). Group B1 isolates were the least common, with statistical significance (0.8%, p < 0.05), and included isolates from 2014 only.
Table 4.
Phylogenetic groups detected in 2014 and 2018.
Comparison of the phylogenetic group distribution in 2014 and 2018 showed that groups B2 and F were more common in 2018, whereas group A was more common in 2014 (Table 4).
A dendrogram was constructed for the most common phylogenetic group, group A (Supplementary Figure S2). All group A isolates were separated into three groups according to the presence of phylogenetic genes: yjaA and arpA (only isolates 1598/2014 and 2142/2018) were detected in the first and second groups, respectively, whereas no genes were detected via quadruplex PCR in the third group. The most common virulence genes in phylogenetic group A were fimH (98.5% of phylogroup A isolates) and fuyA (89% of phylogroup A isolates). The most common virulence gene combination was fimH-fyuA-traT (73.4% of phylogroup A isolates). Moreover, the most common resistance genes were: tetA (100% of phylogroup A isolates), strB (99.5%), sul2 (99.5%), TEM (99%), and NDM (98.5%); the most common resistance gene combination was tetA-strB-sul2-TEM-NDM-strA-AIM-fosA-sul3-CTX-M-9-aadA-IMP (62.1% of phylogroup A isolates).
Moreover, in phylogenetic group B2 (Supplementary Figure S3), the most common virulence genes were fyuA (100% of phylogroup B2 isolates) and fimH (97.5% of phylogroup B2 isolates). The most common virulence gene combination was fyuA-fimH-traT-sat (60% of phylogroup B2 isolates). The most common resistance genes were fosA (100% of phylogroup B2 isolates), sul2 (100%), strA (100%), and tetA (100%); the most common resistance gene combination was fosA-sul2-strA-tetA-TEM-AIM-NDM-strB-aadA (85% of phylogroup B2 isolates).
All isolates of phylogenetic group F (Supplementary Figure S4) had the virulence genes fimH and traT. There was no significant difference among phylogroup F isolates with respect to antibiotic resistance.
3.4. rep-PCR Genotyping Results
The 256 tested isolates exhibited some genetic diversity, as revealed by rep-PCR methods. A total of 235 BOX-PCR profiles were established (Supplementary Figure S5). Dendrograms were constructed using UPGMA, and all profiles were separated into fourteen genotypic clusters. Cluster analysis showed that each cluster contained profiles with a variety of virulence and resistance genes; the presence of certain genes was not restricted to any specific cluster.
Fourteen genotypic clusters were detected. Cluster 9 had the most isolates (n = 52, 20.3% of all isolates), and cluster 5 had the fewest (n = 1, 0.4%). Most of the isolates in cluster 9, the dominant cluster, were collected in 2018 (39 isolates), with fewer isolates collected in 2014 (17 isolates). Only cluster 8 had more isolates from 2014 (18 isolates) than from 2018 (13 isolates). All clusters included isolates from both years of the study.
Cluster 1 lacked virulence genes afaB/C and ibeA, both of which are responsible for pathogenic E. coli adhesion and invasion of host cells. Clusters 5 and 8 lacked the antibiotic resistance genes CTX-M-2 (β-lactam resistance) and mrc-1 (colistin resistance), whereas cluster 7 lacked tetB (tetracycline resistance).
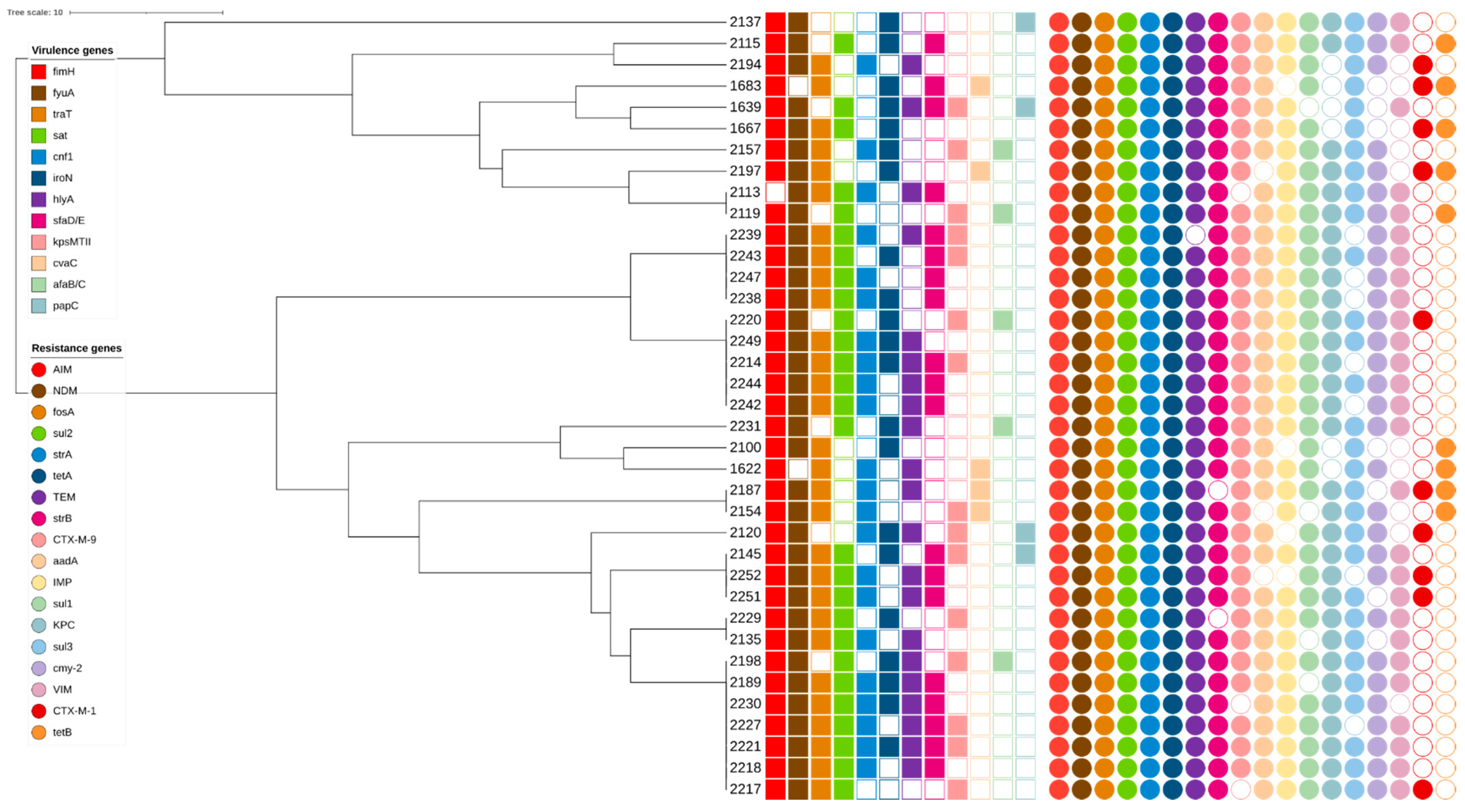
Seventeen BOX-PCR profiles each contained two or three isolates. These profiles were distributed among clusters 1, 9, 10, 12, 13 and 14. No profiles with more than one isolate were found in clusters 2 and 3. All isolates were collected from different healthcare institutions (I, II, III, IV, V, VI, VII, VIII, IX, X, XI, and XII). (GTG)5-PCR analysis indicated that all analyzed isolates were genetically similar; only one isolate, 2137 (hospital XII), was different (Figure 1). The isolates were classified into two phylogenetic groups: A (29 isolates) and B2 (8 isolates). All isolates had different virulence gene profiles (Figure 1). Virulence genes fimH and fyuA were observed in all analyzed isolates except 1683 and 2113, respectively. All isolates also had different resistance gene profiles and gene combinations. The genes aim, fosA, sul2, strA, and tetA, which cause resistance to different types of antibiotic groups, were found in all isolates.
Figure 1.
(GTG)5-PCR dendrogram.
4. Discussion
This study was designed to characterize multidrug-resistant, bacteremia-causing E. coli isolates collected from different healthcare institutions in Lithuania in 2014 and 2018. Several techniques were used to characterize the isolates, namely, resistance and virulence gene determination, phylogenetic group detection, and rep-PCR genotyping, which are important methods for determining inter- and intra-hospital spread.
The most common resistance gene was tetA, responsible for resistance to tetracyclines (observed in all isolates). Domínguez et al. [26] observed that tetA, which encodes an efflux mechanism, is the most common tetracycline resistance gene in E. coli in humans. The most common aminoglycoside resistance genes were aadA (92.6% of isolates), strA (98.4%), and strB (99.2%). These genes are responsible for the production of aminoglycoside-modifying enzymes (strA: adenyltransferase and phosphotransferases; strB; phosphotransferases), which leads to antibiotic deactivation by modification [27].
β-lactam resistance genes were also observed. The most common β-lactamase gene was TEM (98.8% of all isolates). In comparison, in our previous study [10], we observed a lower frequency of this gene (33.9%). CTX-M-9 was the most commonly detected CTX-M group gene (90.6% of all isolates). This result differs from those of previous studies in Lithuania and northern countries [28]; CTX-M-9 was not observed among Lithuanian clinical isolates, and the most common gene in the CTX-M group was CTX-M-15. This difference suggests that this gene emerged in Lithuania between 2012 (when the isolates analyzed by Sepp et al. were collected) and 2014 (when isolates were collected for the present study). Furthermore, we observed a high frequency of two additional metallo-β-lactamase genes in this study: NDM, which was previously detected in Lithuania [10], and AIM, which was not, were detected in 98.4% and 97.3% of all isolates, respectively.
One virulence mechanism responsible for the survival of E. coli strains in the bloodstream is the escape of recognition by the complement system through serum resistance [4]. Multiple virulence factors are involved in serum survival. In this study, 81.3% of isolates had the traT gene. Similar results were found in studies of E. coli bloodstream infections worldwide [29,30,31]. E. coli capsule research showed that almost half (49.2%) of all isolates expressed kpsMTII, which codes for K1 and K5 capsules. These results confirm previous research on bloodstream infections [32].
Siderophores represent another virulence mechanism by which pathogenic bacteria take up iron from host molecules. Siderophores are commonly associated with pathogenic E. coli strains isolated from bacteremia cases [29,31]. In this study, yersiniabactin (fyuA) was the most prevalent siderophore, present in 91.8% of the tested isolates. This fyuA gene product is involved in the efficient uptake of iron from the bloodstream [21] and the bacterial invasion of the bloodstream from the urinary tract [18]. Another siderophore, salmocherin (iroN), is less common (present in 62.5% of isolates) and is mostly associated with invasion of the bloodstream from the urinary tract [18].
Virulence genes related to adhesins were also investigated during this study, including type 1 fimbriae (fimH), S fimbria (staD/E), P fimbria (papC), and Dr binding adhesin (afaB/C). The most common gene was fimH (98.4%), which is a critical virulence factor of uropathogenic E. coli strains, facilitating adhesion to uroepithelial proteins [7]. Bloodstream infections usually occur as a complication of the urinary tract infections promoted by this factor [9].
The virulence genes related to toxin and hemolysin production observed in this study included sat, ibeA, cvaC, hlyA, and cnf1. The most common was sat (53.3% of all isolates), which encodes secreted autotransporter toxin. This toxin has proteolytic activity and influences the vacuolization of urinary epithelial cells [33]. As a result, it is mostly associated with bacteremic urinary tract infection. The second-most common toxin-encoding gene was hlyA (40.3% of all isolates), which encodes α-hemolysin, a pore-forming bacterial exotoxin that may contribute to the virulence of bacteria during bloodstream infections and sepsis [34].
According to the phylogenetic group classification of the E. coli isolates, group A was the most common (79.3%). In the literature, B2 was reported to be the most common phylogenetic group of pathogenic E. coli [4,29], and this was corroborated by the results obtained in previous studies in Lithuania [35,36]. However, in our study, the B2 phylogroup comprised only 15.6% of all isolates, mostly those collected in 2018 (10 isolates from 2014 and 30 isolates from 2018). The same phylogenetic group pattern was observed in Romania, where group A was the dominant group and group B2 was less prevalent [37]. According to that data, isolates of group A are usually strictly commensal strains found in the intestinal microbiota. Another study conducted by Fratamico et al. [38] reported that isolates of groups B2 and D have a higher virulence in humans, allowing them to induce extraintestinal infections in both healthy and immunocompromised hosts. However, an old classification system established by Clermont et al. [22] was used in that study, and there was no distinction between groups D, E, and F; all strains were classified as group D. Conversely, in our study, a newer classification system [5] was used, and group D isolates were absent. However, group F isolates comprised 4.3% of all isolates. They contained virulence genes fimH and traT, which are responsible for effective E. coli adhesion and host serum resistance, respectively.
Based on the BOX-PCR genotyping results, the isolates were divided into fourteen clusters. The most dominant cluster was cluster 9 (20.3%). Cluster analysis showed that all isolates were genetically very similar, with no significant differences associated with collection year or healthcare institution. Furthermore, the genetic profiles of isolates in each cluster contained a variety of virulence and resistance genes; the presence of certain genes was not restricted to any specific cluster. However, in cluster 9, isolates collected in 2018 were more prevalent than those collected in 2014. A possible explanation for these results could be the successful genetic adaptation of bacteria over time and the dissemination of resistance genes, which can occur through transmission via medical staff, contaminated equipment, or patients transferring between healthcare institutions [39]. These data are similar to our previously reported results indicating that clinical isolates of E. coli are spread via intra- and/or inter-hospital dissemination between Lithuanian healthcare institutions [10]. Moreover, 17 BOX-PCR profiles each contained two or three isolates. Further analysis via (GTG)5-PCR showed that all analyzed isolates were genetically very similar; only one isolate, from hospital XII (2137), had a significantly different genetic profile. Moreover, a detailed analysis of the 17 BOX-PCR profiles showed that two virulence genes, fimH and fyuA, were observed in almost all isolates. Resistance genes found in all isolates included AIM, fosA, sul2, strA, and tetA, which are responsible for different types of antibiotic resistance mechanisms.
In summary, the study results indicated that the multidrug resistance of bacteremia-causing E. coli is induced by various combinations of different genetic determinants. The variety of resistance and virulence genes can be explained by genetic plasticity determinants, such as plasmids and transposable elements, and other factors, such as inter- and intra-hospital transmission. Furthermore, 235 BOX-PCR profiles were established, and all isolates exhibited relatively similar genotyping results. However, more studies are needed to evaluate the intra- and inter-hospital transmission of E. coli isolates and analyze their adaptations to antibiotic strategies.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/microorganisms10020449/s1, Table S1: Collected isolates. Figure S1: Dendrogram of phylogenetic group A. Figure S2: Dendrogram of phylogenetic group B2. Figure S3: Dendrogram of phylogenetic group F. Figure S4: BOX-PCR dendrogram.
Author Contributions
Conceptualization, T.K. and N.K.; writing—original draft preparation, T.K. and N.K.; writing—review and editing, T.K.; visualization, T.K.; Supervision, N.K.; data curation, A.M. and I.R. All authors have read and agreed to the published version of the manuscript.
Funding
Study was funded by the PhD student scholarship of the Research Council of Lithuania (Funder: Research Council of Lithuania, Funding number: KD-17072).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mora-Rillo, M.; Fernández-Romero, N.; Navarro-San Francisco, C.; Díez-Sebastián, J.; Romero-Gómez, M.P.; Arnalich Fernández, F.; Arribas López, J.R.; Mingorance, J. Impact of virulence genes on sepsis severity and survival in Escherichia coli bacteremia. Virulence 2015, 6, 93–100. [Google Scholar] [CrossRef]
- Owrangi, B.; Masters, N.; Kuballa, A.; O’Dea, C.; Vollmerhausen, T.L.; Katouli, M. Invasion and translocation of uropathogenic Escherichia coli isolated from urosepsis and patients with community-acquired urinary tract infection. Eur. J. Clin. Microbiol. 2018, 37, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Russo, T.A.; Johnson, J.R. Medical and economic impact of extraintestinal infections due to Escherichia coli: Focus on an increasingly important endemic problem. Microbes Infect. 2003, 5, 449–456. [Google Scholar] [CrossRef]
- Micenková, L.; Beňová, A.; Frankovičová, L.; Bosák, J.; Vrba, M.; Ševčíková, A.; Kmeťová, M.; Šmajs, D. Human Escherichia coli isolates from hemocultures: Septicemia linked to urogenital tract infections is caused by isolates harboring more virulence genes than bacteraemia linked to other conditions. Int. J. Med. Microbiol. 2017, 307, 182–189. [Google Scholar] [CrossRef]
- Clermont, O.; Christenson, J.K.; Denamur, E.; Gordon, D.M. The Clermont Escherichia coli phylo-typing method revisited: Improvement of specificity and detection of new phylo-groups. Environ. Microbiol. Rep. 2013, 5, 58–65. [Google Scholar] [CrossRef]
- Blake, D.; Hillman, K.; Fenlon, D.; Low, J. Transfer of antibiotic resistance between commensal and pathogenic members of the Enterobacteriaceae under ileal conditions. J. Appl. Microbiol. 2003, 95, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Dale, A.P.; Woodford, N. Extra-intestinal pathogenic Escherichia coli (ExPEC): Disease, carriage and clones. J. Infect. 2015, 71, 615–626. [Google Scholar] [CrossRef] [Green Version]
- Jauréguy, F.; Carbonnelle, E.; Bonacorsi, S.; Clec’H, C.; Casassus, P.; Bingen, E.; Picard, B.; Nassif, X.; Lortholary, O. Host and bacterial determinants of initial severity and outcome of Escherichia coli sepsis. Clin. Microbiol. Infect. 2007, 13, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Laupland, K.B. Incidence of bloodstream infection: A review of population-based studies. Clin. Microbiol. Infect. 2013, 19, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Kirtikliene, T.; Naugzemys, D.; Steponkiene, A.; Bogdevic, R.; Vizuje, G.; Zvingila, D.; Kuisiene, N. Evaluation of the Inter- and Intrahospital Spread of Multidrug Resistant Gram-Negative Bacteria in Lithuanian Hospitals. Microb. Drug Resist. 2019, 25, 326–335. [Google Scholar] [CrossRef]
- Zhang, L.-P.; Xue, W.-C.; Meng, D.-Y. First report of New Delhi metallo-β-lactamase 5 (NDM-5)-producing Escherichia coli from blood cultures of three leukemia patients. Int. J. Infect. Dis. 2016, 42, 45–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alyamani, E.J.; Khiyami, A.M.; Booq, R.Y.; Majrashi, M.A.; Bahwerth, F.S.; Rechkina, E. The occurrence of ESBL-producing Escherichia coli carrying aminoglycoside resistance genes in urinary tract infections in Saudi Arabia. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, Q.; Du, Y.; Jiang, X.; Tang, J.; Wang, J.; Li, G.; Jiang, Y. Prevalence of plasmid-mediated AmpC β-lactamases in a Chinese university hospital from 2003 to 2005: First report of CMY-2-type AmpC β-lactamase resistance in China. J. Clin. Microbiol. 2008, 46, 1317–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirel, L.; Walsh, T.R.; Cuvillier, V.; Nordmann, P. Multiplex PCR for detection of acquired carbapenemase genes. Diagn. Microbiol. Infect. Dis. 2011, 70, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Benzerara, Y.; Al, Y.B.E.; Hommeril, B.; Genel, N.; Decré, D.; Rottman, M.; Arlet, G. Emergence of plasmid-mediated fosfomycin-resistance genes among Escherichia coli isolates, France. Emerg. Infect. Dis. 2017, 23, 1564–1567. [Google Scholar] [CrossRef] [Green Version]
- Boerlin, P.; Travis, R.; Gyles, C.L.; Reid-Smith, R.; Lim, N.J.H.; Nicholson, V.; McEwen, S.A.; Friendship, R.; Archambault, M. Antimicrobial resistance and virulence genes of Escherichia coli isolates from swine in Ontario. Appl. Environ. Microbiol. 2005, 71, 6753–6761. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Johnson, J.R.; Stell, A. Extended virulence genotypes of Escherichia coli strains from patients with urosepsis in relation to phylogeny and host compromise. J. Infect. Dis. 2000, 181, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Daigle, F.; Harel, J.; Fairbrother, J.M.; Lebel, P. Expression and detection of pap-, sfa-, and afa-encoded fimbrial adhesin systems among uropathogenic Escherichia coli. Can. J. Microbiol. 1994, 40, 286–291. [Google Scholar] [CrossRef]
- Yamamoto, S.; Terai, A.; Yuri, K.; Kurazono, H.; Takeda, Y.; Yoshida, O. Detection of urovirulence factors in Escherichia coli by multiplex polymerase chain reaction. FEMS Immunol. Med. Microbiol. 1995, 12, 85–90. [Google Scholar] [CrossRef]
- Ananias, M.; Yano, T. Serogroups and virulence genotypes of Escherichia coli isolated from patients with sepsis. Braz. J. Med. Biol. Res. 2008, 41, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Clermont, O.; Bonacorsi, S.; Bingen, E. Rapid and simple determination of the Escherichia coli phylogenetic group. Appl. Environ. Microbiol. 2000, 66, 4555–4558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombek, P.E.; Johnson, L.K.; Zimmerley, S.T.; Sadowsky, M.J. Use of repetitive DNA sequences and the PCR to differentiate Escherichia coli isolates from human and animal sources. Appl. Environ. Microbiol. 2000, 66, 2572–2577. [Google Scholar] [CrossRef] [Green Version]
- Versalovic, J.; Schneider, M.; De Bruijn, F.J.; Lupski, J.R. Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Methods Mol. Cell. Biol. 1994, 5, 25–40. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Domínguez, E.; Zarazaga, M.; Sáenz, Y.; Briñas, L.; Torres, C. Mechanisms of antibiotic resistance in Escherichia coli isolates obtained from healthy children in Spain. Microb. Drug Resist. 2002, 8, 321–327. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updates 2010, 13, 151–171. [Google Scholar] [CrossRef] [Green Version]
- Sepp, E.; Andreson, R.; Balode, A.; Bilozor, A.; Brauer, A.; Egorova, S.; Huik, K.; Ivanova, M.; Kaftyreva, L.; Kõljalg, S.; et al. Phenotypic and molecular epidemiology of ESBL-, AmpC-, and carbapenemase-producing Escherichia coli in Northern and Eastern Europe. Front. Microbiol. 2019, 10, 2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, V.L.; Tomazetto, G.; Cyoia, P.S.; Neves, M.S.; Vidotto, M.C.; Nakazato, G.; Kobayashi, R.K.T. Molecular screening of virulence genes in extraintestinal pathogenic Escherichia coli isolated from human blood culture in Brazil. BioMed Res. Int. 2014, 2014, 465054. [Google Scholar] [CrossRef] [Green Version]
- Miajlovic, H.; Mac Aogáin, M.; Collins, C.J.; Rogers, T.; Smith, S. Characterization of Escherichia coli bloodstream isolates associated with mortality. J. Med. Microbiol. 2016, 65, 71–79. [Google Scholar] [CrossRef]
- Bozcal, E.; Eldem, V.; Aydemir, S.; Skurnik, M. The relationship between phylogenetic classification, virulence and antibiotic resistance of extraintestinal pathogenic Escherichia coli in İzmir province, Turkey. PeerJ 2018, 6, e5470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daga, A.P.; Koga, V.L.; Soncini, J.G.M.; De Matos, C.M.; Perugini, M.R.E.; Pelisson, M.; Kobayashi, R.; Vespero, E.C. Escherichia coli bloodstream infections in patients at a university hospital: Virulence factors and clinical characteristics. Front. Cell. Infect. Microbiol. 2019, 9, 191. [Google Scholar] [CrossRef]
- Sarowska, J.; Futoma-Koloch, B.; Jama-Kmiecik, A.; Frej-Madrzak, M.; Ksiazczyk, M.; Bugla-Ploskonska, G.; Choroszy-Krol, I. Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: Recent reports. Gut Pathog. 2019, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnen, A.F.-P.; Henneke, P. Role of pore-forming toxins in neonatal sepsis. Clin. Dev. Immunol. 2013, 2013, 608456. [Google Scholar] [CrossRef]
- Giedraitienė, A.; Vitkauskienė, A.; Pavilonis, A.; Patamsytė, V.; Genel, N.; Decre, D.; Arlet, G. Prevalence of O25b-ST131 clone among Escherichia coli strains producing CTX-M-15, CTX-M-14 and CTX-M-92 β-lactamases. Infect. Dis. 2017, 49, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Prakapaite, R.; Saab, F.; Planciuniene, R.; Petraitis, V.; Walsh, T.J.; Petraitiene, R.; Semoskaite, R.; Baneviciene, R.; Kalėdienė, L.; Kavaliauskas, P. Molecular characterization of uropathogenic Escherichia coli reveals emergence of drug resistant O15, O22 and O25 serogroups. Medicina 2019, 55, 733. [Google Scholar] [CrossRef] [Green Version]
- Usein, C.-R.; Papagheorghe, R.; Oprea, M.; Condei, M.; Strãuţ, M. Molecular characterization of bacteremic Escherichia coli isolates in Romania. Folia Microbiol. 2015, 61, 221–226. [Google Scholar] [CrossRef]
- Fratamico, P.M.; DebRoy, C.; Liu, Y.; Needleman, D.S.; Baranzoni, G.M.; Feng, P. Advances in molecular serotyping and subtyping of Escherichia coli. Front. Microbiol. 2016, 7, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurieva, T.; Dautzenberg, M.J.D.; Gniadkowski, M.; Derde, L.P.G.; Bonten, M.J.M.; Bootsma, M.C.J. The transmissibility of antibiotic-resistant Enterobacteriaceae in intensive care units. Clin. Infect. Dis. 2017, 66, 489–493. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).