
Clinical Metagenomics Is Increasingly Accurate and Affordable to Detect Enteric Bacterial Pathogens in Stool

 ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
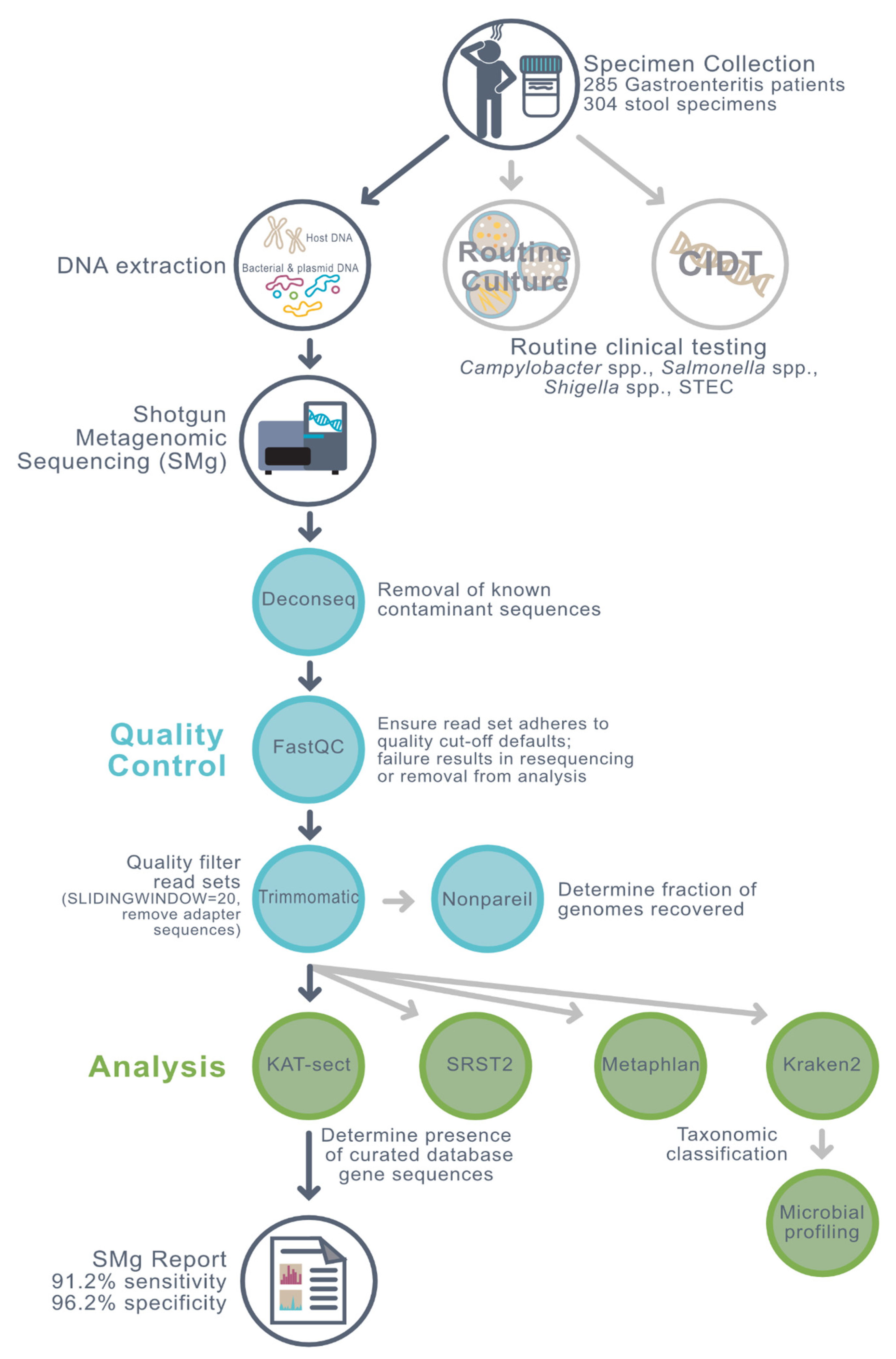
2. Materials and Methods
3. Results and Discussion
3.1. Samples and Conventional Results
3.1.1. Sample Provenance
3.1.2. Clinical Accuracy of PCR Compared to Culture
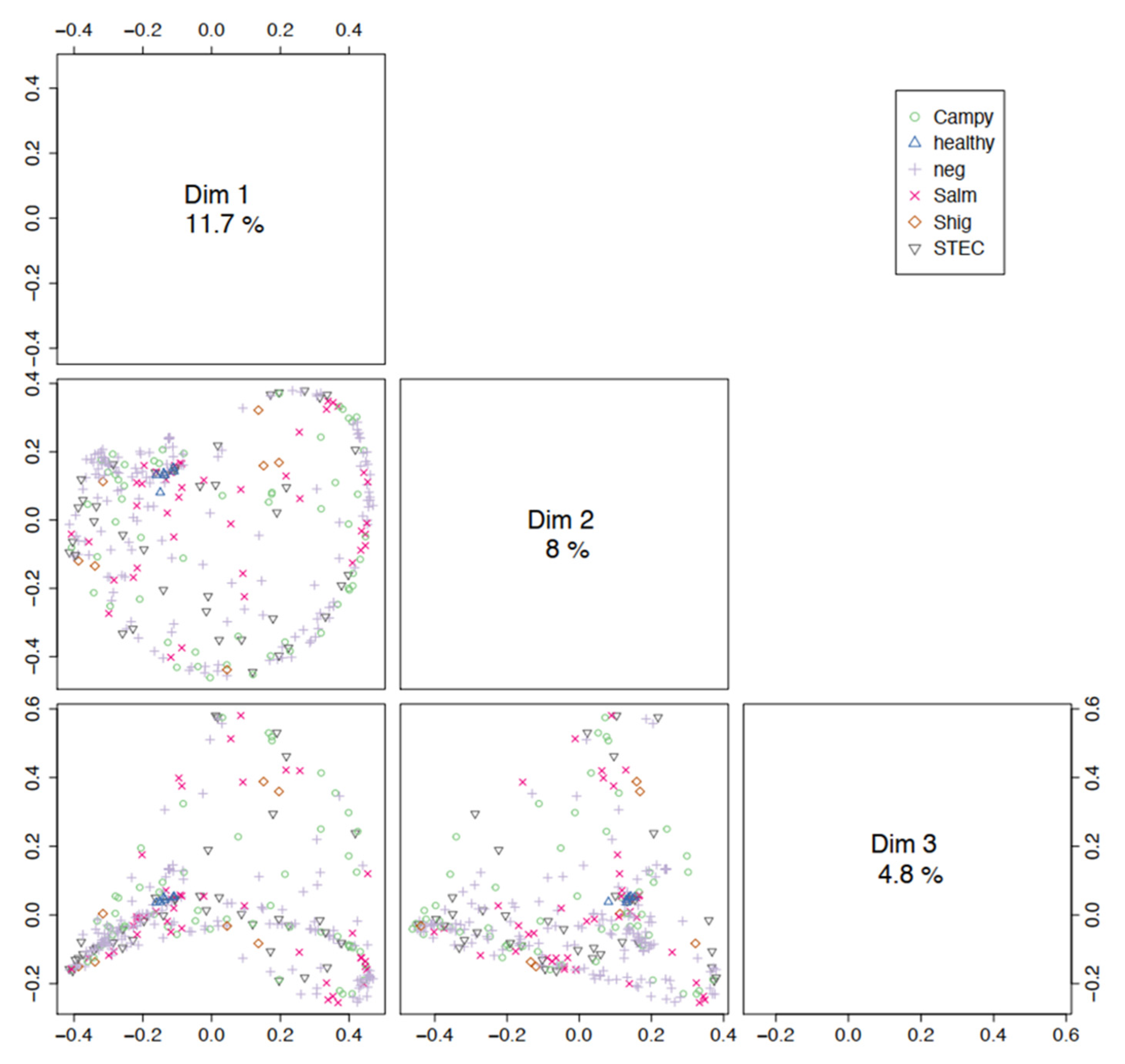
3.2. Microbial Community Profiling
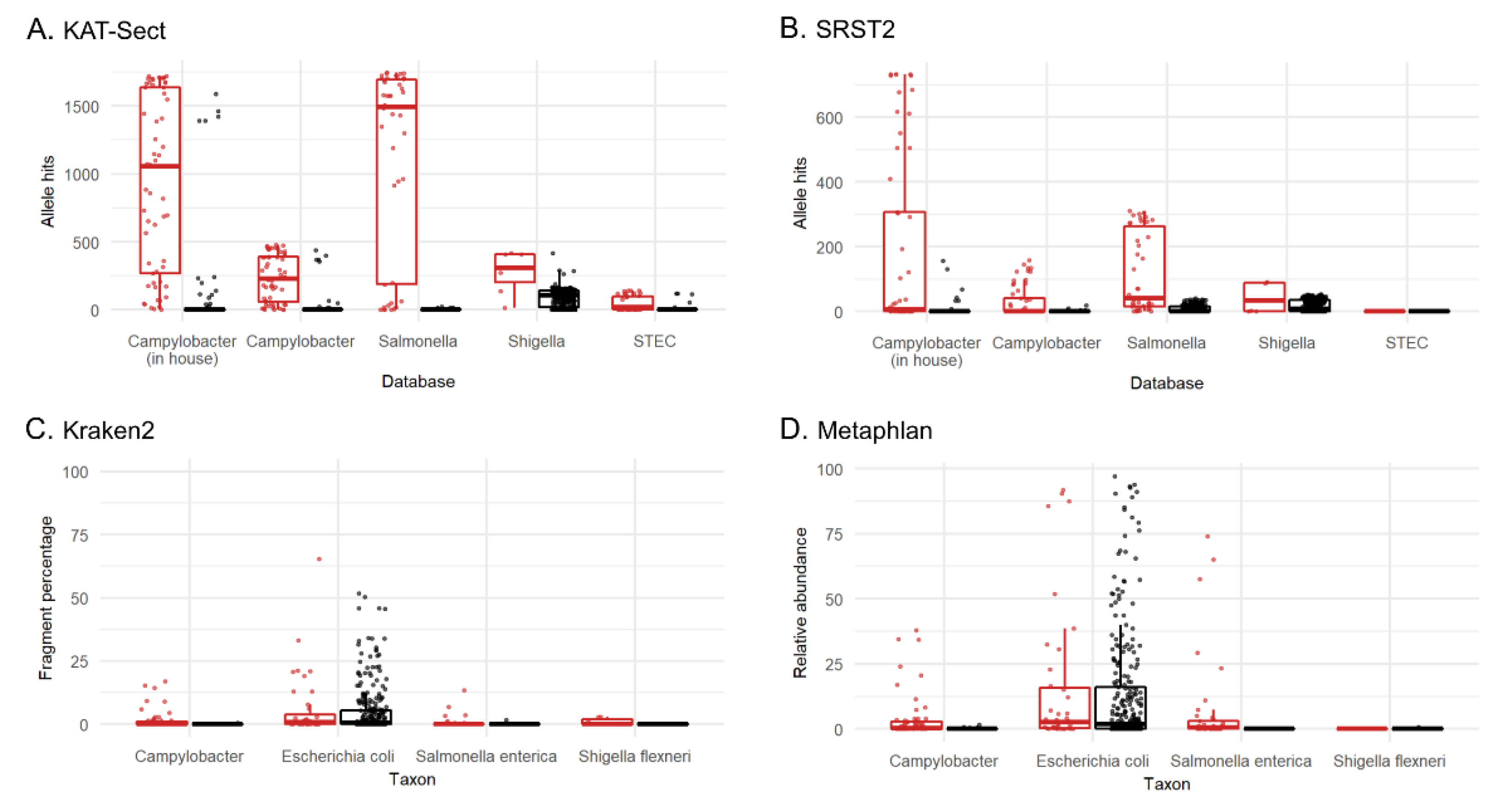
3.3. Pathogen Detection Using Metagenomics
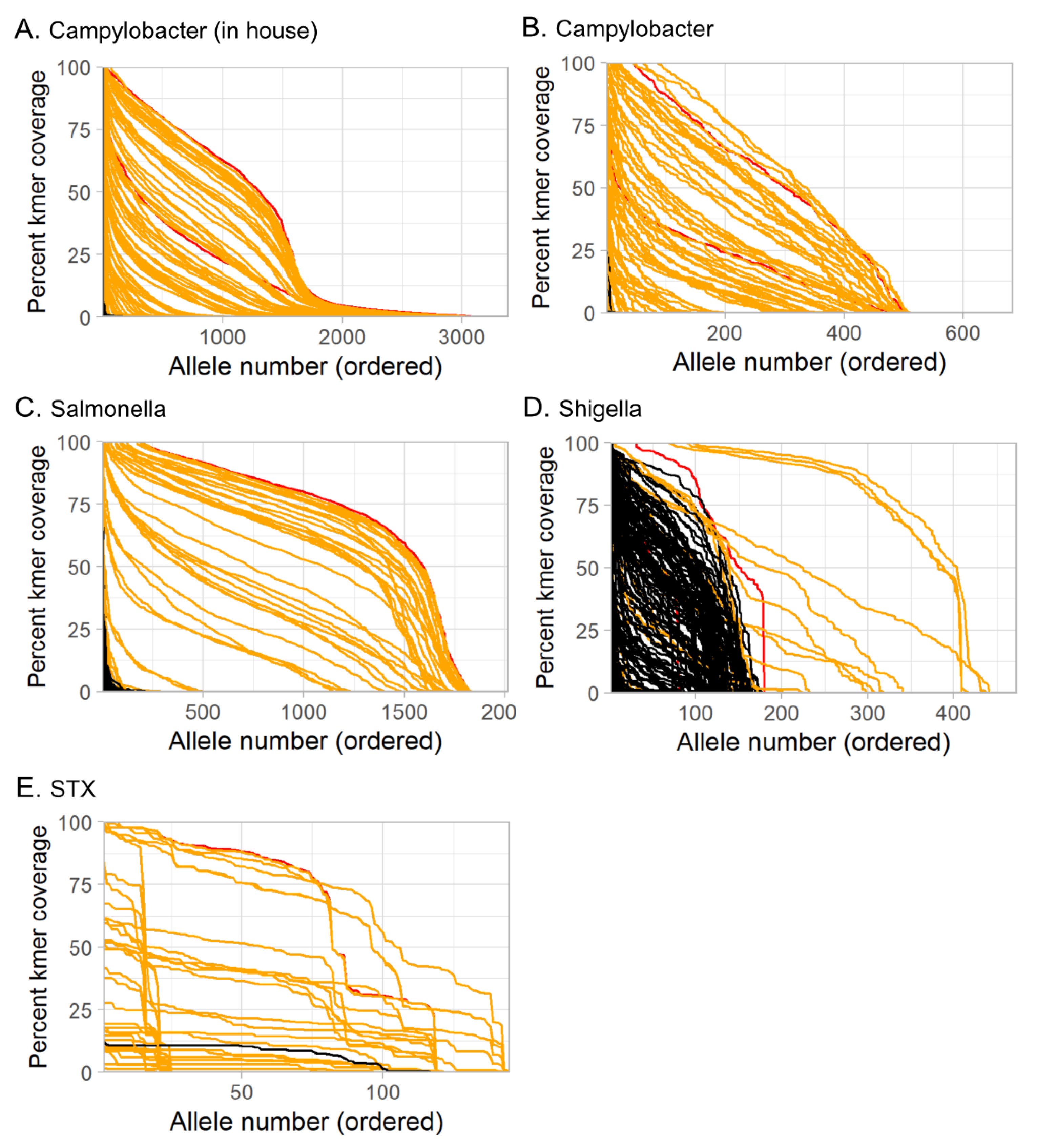
3.3.1. Tool and Method Comparison
3.3.2. Training Set
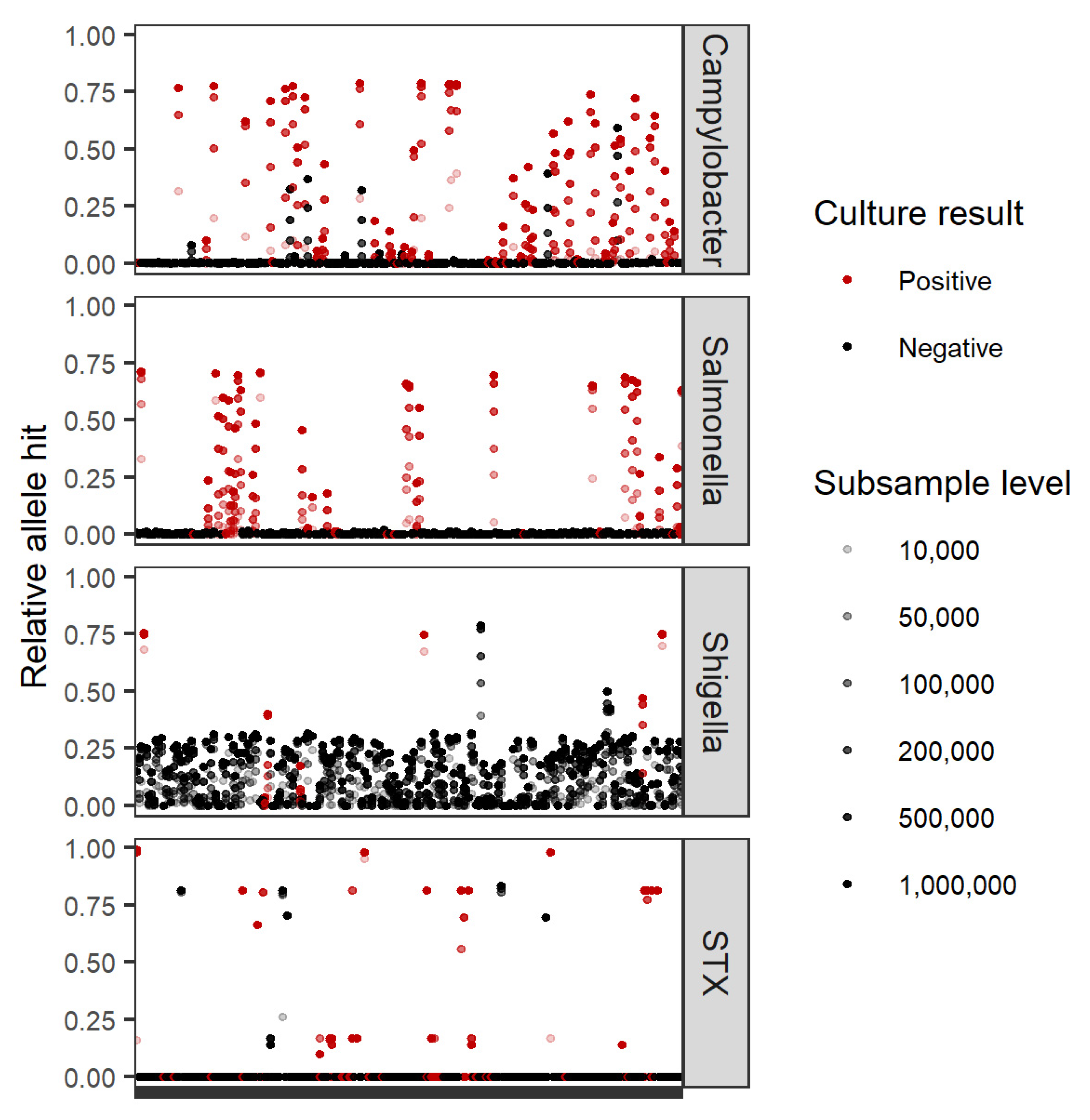
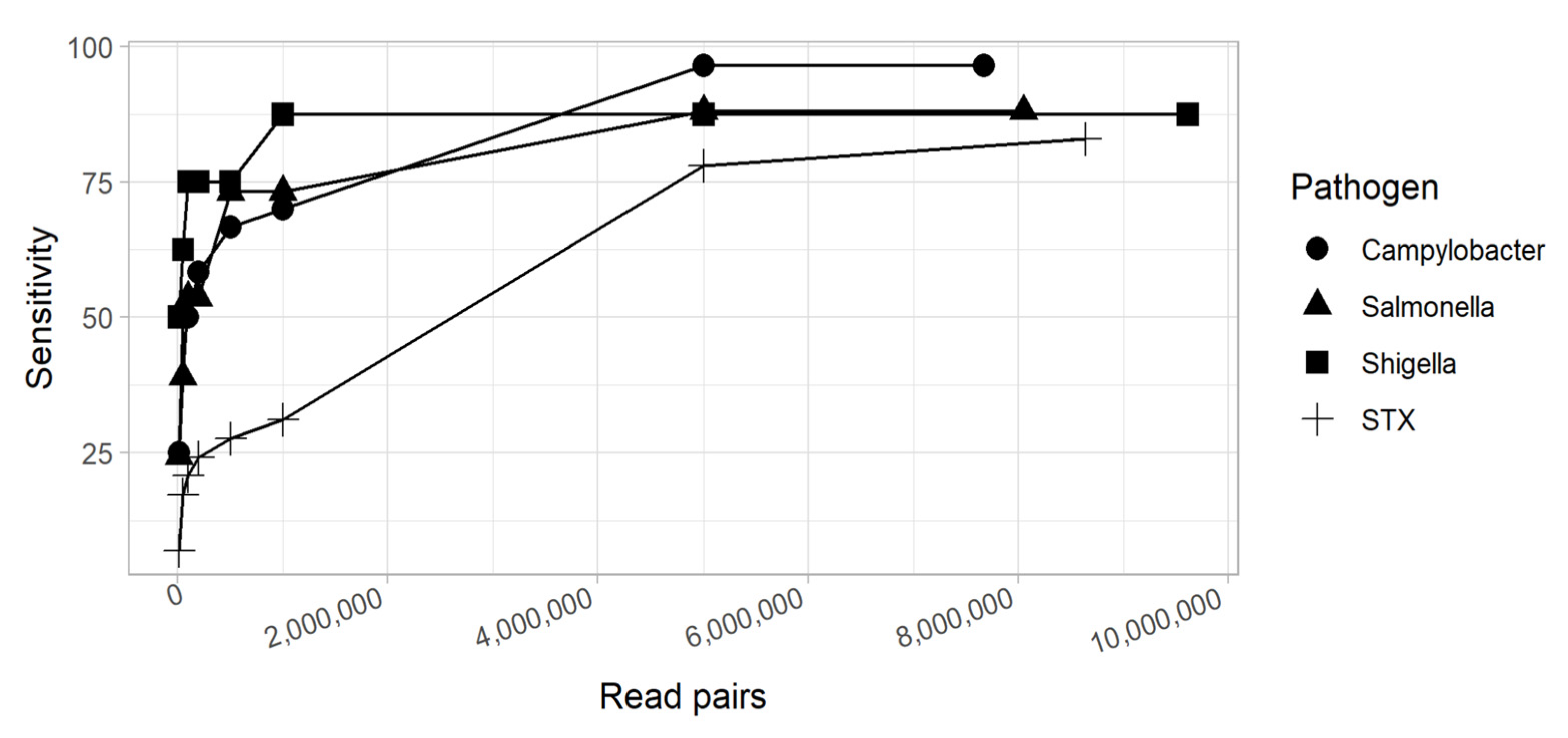
3.3.3. Sensitivity and Specificity of Metagenomics
3.3.4. Factors Affecting Sensitivity of Metagenomics
3.3.5. Factors Affecting Specificity of Metagenomics
3.3.6. Limit of Detection
3.4. Practical Considerations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vernacchio, L.; Vezina, R.M.; Mitchell, A.A.; Lesko, S.M.; Plaut, A.G.; Acheson, D.W.K. Diarrhea in American Infants and Young Children in the Community Setting: Incidence, Clinical Presentation and Microbiology. Pediatr. Infect. Dis. J. 2006, 25, 2–7. [Google Scholar] [CrossRef]
- Berenger, B.; Chui, L.; Reimer, A.R.; Allen, V.; Alexander, D.; Domingo, M.-C.; Haldane, D.; Hoang, L.; Levett, P.; MacKeen, A.; et al. Canadian Public Health Laboratory Network Position Statement: Non-Culture Based Diagnostics for Gastroenteritis and Implications for Public Health Investigations. Can. Commun. Dis. Rep. Releve Mal. Transm. Au Can. 2017, 43, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Carleton, H.A.; Besser, J.; Williams-Newkirk, A.J.; Huang, A.; Trees, E.; Gerner-Smidt, P. Metagenomic Approaches for Public Health Surveillance of Foodborne Infections: Opportunities and Challenges. Foodborne Pathog. Dis. 2019, 16, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Knox, N.C.; Ronholm, J.; Pagotto, F.; Reimer, A. Metagenomics: The Next Culture-Independent Game Changer. Front. Microbiol. 2017, 8, 1069. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.Y.; Miller, S.A. Clinical Metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Miller, S.; Chiu, C.; Rodino, K.G.; Miller, M.B. Point-Counterpoint: Should We Be Performing Metagenomic Next-Generation Sequencing for Infectious Disease Diagnosis in the Clinical Laboratory? J. Clin. Microbiol. 2020, 58, e01739-19. [Google Scholar] [CrossRef] [Green Version]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-Generation Sequencing in the Clinical Laboratory: Exploring the Advantages and Challenges of Nanopore Sequencing. J. Clin. Microbiol. 2019, 58, e01315-19. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.; Holmes, N.; Clarke, T.; Munro, R.; Debebe, B.J.; Loose, M. Readfish Enables Targeted Nanopore Sequencing of Gigabase-Sized Genomes. Nat. Biotechnol. 2021, 39, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Mughini-Gras, L.; Kooh, P.; Fravalo, P.; Augustin, J.-C.; Guillier, L.; David, J.; Thébault, A.; Carlin, F.; Leclercq, A.; Jourdan-Da-Silva, N.; et al. Critical Orientation in the Jungle of Currently Available Methods and Types of Data for Source Attribution of Foodborne Diseases. Front. Microbiol. 2019, 10, 2578. [Google Scholar] [CrossRef]
- Joensen, K.G.; Engsbro, A.L.Ø.; Lukjancenko, O.; Kaas, R.S.; Lund, O.; Westh, H.; Aarestrup, F.M. Evaluating Next-Generation Sequencing for Direct Clinical Diagnostics in Diarrhoeal Disease. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2017, 36, 1325–1338. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Maeda, N.; Miron, I.M.; Yoh, M.; Izutsu, K.; Kataoka, C.; Honda, T.; Yasunaga, T.; Nakaya, T.; Kawai, J.; et al. Metagenomic Diagnosis of Bacterial Infections. Emerg. Infect. Dis. 2008, 14, 1784–1786. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, P.H.H.; Becker, S.L.; Pothier, J.F.; Duffy, B.; N’Goran, E.K.; Beuret, C.; Frey, J.E.; Utzinger, J. Metagenomic Diagnostics for the Simultaneous Detection of Multiple Pathogens in Human Stool Specimens from Côte d’Ivoire: A Proof-of-Concept Study. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 40, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wylie, K.M.; El Feghaly, R.E.; Mihindukulasuriya, K.A.; Elward, A.; Haslam, D.B.; Storch, G.A.; Weinstock, G.M. Metagenomic Approach for Identification of the Pathogens Associated with Diarrhea in Stool Specimens. J. Clin. Microbiol. 2016, 54, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Saltykova, A.; Buytaers, F.E.; Denayer, S.; Verhaegen, B.; Piérard, D.; Roosens, N.H.C.; Marchal, K.; De Keersmaecker, S.C.J. Strain-Level Metagenomic Data Analysis of Enriched In Vitro and In Silico Spiked Food Samples: Paving the Way towards a Culture-Free Foodborne Outbreak Investigation Using STEC as a Case Study. Int. J. Mol. Sci. 2020, 21, 5688. [Google Scholar] [CrossRef]
- Loman, N.J.; Constantinidou, C.; Christner, M.; Rohde, H.; Chan, J.Z.-M.; Quick, J.; Weir, J.C.; Quince, C.; Smith, G.P.; Betley, J.R.; et al. A Culture-Independent Sequence-Based Metagenomics Approach to the Investigation of an Outbreak of Shiga-Toxigenic Escherichia Coli O104:H4. JAMA 2013, 309, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Gigliucci, F.; von Meijenfeldt, F.A.B.; Knijn, A.; Michelacci, V.; Scavia, G.; Minelli, F.; Dutilh, B.E.; Ahmad, H.M.; Raangs, G.C.; Friedrich, A.W.; et al. Metagenomic Characterization of the Human Intestinal Microbiota in Fecal Samples from STEC-Infected Patients. Front. Cell. Infect. Microbiol. 2018, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.D.; Luo, C.; Pena-Gonzalez, A.; Weigand, M.R.; Tarr, C.L.; Konstantinidis, K.T. Metagenomics of Two Severe Foodborne Outbreaks Provides Diagnostic Signatures and Signs of Coinfection Not Attainable by Traditional Methods. Appl. Environ. Microbiol. 2017, 83, e02577-16. [Google Scholar] [CrossRef] [Green Version]
- Finkbeiner, S.R.; Li, Y.; Ruone, S.; Conrardy, C.; Gregoricus, N.; Toney, D.; Virgin, H.W.; Anderson, L.J.; Vinjé, J.; Wang, D.; et al. Identification of a Novel Astrovirus (Astrovirus VA1) Associated with an Outbreak of Acute Gastroenteritis. J. Virol. 2009, 83, 10836–10839. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.S.; Harrach, B.; Ganac, R.D.; Gozum, M.M.A.; Dela Cruz, W.P.; Riedel, B.; Pan, C.; Delwart, E.L.; Schnurr, D.P. New Adenovirus Species Found in a Patient Presenting with Gastroenteritis. J. Virol. 2007, 81, 5978–5984. [Google Scholar] [CrossRef] [Green Version]
- Smits, S.L.; Schapendonk, C.M.E.; van Beek, J.; Vennema, H.; Schürch, A.C.; Schipper, D.; Bodewes, R.; Haagmans, B.L.; Osterhaus, A.D.M.E.; Koopmans, M.P. New Viruses in Idiopathic Human Diarrhea Cases, the Netherlands. Emerg. Infect. Dis. 2014, 20, 1218–1222. [Google Scholar] [CrossRef] [Green Version]
- Greninger, A.L. The Challenge of Diagnostic Metagenomics. Expert Rev. Mol. Diagn. 2018, 18, 605–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negida, A.; Fahim, N.K.; Negida, Y. Sample Size Calculation Guide—Part 4: How to Calculate the Sample Size for a Diagnostic Test Accuracy Study Based on Sensitivity, Specificity, and the Area Under the ROC Curve. Adv. J. Emerg. Med. 2019, 3, e33. [Google Scholar] [CrossRef] [PubMed]
- Charalampous, T.; Kay, G.L.; Richardson, H.; Aydin, A.; Baldan, R.; Jeanes, C.; Rae, D.; Grundy, S.; Turner, D.J.; Wain, J.; et al. Nanopore Metagenomics Enables Rapid Clinical Diagnosis of Bacterial Lower Respiratory Infection. Nat. Biotechnol. 2019, 37, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Raymond, F.; Ouameur, A.A.; Déraspe, M.; Iqbal, N.; Gingras, H.; Dridi, B.; Leprohon, P.; Plante, P.-L.; Giroux, R.; Bérubé, È.; et al. The Initial State of the Human Gut Microbiome Determines Its Reshaping by Antibiotics. ISME J. 2016, 10, 707–720. [Google Scholar] [CrossRef]
- Blankenberg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A. Galaxy Team Manipulation of FASTQ Data with Galaxy. Bioinforma. Oxf. Engl. 2010, 26, 1783–1785. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R. Fast Identification and Removal of Sequence Contamination from Genomic and Metagenomic Datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-R, L.M.; Gunturu, S.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. Nonpareil 3: Fast Estimation of Metagenomic Coverage and Sequence Diversity. mSystems 2018, 3, e00039-18. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating Species Abundance in Metagenomics Data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential Abundance Analysis for Microbial Marker-Gene Surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The R Project for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 30 December 2021).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Community Ecology Package; Vegan: London, UK, 2020. [Google Scholar]
- Arbizu, P.M. PairwiseAdonis; Pairwise Multilevel Comparison Using Adonis. 2021. Available online: https://github.com/pmartinezarbizu/pairwiseAdonis (accessed on 30 December 2021).
- Chen, J.C.-Y.; Tyler, A.D. Systematic Evaluation of Supervised Machine Learning for Sample Origin Prediction Using Metagenomic Sequencing Data. Biol. Direct 2020, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for Enhanced Metagenomic Taxonomic Profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Inouye, M.; Dashnow, H.; Raven, L.-A.; Schultz, M.B.; Pope, B.J.; Tomita, T.; Zobel, J.; Holt, K.E. SRST2: Rapid Genomic Surveillance for Public Health and Hospital Microbiology Labs. Genome Med. 2014, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Mapleson, D.; Garcia Accinelli, G.; Kettleborough, G.; Wright, J.; Clavijo, B.J. KAT: A K-Mer Analysis Toolkit to Quality Control NGS Datasets and Genome Assemblies. Bioinforma. Oxf. Engl. 2017, 33, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Naccache, S.N.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory Validation of a Clinical Metagenomic Sequencing Assay for Pathogen Detection in Cerebrospinal Fluid. Genome Res. 2019, 29, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Here: A Simpler Way to Find Your Files. Available online: https://CRAN.R-project.org/package=here (accessed on 30 December 2021).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open Source Software 2019, 4, 1686. [Google Scholar] [CrossRef]
- Oliver, J.D. The Viable but Nonculturable State in Bacteria. J. Microbiol. 2005, 43, 93–100. [Google Scholar]
- Wilson, M.R.; Naccache, S.N.; Samayoa, E.; Biagtan, M.; Bashir, H.; Yu, G.; Salamat, S.M.; Somasekar, S.; Federman, S.; Miller, S.; et al. Actionable Diagnosis of Neuroleptospirosis by Next-Generation Sequencing. N. Engl. J. Med. 2014, 370, 2408–2417. [Google Scholar] [CrossRef] [Green Version]
- Naccache, S.N.; Peggs, K.S.; Mattes, F.M.; Phadke, R.; Garson, J.A.; Grant, P.; Samayoa, E.; Federman, S.; Miller, S.; Lunn, M.P.; et al. Diagnosis of Neuroinvasive Astrovirus Infection in an Immunocompromised Adult with Encephalitis by Unbiased Next-Generation Sequencing. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2015, 60, 919–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, A.M.; Walk, S.T.; Taube, S.; Taniuchi, M.; Houpt, E.R.; Wobus, C.E.; Young, V.B. Disruption of the Human Gut Microbiota Following Norovirus Infection. PLoS ONE 2012, 7, e48224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Gonzalez, A.; Soto-Girón, M.J.; Smith, S.; Sistrunk, J.; Montero, L.; Páez, M.; Ortega, E.; Hatt, J.K.; Cevallos, W.; Trueba, G.; et al. Metagenomic Signatures of Gut Infections Caused by Different Escherichia Coli Pathotypes. Appl. Environ. Microbiol. 2019, 85, e01820-19. [Google Scholar] [CrossRef] [PubMed]
- Peabody, M.A.; Van Rossum, T.; Lo, R.; Brinkman, F.S.L. Evaluation of Shotgun Metagenomics Sequence Classification Methods Using in Silico and in Vitro Simulated Communities. BMC Bioinform. 2015, 16, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piralla, A.; Lunghi, G.; Ardissino, G.; Girello, A.; Premoli, M.; Bava, E.; Arghittu, M.; Colombo, M.R.; Cognetto, A.; Bono, P.; et al. FilmArrayTM GI Panel Performance for the Diagnosis of Acute Gastroenteritis or Hemorragic Diarrhea. BMC Microbiol. 2017, 17, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-R, L.M.; Konstantinidis, K.T. Nonpareil: A Redundancy-Based Approach to Assess the Level of Coverage in Metagenomic Datasets. Bioinforma. Oxf. Engl. 2014, 30, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, S.-H.; Kuang, Y.-S.; He, J.-R.; Lu, J.-H.; Luo, B.-J.; Jiang, F.-J.; Liu, Y.-Z.; Papasian, C.J.; Xia, H.-M.; et al. Effect of Short-Term Room Temperature Storage on the Microbial Community in Infant Fecal Samples. Sci. Rep. 2016, 6, 26648. [Google Scholar] [CrossRef]
- Doster, E.; Rovira, P.; Noyes, N.R.; Burgess, B.A.; Yang, X.; Weinroth, M.D.; Linke, L.; Magnuson, R.; Boucher, C.; Belk, K.E.; et al. A Cautionary Report for Pathogen Identification Using Shotgun Metagenomics; A Comparison to Aerobic Culture and Polymerase Chain Reaction for Salmonella enterica Identification. Front. Microbiol. 2019, 10, 2499. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Ford, L.; Glass, K.; Williamson, D.A.; Sintchenko, V.; Robson, J.M.B.; Lancsar, E.; Stafford, R.; Kirk, M.D. Cost of Whole Genome Sequencing for Non-Typhoidal Salmonella enterica. PLoS ONE 2021, 16, e0248561. [Google Scholar] [CrossRef]
- Armstrong, G.L.; MacCannell, D.R.; Taylor, J.; Carleton, H.A.; Neuhaus, E.B.; Bradbury, R.S.; Posey, J.E.; Gwinn, M. Pathogen Genomics in Public Health. N. Engl. J. Med. 2019, 381, 2569–2580. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Deng, X.; Lee, M.; Sucu, Y.D.; Arevalo, S.; Stryke, D.; Federman, S.; Gopez, A.; Reyes, K.; Zorn, K.; et al. Rapid Pathogen Detection by Metagenomic Next-Generation Sequencing of Infected Body Fluids. Nat. Med. 2021, 27, 115–124. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterson, C.-L.; Alexander, D.; Chen, J.C.-Y.; Adam, H.; Walker, M.; Ali, J.; Forbes, J.; Taboada, E.; Barker, D.O.R.; Graham, M.; et al. Clinical Metagenomics Is Increasingly Accurate and Affordable to Detect Enteric Bacterial Pathogens in Stool. Microorganisms 2022, 10, 441. https://doi.org/10.3390/microorganisms10020441
Peterson C-L, Alexander D, Chen JC-Y, Adam H, Walker M, Ali J, Forbes J, Taboada E, Barker DOR, Graham M, et al. Clinical Metagenomics Is Increasingly Accurate and Affordable to Detect Enteric Bacterial Pathogens in Stool. Microorganisms. 2022; 10(2):441. https://doi.org/10.3390/microorganisms10020441
Chicago/Turabian StylePeterson, Christy-Lynn, David Alexander, Julie Chih-Yu Chen, Heather Adam, Matthew Walker, Jennifer Ali, Jessica Forbes, Eduardo Taboada, Dillon O. R. Barker, Morag Graham, and et al. 2022. "Clinical Metagenomics Is Increasingly Accurate and Affordable to Detect Enteric Bacterial Pathogens in Stool" Microorganisms 10, no. 2: 441. https://doi.org/10.3390/microorganisms10020441
APA StylePeterson, C.-L., Alexander, D., Chen, J. C.-Y., Adam, H., Walker, M., Ali, J., Forbes, J., Taboada, E., Barker, D. O. R., Graham, M., Knox, N., & Reimer, A. R. (2022). Clinical Metagenomics Is Increasingly Accurate and Affordable to Detect Enteric Bacterial Pathogens in Stool. Microorganisms, 10(2), 441. https://doi.org/10.3390/microorganisms10020441