
Prion-like Domains in Spike Protein of SARS-CoV-2 Differ across Its Variants and Enable Changes in Affinity to ACE2


Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Sequences
2.2. Identification of PrDs in Viral Proteomes
2.3. Statistical Analysis
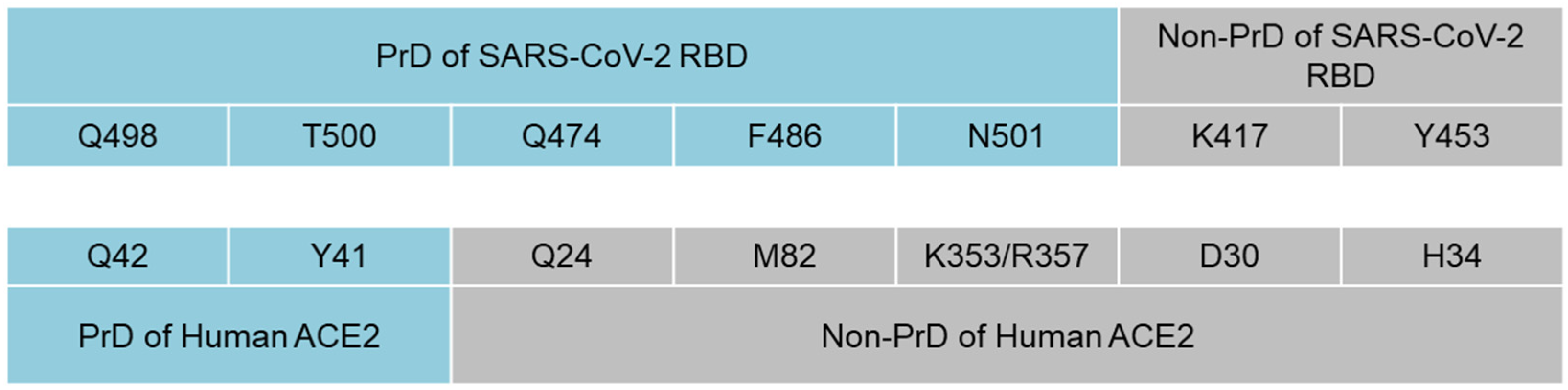
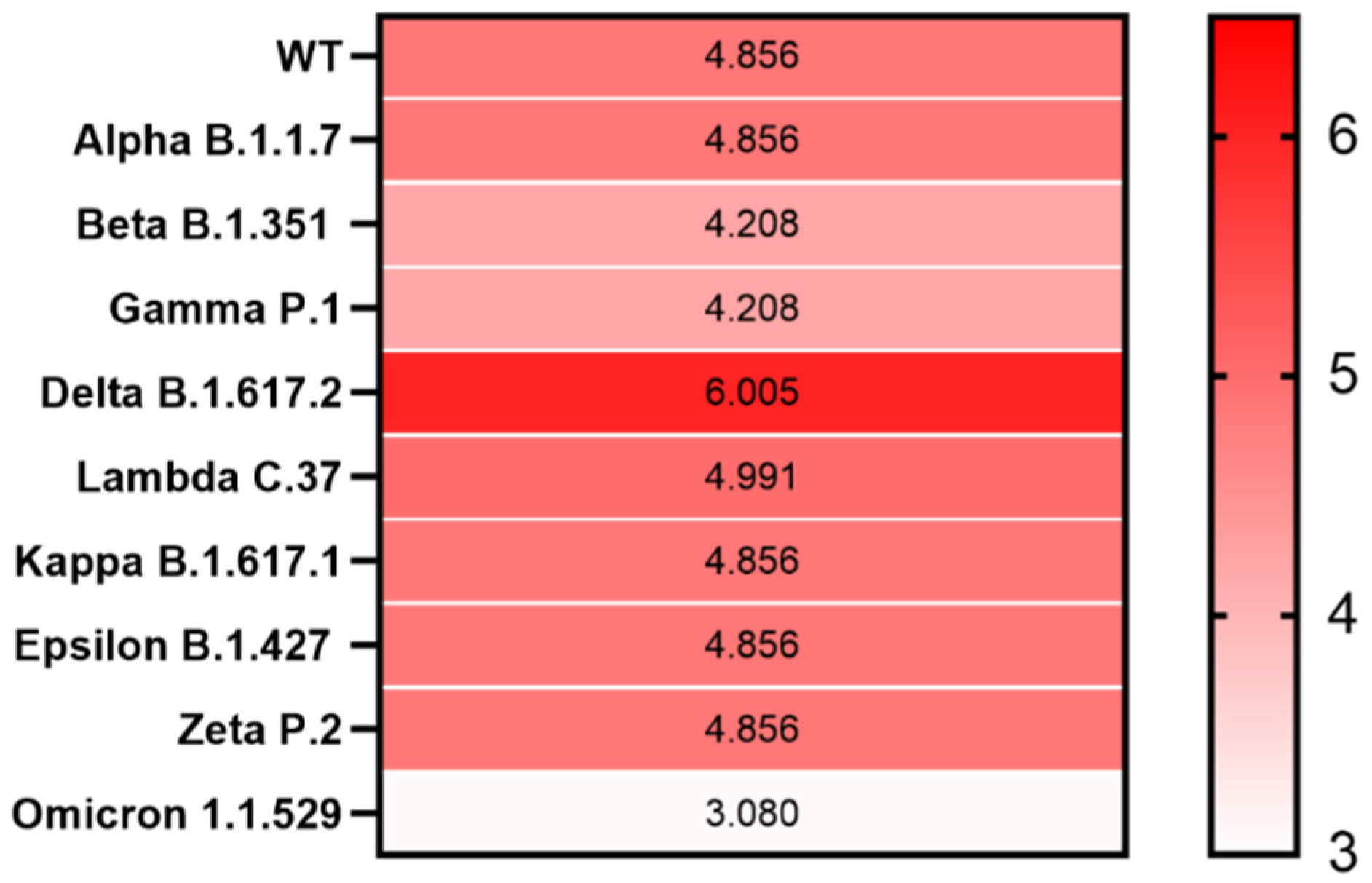
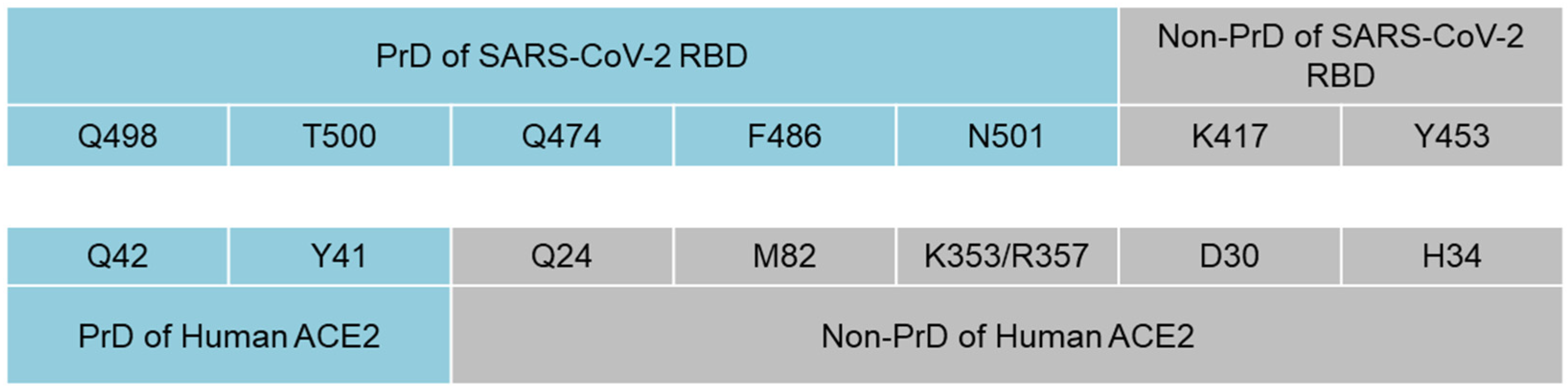
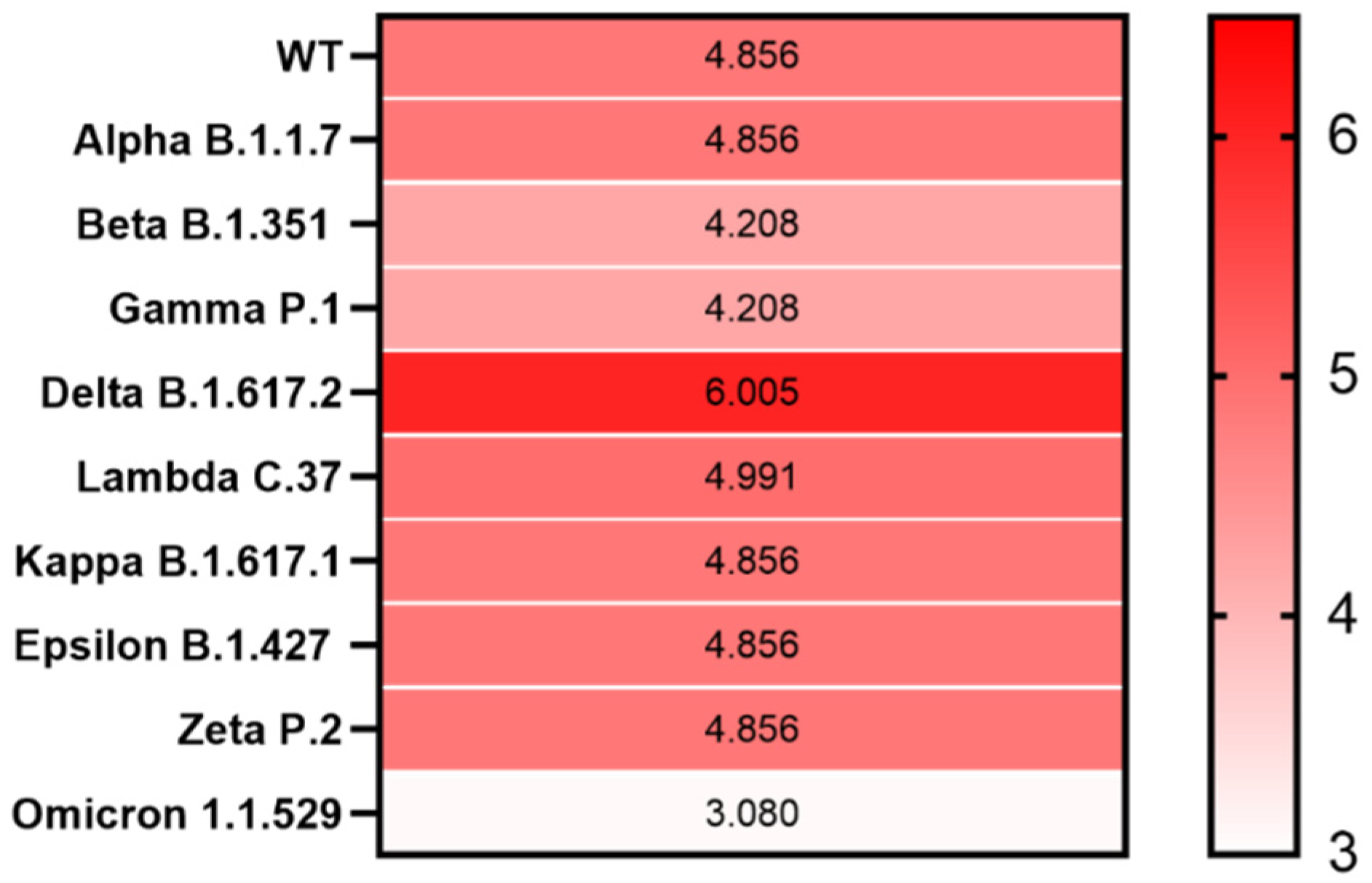
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lipsitch, M.; Swerdlow, D.L.; Finelli, L. Defining the epidemiology of COVID-19—Studies needed. N. Engl. J. Med. 2020, 382, 1194–1196. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhao, S.; Teng, T.; Abdalla, A.E.; Zhu, W.; Xie, L.; Wang, Y.; Guo, X. Systematic Comparison of Two Animal-to-Human Transmitted Human Coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 2020, 12, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.W.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses; Humana Press: New York, NY, USA, 2015; pp. 1–23. [Google Scholar]
- Memish, Z.A.; Perlman, S.; Van Kerkhove, M.D.; Zumla, A. Middle East respiratory syndrome. Lancet 2020, 395, 1063–1077. [Google Scholar] [CrossRef]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining ‘host jump’of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef] [Green Version]
- Shang, W.; Yang, Y.; Rao, Y.; Rao, X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. NPJ Vaccines 2020, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Xiao, X.; Aierken, A.; Liao, M.; Hua, J. The ACE2 Expression in Sertoli cells and Germ cells may cause male reproductive disorder after SARS-CoV-2 Infection. J. Cell. Mol. Med. 2020, 24, 9472–9477. [Google Scholar] [CrossRef]
- Inchingolo, A.D.; Inchingolo, A.M.; Bordea, I.R.; Malcangi, G.; Xhajanka, E.; Scarano, A.; Lorusso, F.; Farronato, M.; Tartaglia, G.M.; Isacco, C.G.; et al. SARS-CoV-2 disease adjuvant therapies and supplements breakthrough for the infection prevention. Microorganisms 2021, 9, 525. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Bhattacharyya, R.; Sengupta, J. Dynamics and electrostatics define an allosteric druggable site within the receptor-binding domain of SARS-CoV-2 spike protein. FEBS Lett. 2021, 595, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Celik, I.; Yadav, R.; Duzgun, Z.; Albogami, S.; El-Shehawi, A.M.; Idroes, R.; Tallei, T.E.; Emran, T.B. Interactions of the receptor binding domain of SARS-CoV-2 variants with hACE2: Insights from molecular docking analysis and molecular dynamic simulation. Biology 2021, 10, 880. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Tzou, P.; Nouhin, J.; Gupta, R.; de Oliveira, T.; Kosakovsky Pond, S.; Fera, D.; Shafer, R. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.; Abbott, S.; Barnard, R.; Jarvis, C.; Kucharski, A.; Munday, J.; Pearson, C.; Russell, T.; Tully, D.; Washburne, A.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B. 1.1. 7 in England. Science 2021, 372, 6538. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Gilby, N.B.; Wei, G.W. Omicron (B. 1.1. 529): Infectivity, vaccine breakthrough, and antibody resistance. arXiv 2021, arXiv:2112.01318. [Google Scholar]
- Tetz, G.; Tetz, V. Prion-Like Domains in Phagobiota. Front. Microbiol. 2017, 8, 2239. [Google Scholar] [CrossRef]
- Tetz, G.; Tetz, V. Prion-like domains in eukaryotic viruses. Sci. Rep. 2018, 8, 8931. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Tetz, G.; Brown, S.M.; Hao, Y.; Tetz, V. Type 1 diabetes: An association between autoimmunity, the dynamics of gut amyloid-producing E. coli and their phages. Sci. Rep. 2019, 9, 9685. [Google Scholar] [CrossRef] [Green Version]
- Tetz, G.; Pinho, M.; Pritzkow, S.; Mendez, N.; Soto, C.; Tetz, V. Bacterial DNA promotes Tau aggregation. Sci. Rep. 2020, 10, 2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, D.; Koulov, A.; Balch, W.; Kelly, J. Functional amyloid—From bacteria to humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Michelitsch, M.; Weissman, J. A census of glutamine/asparagine-rich regions: Implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. USA 2000, 9, 11910–11915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, E.; MacLea, K.; Anderson, C.; Ben-Hur, A. A Bioinformatics Method for Identifying Q/N-Rich Prion-Like Domains in Proteins. Methods Mol. Biol. 2013, 1017, 219–228. [Google Scholar] [PubMed]
- Lancaster, A.; Nutter-Upham, A.; Lindquist, S.; King, O. PLAAC: A web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 2014, 30, 2501–2502. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Guo, Y.; Xia, L.; Zhou, Q. Structural basis for the recognition of the 2019-nCoV by human ACE2. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Gil-Garcia, M.; Iglesias, V.; Pallarès, I.; Ventura, S. Prion-like proteins: From computational approaches to proteome-wide analysis. FEBS Open Bio 2021, 11, 2400. [Google Scholar] [CrossRef]
- Brielle, E.S.; Schneidman-Duhovny, D.; Linial, M. The SARS-CoV-2 exerts a distinctive strategy for interacting with the ACE2 human receptor. Viruses 2020, 12, 497. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Damas, J.; Hughes, G.M.; Keough, K.C.; Painter, C.A.; Persky, N.S.; Corbo, M.; Hiller, M.; Koepfli, K.P.; Pfenning, A.R.; Zhao, H.; et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc. Natl. Acad. Sci. USA 2020, 117, 22311–22322. [Google Scholar] [CrossRef]
- Lim, H.; Baek, A.; Kim, J.; Kim, M.S.; Liu, J.; Nam, K.Y.; Yoon, J.; No, K.T. Hot spot profiles of SARS-CoV-2 and human ACE2 receptor protein protein interaction obtained by density functional tight binding fragment molecular orbital method. Sci. Rep. 2020, 10, 16862. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.; Archer, B.; Laurenson-Schafer, H.; Jinnai, Y.; Konings, F.; Batra, N.; Pavlin, B.; Vandemaele, K.; Van Kerkhove, M.; Jombart, T.; et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Eurosurveillance 2021, 26, 2100509. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell. Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Wei, D.Q.; Kousar, K.; Abubaker, J.; Ahmad, S.; Ali, J.; Al-Mulla, F.; Ali, S.S.; Nizam-Uddin, N.; Sayaf, A.M.; et al. Preliminary Structural Data Revealed That the SARS-CoV-2 B. 1.617 Variant’s RBD Binds to ACE2 Receptor Stronger Than the Wild Type to Enhance the Infectivity. ChemBioChem 2021, 22, 2641. [Google Scholar] [CrossRef]
- Khan, A.; Gui, J.; Ahmad, W.; Haq, I.; Shahid, M.; Khan, A.A.; Shah, A.; Khan, A.; Ali, L.; Anwar, Z.; et al. The SARS-CoV-2 B. 1.618 variant slightly alters the spike RBD–ACE2 binding affinity and is an antibody escaping variant: A computational structural perspective. RSC Adv. 2021, 11, 30132–30147. [Google Scholar] [CrossRef]
- UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2011, 40, D71–D75. [Google Scholar]
- NCBI Protein Database [Internet]. National Library of Medicine (US), National Center for Biotechnology Information: Bethesda, MD, USA. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 12 December 2021).
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Deng, A.; Li, K.; Hu, Y.; Li, Z.; Xiong, Q.; Liu, Z.; Guo, Q.; Zou, L.; Zhang, H.; et al. Viral infection and transmission in a large well-traced outbreak caused by the Delta SARS-CoV-2 variant. MedRxiv 2021. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| S Protein | |||
|---|---|---|---|
| Domain | Prion-like Domain AA Position | LLR Score | |
| SARS-CoV-2 | RBD | 473–510 | 4.856 |
| SARS-CoV | HR1 | 900–910 | 4.426 |
| MERS-CoV | NA | Non-detectable | 4.49 |
| HCoV-OC43 | NA | Non-detectable | 2.828 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tetz, G.; Tetz, V. Prion-like Domains in Spike Protein of SARS-CoV-2 Differ across Its Variants and Enable Changes in Affinity to ACE2. Microorganisms 2022, 10, 280. https://doi.org/10.3390/microorganisms10020280
Tetz G, Tetz V. Prion-like Domains in Spike Protein of SARS-CoV-2 Differ across Its Variants and Enable Changes in Affinity to ACE2. Microorganisms. 2022; 10(2):280. https://doi.org/10.3390/microorganisms10020280
Chicago/Turabian StyleTetz, George, and Victor Tetz. 2022. "Prion-like Domains in Spike Protein of SARS-CoV-2 Differ across Its Variants and Enable Changes in Affinity to ACE2" Microorganisms 10, no. 2: 280. https://doi.org/10.3390/microorganisms10020280
APA StyleTetz, G., & Tetz, V. (2022). Prion-like Domains in Spike Protein of SARS-CoV-2 Differ across Its Variants and Enable Changes in Affinity to ACE2. Microorganisms, 10(2), 280. https://doi.org/10.3390/microorganisms10020280