
Water Column Microbial Communities Vary along Salinity Gradients in the Florida Coastal Everglades Wetlands

 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Flow Cytometry
2.3. Molecular Methods
2.4. Bioinformatics
2.5. Preprocessing of the Dataset
3. Results
3.1. General Overview of the Datasets
3.2. Freshwater Marsh Communities
3.3. Ecotone
3.4. Florida Bay
4. Discussion
4.1. Freshwater Marsh Communities
4.2. Ecotone
4.3. Florida Bay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Costanza, R.; d’Arge, R.; de Groot, R.; Farber, S.; Grasso, M.; Hannon, B.; Limburg, K.; Naeem, S.; ONeill, R.; Paruelo, J.; et al. The value of the world’s ecosystem services and natural capital. Nature 1997, 387, 253–260. [Google Scholar] [CrossRef]
- de Groot, R.; Brander, L.; van der Ploeg, S.; Costanza, R.; Bernard, F.; Braat, L.; Christie, M.; Crossman, N.; Ghermandi, A.; Hein, L.; et al. Global estimates of the value of ecosystems and their services in monetary units. Ecosyst. Serv. 2012, 1, 50–61. [Google Scholar] [CrossRef]
- Davidson, N.C. How much wetland has the world lost? Long-term and recent trends in global wetland area. Mar. Freshw. Res. 2014, 65, 934–941. [Google Scholar] [CrossRef]
- Sklar, F.; McVoy, C.; Van Zee, R.; Gawlik, D.; Swift, D.; Park, W.; Fitz, C.; Wu, Y.; Rudnick, D.; Fontaine, T.; et al. Hydrologic needs: The effects of altered hydrology on the everglades. In Everglades Interim Report; SFWMD: West Palm Beach, FL, USA, 1999. [Google Scholar]
- McVoy, C.; Park Said, W.; Obeysekera, J.; VanArman, J.A.; Dreschel, T.W. Landscapes and Hydrology of the Predrainage Everglades; University Press of Florida: Gainesville, FL, USA, 2011. [Google Scholar]
- Troxler, T.; Gaiser, E.; Barr, J.; Fuentes, J.; Jaffe, R.; Childers, D.; Collado-Vides, L.; Rivera-Monroy, V.; Castaneda-Moya, E.; Anderson, W.; et al. Integrated carbon budget models for the everglades terrestrial-coastal-oceanic gradient current status and needs for inter-site comparisons. Oceanography 2013, 26, 98–107. [Google Scholar] [CrossRef]
- Titus, J.; Richman, C. Maps of lands vulnerable to sea level rise: Modeled elevations along the US Atlantic and Gulf coasts. Clim. Res. 2001, 18, 205–228. [Google Scholar] [CrossRef]
- Church, J.; Clark, P.; Cazenave, A.; Gregory, J.; Jevrejeva, S.; Levermann, A.; Merrifield, M.; Milne, G.; Nerem, R.; Nunn, P.; et al. Sea-level rise by 2100. Science 2013, 342, 1445. [Google Scholar] [CrossRef] [PubMed]
- Haigh, I.; Wahl, T.; Rohling, E.; Price, R.; Pattiaratchi, C.; Calafat, F.; Dangendorf, S. Timescales for detecting a significant acceleration in sea level rise. Nat. Commun. 2014, 5, 3635. [Google Scholar] [CrossRef] [PubMed]
- Flower, H.; Rains, M.; Fitz, C. Visioning the future: Scenarios modeling of the florida coastal everglades. Environ. Manag. 2017, 60, 989–1009. [Google Scholar] [CrossRef]
- Yao, Q.; Liu, K. Dynamics of marsh-mangrove ecotone since the mid-holocene: A palynological study of mangrove encroachment and sea level rise in the shark river estuary, Florida. PLoS ONE 2017, 12, e0173670. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J. Ecological assessments with algae: A review and synthesis. J. Phycol. 2014, 50, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.; Brown, M. Diatoms as indicators of isolated herbaceous wetland condition in Florida, USA. Ecol. Indic. 2007, 7, 521–540. [Google Scholar] [CrossRef]
- Gaiser, E. Periphyton as an indicator of restoration in the Florida everglades. Ecol. Indic. 2009, 9, S37–S45. [Google Scholar] [CrossRef]
- Gaiser, E.; McCormick, P.; Hagerthey, S.; Gottlieb, A. Landscape patterns of periphyton in the Florida Everglades. Crit. Rev. Environ. Sci. Technol. 2011, 41, 92–120. [Google Scholar] [CrossRef]
- Caruso, G.; Azzaro, M.; Caroppo, C.; Decembrini, F.; Monticelli, L.; Leonardi, M.; Maimone, G.; Zaccone, R.; La Ferla, R. Microbial community and its potential as descriptor of environmental status. ICES J. Mar. Sci. 2016, 73, 2174–2177. [Google Scholar] [CrossRef]
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 2007, 5, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Gasol, J.; Pinhassi, J.; Alonso-Saez, L.; Ducklow, H.; Herndl, G.; Koblizek, M.; Labrenz, M.; Luo, Y.; Moran, X.; Reinthaler, T.; et al. Towards a better understanding of microbial carbon flux in the sea. Aquat. Microb. Ecol. 2008, 53, 21–38. [Google Scholar] [CrossRef]
- Henson, M.; Hanssen, J.; Spooner, G.; Fleming, P.; Pukonen, M.; Stahr, F.; Thrash, J. Nutrient dynamics and stream order influence microbial community patterns along a 2914 kilometer transect of the Mississippi river. Limnol. Oceanogr. 2018, 63, 1837–1855. [Google Scholar] [CrossRef]
- Markussen, T.; Happel, E.; Teikari, J.; Huchaiah, V.; Alneberg, J.; Andersson, A.; Sivonen, K.; Riemann, L.; Middelboe, M.; Kisand, V. Coupling biogeochemical process rates and metagenomic blueprints of coastal bacterial assemblages in the context of environmental change. Environ. Microbiol. 2018, 20, 3083–3099. [Google Scholar] [CrossRef]
- Ikenaga, M.; Guevara, R.; Dean, A.L.; Pisani, C.; Boyer, J.N. Changes in community structure of sediment bacteria along the florida coastal everglades marsh-mangrove-seagrass salinity gradient. Microb. Ecol. 2010, 59, 284–295. [Google Scholar] [CrossRef]
- Lebaron, P.; Servais, P.; Agogue, H.; Courties, C.; Joux, F. Does the high nucleic acid content of individual bacterial cells allow us to discriminate between active cells and inactive cells in aquatic systems? Appl. Environ. Microbiol. 2001, 67, 1775–1782. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.; Lauber, C.; Walters, W.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the illumina Hiseq and Miseq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Amaral-Zettler, L.; McCliment, E.; Ducklow, H.; Huse, S. A method for studying protistan diversity using massively parallel sequencing of v9 hypervariable regions of small-subunit ribosomal RNA genes. PLoS ONE 2009, 4, e6372. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.; Dillon, M.; Bokulich, N.; Abnet, C.; Al-Ghalith, G.; Alexander, H.; Alm, E.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.; McMurdie, P.; Rosen, M.; Han, A.; Johnson, A.; Holmes, S. DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Bokulich, N.; Kaehler, B.; Rideout, J.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.; Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.; Simpson, G.; Solymos, P. Vegan: Community Ecology Package. R Package Version 2.5-6. 2019. 2020. Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 14 December 2021).
- Herlemann, D.P.; Woelk, J.; Labrenz, M.; Jürgens, K. Diversity and abundance of “Pelagibacterales” (SAR11) in the Baltic Sea salinity gradient. Syst. Appl. Microbiol. 2014, 37, 601–604. [Google Scholar] [CrossRef]
- Fortunato, C.S.; Eiler, A.; Herfort, L.; Needoba, J.A.; Peterson, T.D.; Crump, B.C. Determining indicator taxa across spatial and seasonal gradients in the columbia river coastal margin. ISME J. 2013, 7, 1899–1911. [Google Scholar] [CrossRef]
- Ewe, S.; Gaiser, E.; Childers, D.; Iwaniec, D.; Rivera-Monroy, V.; Twilley, R. Spatial and temporal patterns of aboveground net primary productivity (anpp) along two freshwater-estuarine transects in the florida coastal everglades. Hydrobiologia 2006, 569, 459–474. [Google Scholar] [CrossRef]
- Hwang, C.; Cho, B. Cohaesibacter gelatinilyticus gen. Nov., sp nov., a marine bacterium that forms a distinct branch in the order Rhizobiales, and proposal of Cohaesibacteraceae fam. Nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 267–277. [Google Scholar] [CrossRef]
- Boyer, J.; Dailey, S.; Gibson, P.; Rogers, M.; Mir-Gonzalez, D. The role of dissolved organic matter bioavailability in promoting phytoplankton blooms in Florida bay. Hydrobiologia 2006, 569, 71–85. [Google Scholar] [CrossRef]
- Jeong, H. The ecological roles of heterotrophic dinoflagellates in marine planktonic community. J. Eukaryot. Microbiol. 1999, 46, 390–396. [Google Scholar] [CrossRef]
- Kominoski, J.; Gaiser, E.; Castaneda-Moya, E.; Davis, S.; Dessu, S.; Julian, P.; Lee, D.; Marazzi, L.; Rivera-Monroy, V.; Sola, A.; et al. Disturbance legacies increase and synchronize nutrient concentrations and bacterial productivity in coastal ecosystems. Ecology 2020, 101, e02988. [Google Scholar] [CrossRef]
- Zwart, G.; van Hannen, E.J.; Kamst-van Agterveld, M.P.; Van der Gucht, K.; Lindström, E.S.; Van Wichelen, J.; Lauridsen, T.; Crump, B.C.; Han, S.K.; Declerck, S. Rapid screening for freshwater bacterial groups by using reverse line blot hybridization. Appl. Environ. Microbiol. 2003, 69, 5875–5883. [Google Scholar] [CrossRef]
- Zwisler, W.; Selje, N.; Simon, M. Seasonal patterns of the bacterioplankton community composition in a large mesotrophic lake. Aquat. Microb. Ecol. 2003, 31, 211–225. [Google Scholar] [CrossRef][Green Version]
- Buck, U.; Grossart, H.; Amann, R.; Pernthaler, J. Substrate incorporation patterns of bacterioplankton populations in stratified and mixed waters of a Humic Lake. Environ. Microbiol. 2009, 11, 1854–1865. [Google Scholar] [CrossRef]
- Wu, Q.; Zwart, G.; Schauer, M.; Kamst-van Agterveld, M.; Hahn, M. Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan plateau, China. Appl. Environ. Microbiol. 2006, 72, 5478–5485. [Google Scholar] [CrossRef] [PubMed]
- Percent, S.F.; Frischer, M.E.; Vescio, P.A.; Duffy, E.B.; Milano, V.; McLellan, M.; Stevens, B.M.; Boylen, C.W.; Nierzwicki-Bauer, S.A. Bacterial community structure of acid-impacted lakes: What controls diversity? Appl. Environ. Microbiol. 2008, 74, 1856–1868. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jezberova, J.; Jezbera, J.; Brandt, U.; Lindstrom, E.; Langenheder, S.; Hahn, M. Ubiquity of Polynucleobacter necessarius ssp. asymbioticus in lentic freshwater habitats of a heterogenous 2000 km2 area. Environ. Microbiol. 2010, 12, 3302. [Google Scholar]
- Newton, R.; Jones, S.; Eiler, A.; McMahon, K.; Bertilsson, S. A guide to the natural history of freshwater lake bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 14–49. [Google Scholar] [CrossRef]
- Wu, Q.; Hahn, M. High predictability of the seasonal dynamics of a species-like Polynucleobacter population in a freshwater lake. Environ. Microbiol. 2006, 8, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R.; Rodriguez-Valera, F.; McMahon, K.D.; Toyama, D.; Rinke, R.; Cristina Souza de Oliveira, T.; Wagner Garcia, J.; Pellon de Miranda, F.; Henrique-Silva, F. Metagenomics of the water column in the pristine upper course of the amazon river. PLoS ONE 2011, 6, e23785. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.; Koll, U.; Jezberova, J.; Camacho, A. Global phylogeography of pelagic polynucleobacter bacteria: Restricted geographic distribution of subgroups, isolation by distance and influence of climate. Environ. Microbiol. 2015, 17, 829–840. [Google Scholar] [CrossRef]
- Hahn, M.; Scheuerl, T.; Jezberova, J.; Koll, U.; Jezbera, J.; Simek, K.; Vannini, C.; Petroni, G.; Wu, Q. The passive yet successful way of planktonic life: Genomic and experimental analysis of the ecology of a free-living Polynucleobacter population. PLoS ONE 2012, 7, e32772. [Google Scholar]
- Hoetzinger, M.; Schmidt, J.; Jezberova, J.; Koll, U.; Hahn, M. Microdiversification of a pelagic Polynucleobacter species is mainly driven by acquisition of genomic islands from a partially interspecific gene pool. Appl. Environ. Microbiol. 2017, 83, e02266-16. [Google Scholar] [CrossRef]
- Qualls, R.; Richardson, C. Factors controlling concentration, export, and decomposition of dissolved organic nutrients in the everglades of Florida. Biogeochemistry 2003, 62, 197–229. [Google Scholar] [CrossRef]
- Kolmonen, E.; Sivonen, K.; Rapala, J.; Haukka, K. Diversity of cyanobacteria and heterotrophic bacteria in cyanobacterial blooms in lake joutikas, finland. Aquat. Microb. Ecol. 2004, 36, 201–211. [Google Scholar] [CrossRef][Green Version]
- Kolmonen, E.; Haukka, K.; Rantala-Ylinen, A.; Rajaniemi-Wacklin, P.; Lepisto, L.; Sivonen, K. Bacterioplankton community composition in 67 finnish lakes differs according to trophic status. Aquat. Microb. Ecol. 2011, 62, 241–250. [Google Scholar] [CrossRef]
- Arnds, J.; Knittel, K.; Buck, U.; Winkel, M.; Amann, R. Development of a 16S rRNA-targeted probe set for Verrucomicrobia and its application for fluorescence in situ hybridization in a humic lake. Syst. Appl. Microbiol. 2010, 33, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Hugerth, L.; Larsson, J.; Alneberg, J.; Lindh, M.; Legrand, C.; Pinhassi, J.; Andersson, A. Metagenome-assembled genomes uncover a global brackish microbiome. Genome Biol. 2015, 16, 279. [Google Scholar] [CrossRef] [PubMed]
- Parveen, B.; Mary, I.; Vellet, A.; Ravet, V.; Debroas, D. Temporal dynamics and phylogenetic diversity of free-living and particle-associated verrucomicrobia communities in relation to environmental variables in a mesotrophic lake. FEMS Microbiol. Ecol. 2013, 83, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Unno, T.; Gould, T.; Jarvis, B.; Phillips, J.; Cotner, J.; Sadowsky, M. Application of illumina next-generation sequencing to characterize the bacterial community of the upper Mississippi river. J. Appl. Microbiol. 2013, 115, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Rodibaugh, K.; Hahn, D.; Nowlin, W. Bacterial community composition and carbon metabolism in a subtropical riverscape. Hydrobiologia 2017, 792, 209–226. [Google Scholar] [CrossRef]
- Marshall, W.; Laybourn-Parry, J. The balance between photosynthesis and grazing in antarctic mixotrophic cryptophytes during summer. Freshw. Biol. 2002, 47, 2060–2070. [Google Scholar] [CrossRef]
- Laybourn-Parry, J.; Marshall, W.; Marchant, H. Flagellate nutritional versatility as a key to survival in two contrasting antarctic saline lakes. Freshw. Biol. 2005, 50, 830–838. [Google Scholar] [CrossRef]
- Williams, A.; Trexler, J. A preliminary analysis of the correlation of food-web characteristics with hydrology and nutrient gradients in the southern everglades. Hydrobiologia 2006, 569, 493–504. [Google Scholar] [CrossRef]
- Scherwass, A.; Fischer, Y.; Arndt, H. Detritus as a potential food source for protozoans: Utilization of fine particulate plant detritus by a heterotrophic flagellate, Chilomonas paramecium, and a ciliate, Tetrahymena pyriformis. Aquat. Ecol. 2005, 39, 439–445. [Google Scholar] [CrossRef]
- Kuehn, K.; Francoeur, S.; Findlay, R.; Neely, R. Priming in the microbial landscape: Periphytic algal stimulation of litter-associated microbial decomposers. Ecology 2014, 95, 749–762. [Google Scholar] [CrossRef]
- Kark, S.; van Rensburg, B. Ecotones: Marginal or central areas of transition? Isr. J. Ecol. Evol. 2006, 52, 29–53. [Google Scholar] [CrossRef]
- Chen, R.; Twilley, R. Patterns of mangrove forest structure and soil nutrient dynamics along the shark River estuary, Florida. Estuaries 1999, 22, 955–970. [Google Scholar] [CrossRef]
- Davis, S.; Cable, J.; Childers, D.; Coronado-Molina, C.; Day, J.; Hittle, C.; Madden, C.; Reyes, E.; Rudnick, D.; Sklar, F. Importance of storm events in controlling ecosystem structure and function in a Florida gulf coast estuary. J. Coast. Res. 2004, 20, 1198–1208. [Google Scholar] [CrossRef]
- Sutula, M.; Perez, B.; Reyes, E.; Childers, D.; Davis, S.; Day, J.; Rudnick, D.; Sklar, F. Factors affecting spatial and temporal variability in material exchange between the southern everglades wetlands and Florida bay (USA). Estuar. Coast. Shelf Sci. 2003, 57, 757–781. [Google Scholar] [CrossRef]
- Ross, M.; Meeder, J.; Sah, J.; Ruiz, P.; Telesnicki, G. The southeast saline everglades revisited: 50 years of coastal vegetation change. J. Veg. Sci. 2000, 11, 101–112. [Google Scholar] [CrossRef]
- Andersen, F.; Kristensen, E. Oxygen microgradients in the rhizosphere of the mangrove Avicennia marina. Mar. Ecol. Prog. Ser. 1988, 44, 201–204. [Google Scholar] [CrossRef]
- Alongi, D.; Clough, B.; Robertson, A. Nutrient-use efficiency in arid-zone forests of the mangroves Rhizophora stylosa and Avicennia marina. Aquat. Bot. 2005, 82, 121–131. [Google Scholar] [CrossRef]
- Reef, R.; Feller, I.; Lovelock, C. Nutrition of mangroves. Tree Physiol. 2010, 30, 1148–1160. [Google Scholar] [CrossRef] [PubMed]
- Holguin, G.; Vazquez, P.; Bashan, Y. The role of sediment microorganisms in the productivity, conservation, and rehabilitation of mangrove ecosystems: An overview. Biol. Fertil. Soils 2001, 33, 265–278. [Google Scholar] [CrossRef]
- Teeling, H.; Fuchs, B.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.; Kassabgy, M.; Huang, S.; Mann, A.; Waldmann, J.; et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, M.; Kirchman, D. Natural assemblages of marine proteobacteria and members of the cytophaga-flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl. Environ. Microbiol. 2000, 66, 1692–1697. [Google Scholar] [CrossRef]
- Kirchman, D. The ecology of cytophaga-flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 2002, 39, 91–100. [Google Scholar] [CrossRef]
- Taylor, J.; Cottingham, S.; Billinge, J.; Cunliffe, M. Seasonal microbial community dynamics correlate with phytoplankton-derived polysaccharides in surface coastal waters. ISME J. 2014, 8, 245–248. [Google Scholar] [CrossRef]
- Hall, A.; Wells, R.; Sweeney, J.; Townsend, F.; Balmer, B.; Hohn, A.; Rhinehart, H. Annual, seasonal and individual variation in hematology and clinical blood chemistry profiles in bottlenose dolphins (Tursiops Truncatus) from Sarasota Bay, Florida. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2007, 148, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Fourqurean, J.; Zieman, J.; Powell, G. Phosphorus limitation of primary production in Florida Bay—Evidence from C-N-P ratios of the dominant seagrass Thalassia testudinum. Limnol. Oceanogr. 1992, 37, 162–171. [Google Scholar] [CrossRef]
- Briceno, H.; Boyer, J. Climatic controls on phytoplankton biomass in a sub-tropical estuary, Florida Bay, USA. Estuaries Coasts 2010, 33, 541–553. [Google Scholar] [CrossRef]
- Briceño, H.O.; Boyer, J.N.; Castro, J.; Harlem, P. Biogeochemical classification of south Florida’s estuarine and coastal waters. Mar. Pollut. Bull. 2013, 75, 187–204. [Google Scholar] [CrossRef]
- Klindworth, A.; Mann, A.J.; Huang, S.; Wichels, A.; Quast, C.; Waldmann, J.; Teeling, H.; Glöckner, F.O. Diversity and activity of marine bacterioplankton during a diatom bloom in the North Sea assessed by total RNA and pyrotag sequencing. Mar. Genom. 2014, 18, 185–192. [Google Scholar] [CrossRef]
- King, G.; Smith, C.; Tolar, B.; Hollibaugh, J. Analysis of compostion and structure of coastal to mesopelagic bacterioplankton communities in the northern Gulf of Mexico. Front. Microbiol. 2013, 3, 438. [Google Scholar] [CrossRef]
- Pisani, O.; Yamashita, Y.; Jaffe, R. Photo-dissolution of flocculent, detrital material in aquatic environments: Contributions to the dissolved organic matter pool. Water Res. 2011, 45, 3836–3844. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
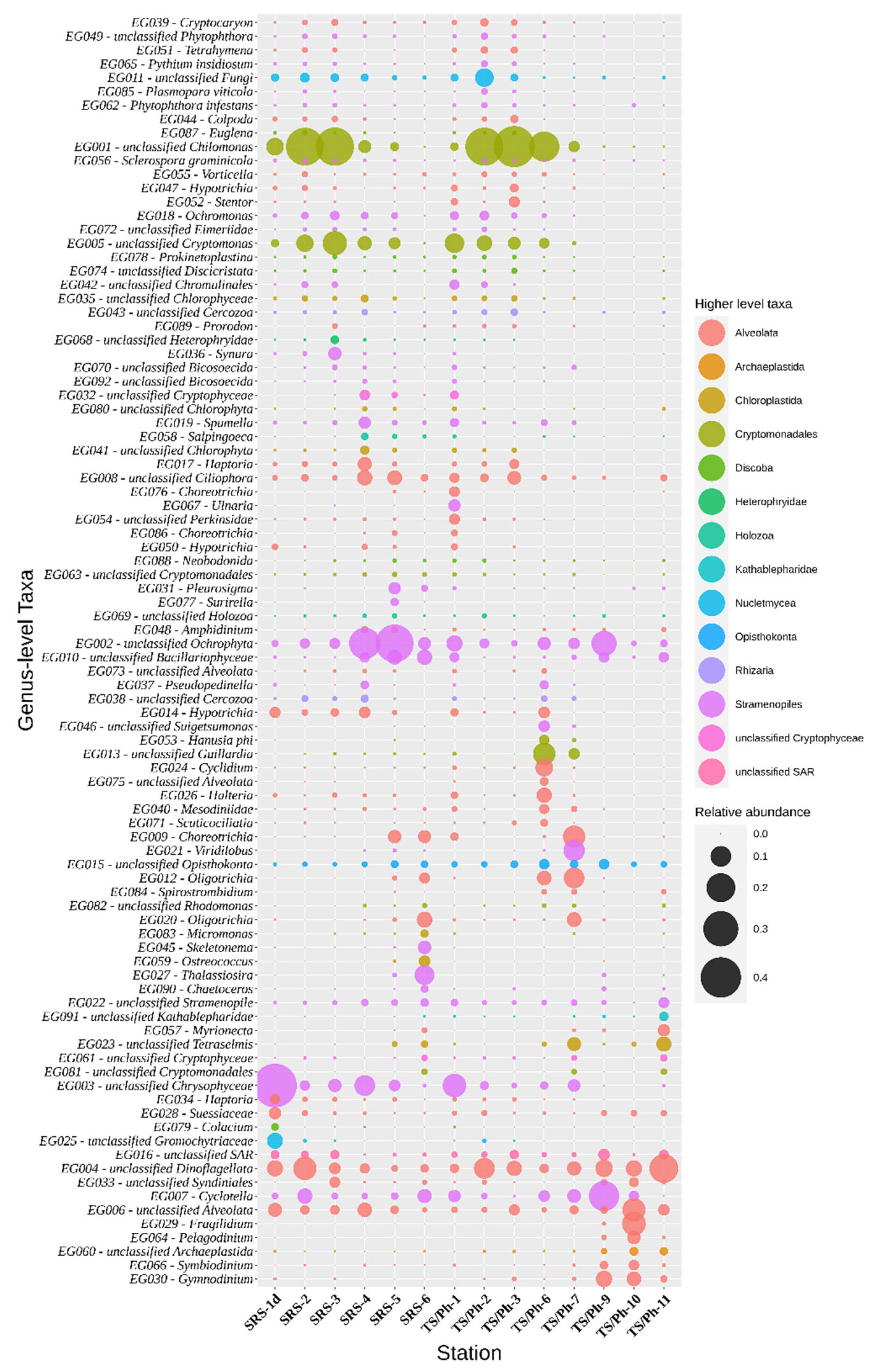{kind=link}
| Station | BA (cells mL−1) | BP (μg C L−1 d−1) | Chl a (μg L−1) | Salinity (PSU) | TN (μM) | TP (μM) | DOC (μM) |
|---|---|---|---|---|---|---|---|
| TS/Ph-1 | 1,206,442 | 36.53 | 2.89 | 0.2 | 33.67 | 0.6 | 851.67 |
| TS/Ph-2 | 1,200,550 | 29.99 | 0.11 | 0.2 * | 43.70 * | NA | 746.67 |
| TS/Ph-3 | 1,233,549 | 6.54 | 2.71 | 0.2 | 66.23 | 0.53 | 893.33 |
| TS/Ph-6 | 2,586,481 | 71.87 | 1.16 | 6.1 * | 75.40 * | 0.4 * | 1254.17 |
| TS/Ph-7 | 3,884,709 | 22.58 | 1.56 | 14.8 * | 68.84 * | 0.41* | 1085.83 |
| TS/Ph-9 | 1,493,439 | 3.98 | 0.07 | 39.50 | 45.39 | 0.23 | 460.42 ** |
| TS/Ph-10 | 2,811,578 | 13.82 | 0.12 | 41.40 | 53.62 | 0.23 | 631.67 ** |
| TS/Ph-11 | 2,455,006 | 7.58 | 0.06 | 39.49 | 23.25 | 0.30 | 270 ** |
| SRS-1d | 1,048,253 | 5.11 | 2.49 | 0.2 | 70.29 | 0.54 | 1268.33 |
| SRS-2 | 1,390,989 | 9.51 | 1.63 | 0.1 | 55.93 | 0.37 | 1148.08 |
| SRS3 | 1,127,995 | 95.80 | 1.25 | 0.2 | 53.25 | 1.00 | 1243.33 |
| SRS4 | 2,272,470 | 42.85 | 1.79 | 0.4 | 63.76 | 0.71 | 1157.33 |
| SRS5 | 2,381,611 | 29.66 | 1.61 | 2.1 | 51.76 | 0.63 | 1066.67 |
| SRS6 | 3,272,441 | 22.64 | 1.3 | 19.3 | 37.94 | 0.70 | 929.25 |
| Station | Sequences (bac.) | SV (bac.) | Genus-Level Taxa (bac.) | H’ (bac.) | Sequences (euk.) | SV (euk.) | Genus-Level Taxa (euk.) | H’ (euk.) |
|---|---|---|---|---|---|---|---|---|
| TS/Ph-1 | 80125 | 281 | 151 | 6.36 | 173981 | 1654 | 213 | 8.41 |
| TS/Ph-2 | 70198 | 1302 | 165 | 4.58 | 269629 | 1302 | 148 | 7.02 |
| TS/Ph-3 | 90170 | 1227 | 145 | 4.19 | 198954 | 1227 | 135 | 7.02 |
| TS/Ph-6 | 68729 | 752 | 87 | 4.75 | 305749 | 752 | 115 | 6.02 |
| TS/Ph-7 | 50720 | 1008 | 75 | 4.61 | 208940 | 1008 | 154 | 6.71 |
| TS/Ph-9 | 56338 | 488 | 50 | 4.51 | 77764 | 488 | 103 | 4.83 |
| TS/Ph-10 | 53636 | 1084 | 64 | 3.85 | 102902 | 281 | 52 | 5.50 |
| TS/Ph-11 | 170185 | 1654 | 128 | 5.82 | 135226 | 1084 | 146 | 6.81 |
| SRS-1d | 56789 | 1174 | 104 | 4.66 | 377682 | 1174 | 130 | 5.34 |
| SRS-2 | 58820 | 1029 | 86 | 4.42 | 183907 | 1029 | 141 | 6.42 |
| SRS-3 | 76305 | 1358 | 168 | 5.81 | 273752 | 1358 | 172 | 6.20 |
| SRS-4 | 97050 | 490 | 139 | 5.88 | 389092 | 490 | 95 | 6.65 |
| SRS-5 | 149420 | 1748 | 188 | 5.71 | 315255 | 1748 | 203 | 6.60 |
| SRS-6 | 129150 | 1252 | 133 | 4.44 | 188495 | 1252 | 188 | 6.77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laas, P.; Ugarelli, K.; Travieso, R.; Stumpf, S.; Gaiser, E.E.; Kominoski, J.S.; Stingl, U. Water Column Microbial Communities Vary along Salinity Gradients in the Florida Coastal Everglades Wetlands. Microorganisms 2022, 10, 215. https://doi.org/10.3390/microorganisms10020215
Laas P, Ugarelli K, Travieso R, Stumpf S, Gaiser EE, Kominoski JS, Stingl U. Water Column Microbial Communities Vary along Salinity Gradients in the Florida Coastal Everglades Wetlands. Microorganisms. 2022; 10(2):215. https://doi.org/10.3390/microorganisms10020215
Chicago/Turabian StyleLaas, Peeter, Kelly Ugarelli, Rafael Travieso, Sandro Stumpf, Evelyn E. Gaiser, John S. Kominoski, and Ulrich Stingl. 2022. "Water Column Microbial Communities Vary along Salinity Gradients in the Florida Coastal Everglades Wetlands" Microorganisms 10, no. 2: 215. https://doi.org/10.3390/microorganisms10020215
APA StyleLaas, P., Ugarelli, K., Travieso, R., Stumpf, S., Gaiser, E. E., Kominoski, J. S., & Stingl, U. (2022). Water Column Microbial Communities Vary along Salinity Gradients in the Florida Coastal Everglades Wetlands. Microorganisms, 10(2), 215. https://doi.org/10.3390/microorganisms10020215