
The Spike Protein of SARS-coV2 19B (S) Clade Mirrors Critical Features of Viral Adaptation and Coevolution

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Viral Genome Sequences
2.3. Sequences Analysis
2.4. Phylogenetic Analysis
2.5. Evolutionary Distance and Neutrality Testing
2.6. 19B (S) A.29 Lineage Analysis
2.7. Data Visualization
3. Results
3.1. The Variation of the 19 (S) Clade across Continents
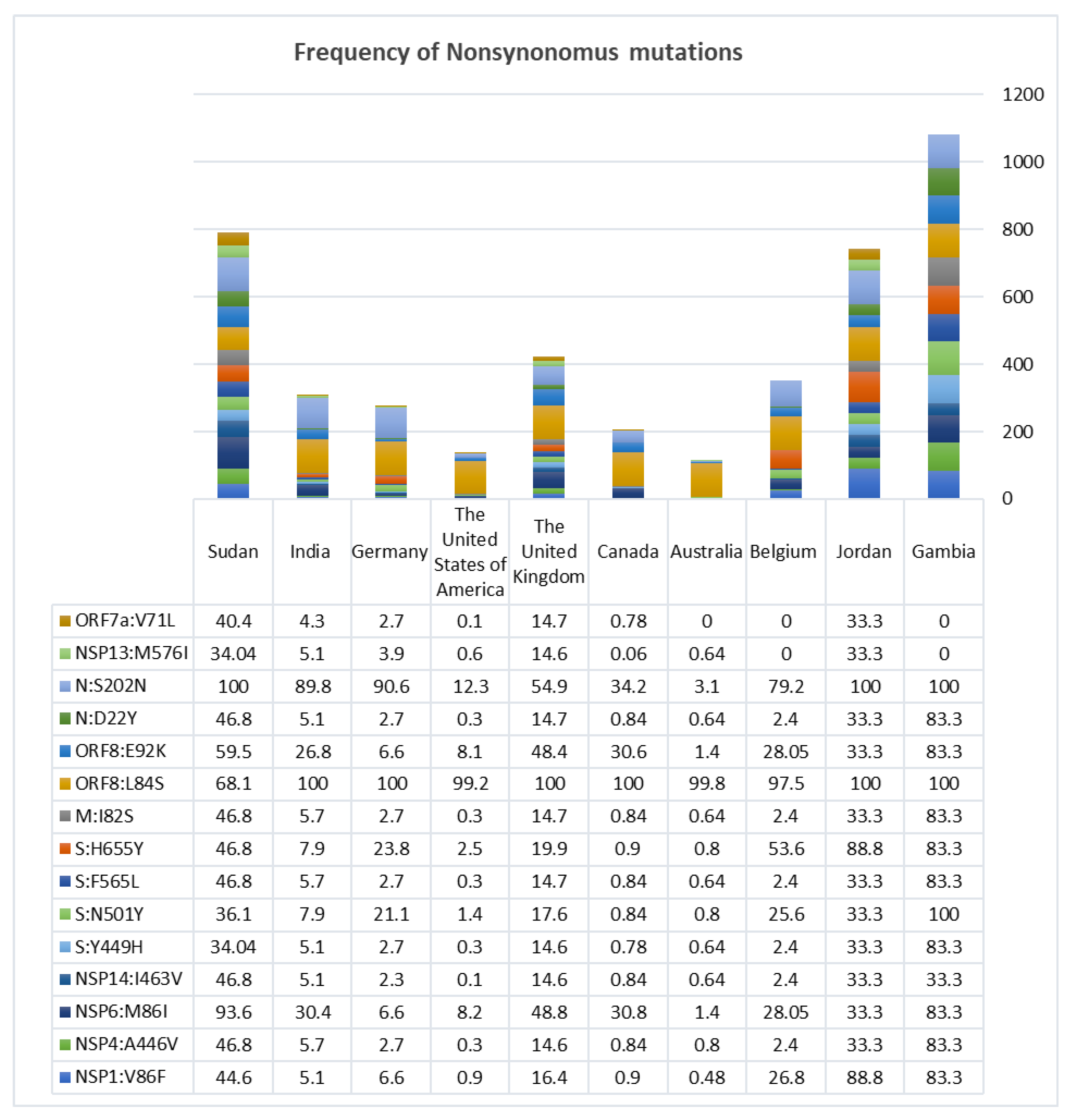
3.2. Nonsynonymous Mutations Found to Be Exceeding in Africa
3.3. Tajima’s Neutrality Test Returned a Negative D Value Hence Directional Selection
3.4. Mutational Variation in the A.29 Lineage of the 19B (S) Clade and the Spike Protein Structural Variation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Li, L.L.; Wang, J.L.; Ma, X.H.; Sun, X.M.; Li, J.S.; Yang, X.F.; Shi, W.F.; Duan, Z.J. A Novel SARS-CoV-2 Related Coronavirus with Complex Recombination Isolated from Bats in Yunnan Province, China. Emerg. Microbes Infect. 2021, 10, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.C.; Brisse, S.; Brosch, R.; Fabre, M.; Omaïs, B.; Marmiesse, M.; Supply, P.; Vincent, V. Ancient Origin and Gene Mosaicism of the Progenitor of Mycobacterium Tuberculosis. PLoS Pathog. 2005, 1, e5. [Google Scholar] [CrossRef]
- Wiens, K.E.; Woyczynski, L.P.; Ledesma, J.R.; Ross, J.M.; Zenteno-Cuevas, R.; Goodridge, A.; Ullah, I.; Mathema, B.; Djoba Siawaya, J.F.; Biehl, M.H.; et al. Global Variation in Bacterial Strains That Cause Tuberculosis Disease: A Systematic Review and Meta-Analysis. BMC Med. 2018, 16, 196. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.E.; Barker, D.C. The Origin and Evolution of the Leishmania Donovani Complex as Inferred from a Mitochondrial Cytochrome Oxidase II Gene Sequence. Infect. Genet. Evol. 2001, 1, 61–68. [Google Scholar] [CrossRef]
- Falush, D.; Wirth, T.; Linz, B.; Pritchard, J.K.; Stephens, M.; Kidd, M.; Blaser, M.J.; Graham, D.Y.; Vacher, S.; Perez-Perez, G.I.; et al. Traces of Human Migrations in Helicobacter Pylori Populations. Science 2003, 299, 1582–1585. [Google Scholar] [CrossRef] [Green Version]
- CDC. SARS-CoV-2 Variant Classifications and Definitions. CDC. 13 July 2021; pp. 1–12. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html#Interest (accessed on 15 August 2022).
- Letko, M.; Marzi, A.; Munster, V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereson, M.J.; Flichman, D.M.; Martínez, A.P.; Baré, P.; Garcia, G.H.; Di Lello, F.A. Evolutionary Analysis of SARS-CoV-2 Spike Protein for Its Different Clades. J. Med. Virol. 2021, 93, 3000–3006. [Google Scholar] [CrossRef] [PubMed]
- Hussman, J.P. Cellular and Molecular Pathways of COVID-19 and Potential Points of Therapeutic Intervention. Front. Pharmacol. 2020, 11, 1169. [Google Scholar] [CrossRef]
- Alejandra Tortorici, M.; Walls, A.C.; Lang, Y.; Wang, C.; Li, Z.; Koerhuis, D.; Boons, G.J.; Bosch, B.J.; Rey, F.A.; de Groot, R.J.; et al. Structural Basis for Human Coronavirus Attachment to Sialic Acid Receptors. Nat. Struct. Mol. Biol. 2019, 26, 481–489. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.; Denison, M.R. Implications of Altered Replication Fidelity on the Evolution and Pathogenesis of Coronaviruses. Curr. Opin. Virol. 2012, 2, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent Emergence of SARS-CoV-2 Spike Deletion H69/V70 and Its Role in the Alpha Variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Leigh Brown, A.J.; Hepler, N.L.; Mehta, S.R.; Richman, D.D.; Smith, D.M.; Kosakovsky Pond, S.L. The Global Transmission Network of HIV-1. J. Infect. Dis. 2014, 209, 304–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.; Neher, R. Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- GISAID—Gisaid. 2021. Available online: https://gisaid.org/ (accessed on 15 August 2022).
- Mercatelli, D.; Triboli, L.; Fornasari, E.; Ray, F.; Giorgi, F.M. Coronapp: A Web Application to Annotate and Monitor SARS-CoV-2 Mutations. J. Med. Virol. 2021, 93, 3238–3245. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Kimura, M. Diffusion Models in Population Genetics. J. Appl. Probab. 1964, 1, 177–232. [Google Scholar] [CrossRef] [Green Version]
- Chudalayandi, S. Bioinformatics_Workbook. 2021. Available online: https://bioinformaticsworkbook.org/about.html#gsc.tab=0 (accessed on 15 August 2022).
- Format, V.; Format, C.; Converter, I.; Explorer, I.; Svg, A. RNAfold Web Server. Monatshefte. 2011, pp. 8–10. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi (accessed on 15 August 2022).
- SIB Swiss Institute of Bioinformatics ExPASy-Translate Tool 2011. Available online: https://www.expasy.org/ (accessed on 15 August 2022).
- Swiss Institue of Bioinformatics SWISS-MODEL Interactive Workspace. Database 2021. Available online: https://swissmodel.expasy.org/ (accessed on 15 August 2022).
- CoVariants. Available online: https://covariants.org/ (accessed on 15 August 2022).
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The Proximal Origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Tonkin-Hill, G.; Martincorena, I.; Amato, R.; Lawson, A.R.; Gerstung, M.; Johnston, I.; Jackson, D.K.; Park, N.; Lensing, S.V.; Quail, M.A.; et al. Patterns of Within-Host Genetic Diversity in SARS-CoV-2. Elife 2021, 10, e66857. [Google Scholar] [CrossRef]
- Elhassan, N.; Gebremeskel, E.I.; Elnour, M.A.; Isabirye, D.; Okello, J.; Hussien, A.; Kwiatksowski, D.; Hirbo, J.; Tishkoff, S.; Ibrahim, M.E. The Episode of Genetic Drift Defining the Migration of Humans out of Africa Is Derived from a Large East African Population Size. PLoS ONE 2014, 9, e97674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.C.; Tishkoff, S.A. African Genetic Diversity: Implications for Human Demographic History, Modern Human Origins, and Complex Disease Mapping. Annu. Rev. Genomics Hum. Genet. 2008, 9, 403–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirugo, G.; Hennig, B.J.; Adeyemo, A.A.; Matimba, A.; Newport, M.J.; Ibrahim, M.E.; Ryckman, K.K.; Tacconelli, A.; Mariani-Costantini, R.; Novelli, G.; et al. Genetic Studies of African Populations: An Overview on Disease Susceptibility and Response to Vaccines and Therapeutics. Hum. Genet. 2008, 123, 557–598. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escalera, A.; Gonzalez-Reiche, A.S.; Aslam, S.; Mena, I.; Laporte, M.; Pearl, R.L.; Fossati, A.; Rathnasinghe, R.; Alshammary, H.; van de Guchte, A.; et al. Mutations in SARS-CoV-2 Variants of Concern Link to Increased Spike Cleavage and Virus Transmission. Cell Host Microbe 2022, 30, 373–387.e7. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hannon, W.W.; Loes, A.N.; Hauser, K.; Dillen, J.R.; Ferri, E.; Farrell, A.G.; Dadonaite, B.; McCallum, M.; et al. Shifting Mutational Constraints in the SARS-CoV-2 Receptor-Binding Domain during Viral Evolution. Science 2022, 424, eabo7896. [Google Scholar] [CrossRef]
- Klinakis, A.; Cournia, Z.; Rampias, T. N-Terminal Domain Mutations of the Spike Protein Are Structurally Implicated in Epitope Recognition in Emerging SARS-CoV-2 Strains. Comput. Struct. Biotechnol. J. 2021, 19, 5556–5567. [Google Scholar] [CrossRef]
- Burch, C.L.; Chao, L. Evolution by Small Steps and Rugged Landscapes in the RNA Virus Φ6. Genetics 1999, 151, 921–927. [Google Scholar] [CrossRef]
- Xue, S.-A.; Jones, M.D.; Lu, Q.-L.; Middeldorp, J.M.; Griffin, B.E. Genetic Diversity: Frameshift Mechanisms Alter Coding of a Gene (Epstein-Barr Virus LF3 Gene) That Contains Multiple 102-Base-Pair Direct Sequence Repeats. Mol. Cell. Biol. 2003, 23, 2192–2201. [Google Scholar] [CrossRef] [Green Version]
- Mathur, P.; Goyal, P.; Verma, G.; Yadav, P. Entropy Based Analysis of SARS-CoV-2 Spread in India Using Informative Subtype Markers. Sci. Rep. 2021, 11, 15972. [Google Scholar] [CrossRef]
- Sanjuán, R.; Thoulouze, M.I. Why Viruses Sometimes Disperse in Groups. Virus Evol. 2019, 5, vez014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, E.; Sheldon, J.; Perales, C. Viral Quasispecies Evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaskov, J.; Buzacott, K.; Thu, H.M.; Lowry, K.; Holmes, E.C. Long-Term Transmission of Defective RNA Viruses in Humans and Aedes Mosquitoes. Science 2006, 311, 236–238. [Google Scholar] [CrossRef]
- Park, D.; Hahn, Y. Rapid Protein Sequence Evolution via Compensatory Frameshift Is Widespread in RNA Virus Genomes. BMC Bioinform. 2021, 22, 251. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, R.N.; Malik, H.S. Genetic Conflicts: The Usual Suspects and Beyond. J. Exp. Biol. 2017, 220, 6–17. [Google Scholar] [CrossRef] [Green Version]
- Kaján, G.L.; Doszpoly, A.; Tarján, Z.L.; Vidovszky, M.Z.; Papp, T. Virus–Host Coevolution with a Focus on Animal and Human DNA Viruses. J. Mol. Evol. 2020, 88, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Campo, D.S.; Roh, H.J.; Pearlman, B.L.; Fierer, D.S.; Ramachandran, S.; Vaughan, G.; Hinds, A.; Dimitrova, Z.; Skums, P.; Khudyakov, Y. Increased Mitochondrial Genetic Diversity in Persons Infected With Hepatitis C Virus. Cmgh 2016, 2, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.; Salih, A. The Y Chromosome Ancestry Marker R1b1b2: A Surrogate of the SARS-CoV-2 Population Affinity. Hum. Genome Var. 2021, 8, 11. [Google Scholar] [CrossRef]
- Zhang, C.; Verma, A.; Feng, Y.; Melo, M.C.R.; McQuillan, M.; Hansen, M.; Lucas, A.; Park, J.; Ranciaro, A.; Thompson, S.; et al. Impact of Natural Selection on Global Patterns of Genetic Variation and Association with Clinical Phenotypes at Genes Involved in SARS-CoV-2 Infection. Proc. Natl. Acad. Sci. USA 2022, 119, e2123000119. [Google Scholar] [CrossRef]
- Cele, S.; Karim, F.; Lustig, G.; San, J.E.; Hermanus, T.; Tegally, H.; Snyman, J.; Moyo-Gwete, T.; Wilkinson, E.; Bernstein, M.; et al. SARS-CoV-2 Prolonged Infection during Advanced HIV Disease Evolves Extensive Immune Escape. Cell Host Microbe 2022, 30, 154–162.e5. [Google Scholar] [CrossRef]
- Maponga, T.G.; Jeffries, M.; Tegally, H.; Sutherland, A.; Wilkinson, E.; Lessells, R.J.; Msomi, N.; van Zyl, G.; de Oliveira, T.; Preiser, W. Persistent Severe Acute Respiratory Syndrome Coronavirus 2 Infection With Accumulation of Mutations in a Patient with Poorly Controlled Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2022, ciac548. [Google Scholar] [CrossRef] [PubMed]
- Gesesew, H.A.; Koye, D.N.; Fetene, D.M.; Woldegiorgis, M.; Kinfu, Y.; Geleto, A.B.; Melaku, Y.A.; Mohammed, H.; Alene, K.A.; Awoke, M.A.; et al. Risk Factors for COVID-19 Infection, Disease Severity and Related Deaths in Africa: A Systematic Review. BMJ Open 2021, 11, e044618. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; San, J.E.; Cotten, M.; Tegomoh, B.; Mboowa, G.; Martin, D.P.; Baxter, C.; Moir, M.; Lambisia, A.; Diallo, A.; et al. The Evolving SARS-CoV-2 Epidemic in Africa: Insights from Rapidly Expanding Genomic Surveillance. medRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Giri, B.; Guzmán, M.G.; Jiang, H.; Abbès, S. Establishment of Regional Genomic Surveillance Networks in Lower and Lower-Middle Income Countries. 2022; preprints. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Continent | Total Number of 19B (S) | NS-WG | S-WG | Total Substitutions | NS-Spike | S-Spike | Total Substitution in the Spike | Deletion in WG | Deletion–Frameshift In WG | Percentages of Deletion–Frameshift |
|---|---|---|---|---|---|---|---|---|---|---|
| Africa | 1147 | 1468 | 1013 | 2481 | 205 | 132 | 337 | 28 | 18 | ~1.6% |
| Asia | 1349 | 1078 | 718 | 1796 | 150 | 152 | 302 | 18 | 27 | ~2% |
| Europe | 3546 | 1985 | 1205 | 3190 | 277 | 154 | 431 | 39 | 45 | ~1.3% |
| North America | 8313 | 3822 | 2225 | 6047 | 546 | 299 | 845 | 55 | 55 | ~0.66% |
| South America | 244 | 342 | 241 | 583 | 47 | 29 | 76 | 3 | 5 | ~2.1% |
| Oceania | 938 | 323 | 198 | 521 | 55 | 30 | 85 | 11 | 11 | ~1.2% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussein, B.K.; Ibrahium, O.M.; Alamin, M.F.; Ahmed, L.A.M.; Abuswar, S.A.E.; Abdelraheem, M.H.; Ibrahim, M.E. The Spike Protein of SARS-coV2 19B (S) Clade Mirrors Critical Features of Viral Adaptation and Coevolution. Microorganisms 2022, 10, 2017. https://doi.org/10.3390/microorganisms10102017
Hussein BK, Ibrahium OM, Alamin MF, Ahmed LAM, Abuswar SAE, Abdelraheem MH, Ibrahim ME. The Spike Protein of SARS-coV2 19B (S) Clade Mirrors Critical Features of Viral Adaptation and Coevolution. Microorganisms. 2022; 10(10):2017. https://doi.org/10.3390/microorganisms10102017
Chicago/Turabian StyleHussein, Bidour K., Omnia M. Ibrahium, Marwa F. Alamin, Lamees A. M. Ahmed, Safa A. E. Abuswar, Mohammed H. Abdelraheem, and Muntaser E. Ibrahim. 2022. "The Spike Protein of SARS-coV2 19B (S) Clade Mirrors Critical Features of Viral Adaptation and Coevolution" Microorganisms 10, no. 10: 2017. https://doi.org/10.3390/microorganisms10102017
APA StyleHussein, B. K., Ibrahium, O. M., Alamin, M. F., Ahmed, L. A. M., Abuswar, S. A. E., Abdelraheem, M. H., & Ibrahim, M. E. (2022). The Spike Protein of SARS-coV2 19B (S) Clade Mirrors Critical Features of Viral Adaptation and Coevolution. Microorganisms, 10(10), 2017. https://doi.org/10.3390/microorganisms10102017