
The SARS-CoV-2 S1 Spike Protein Promotes MAPK and NF-kB Activation in Human Lung Cells and Inflammatory Cytokine Production in Human Lung and Intestinal Epithelial Cells

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Experimental Manipulation
2.3. Western Blotting Analysis
2.4. Meso Scale Analysis of Cytokines
2.5. Statistical Analysis
3. Results
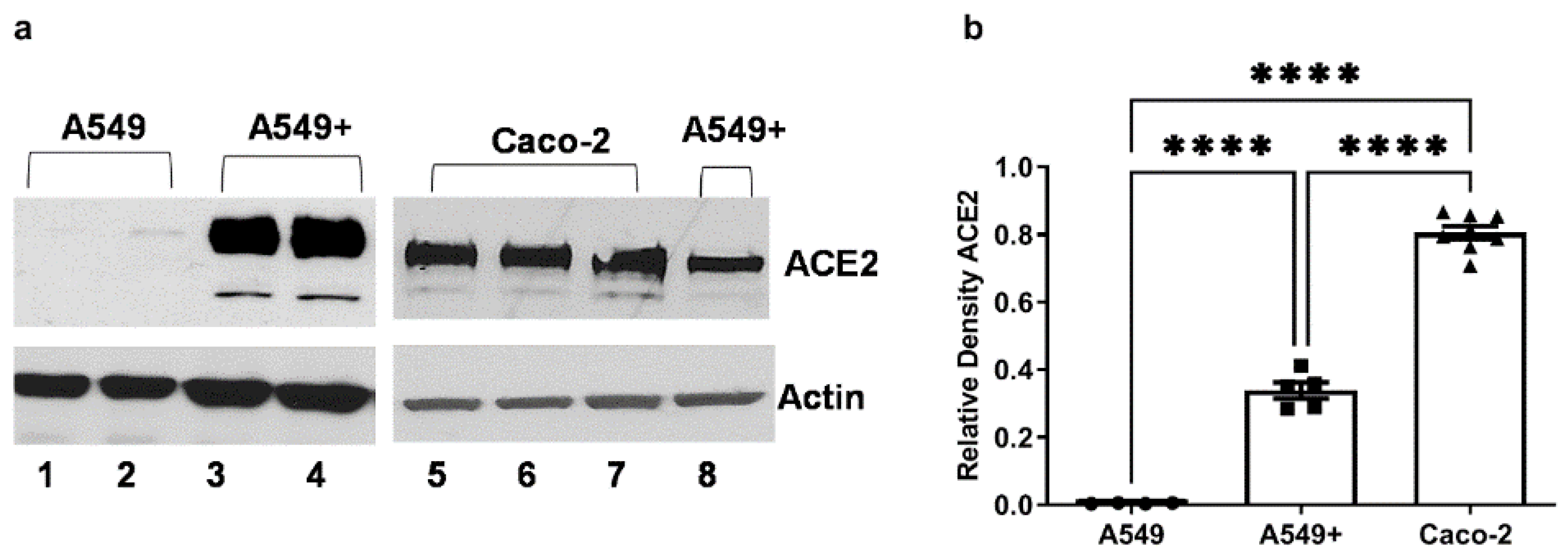
3.1. Expression of SARS-CoV-2 Receptor ACE2 in A549, A549+, and Caco-2 Cell Lines
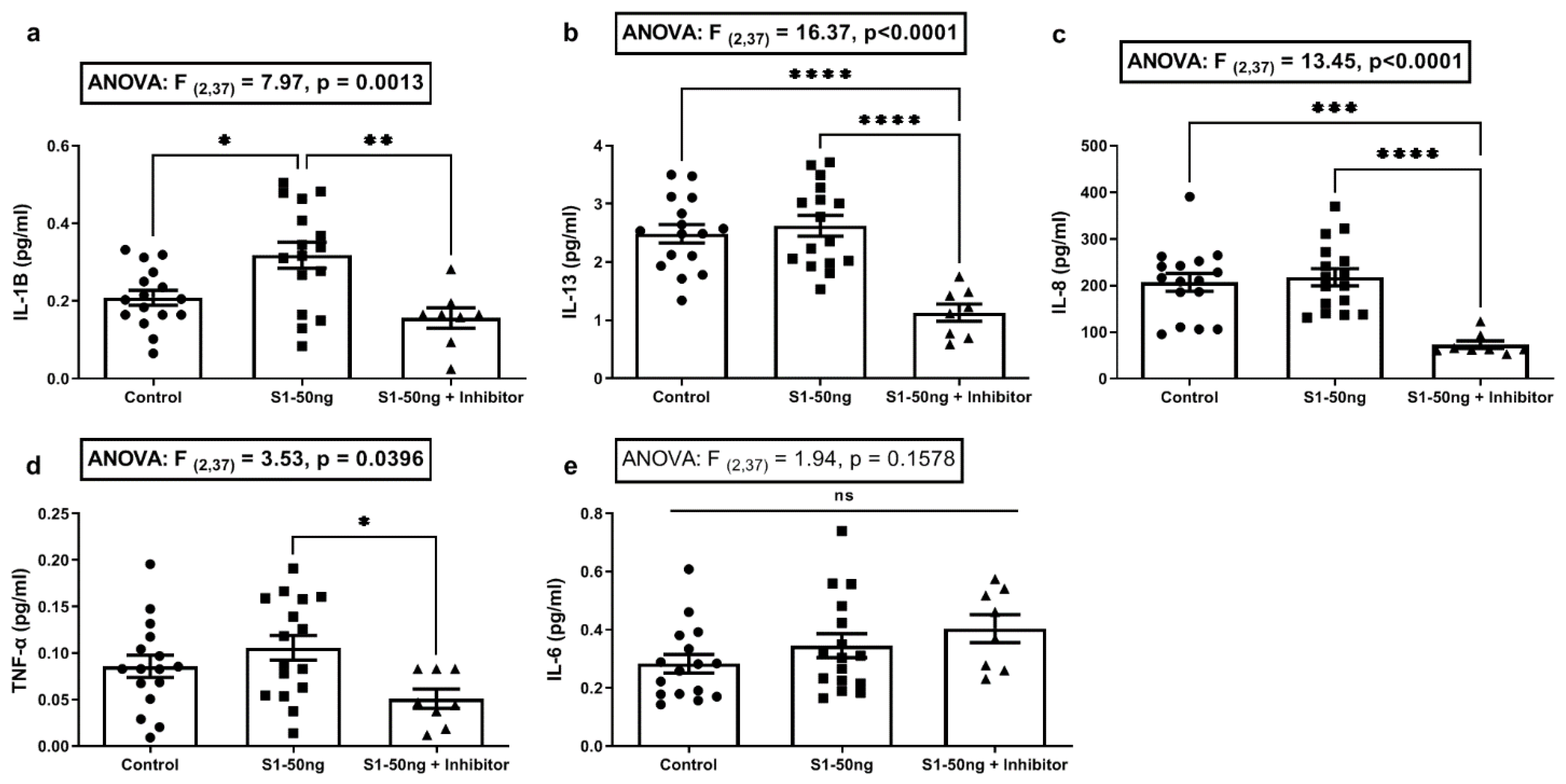
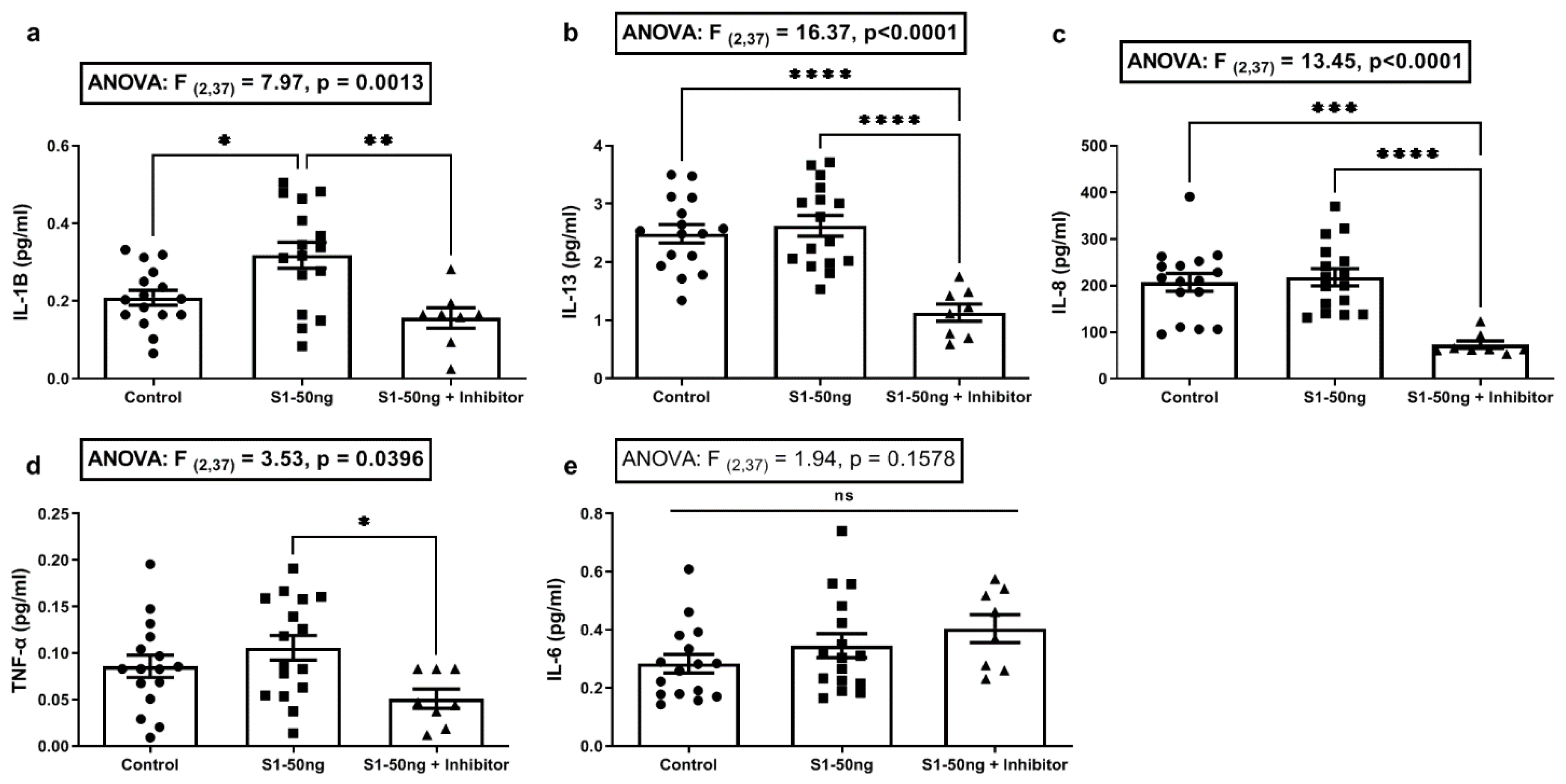
3.2. The SARS-CoV-2 S1 Spike Protein Stimulates Production of the Key COVID-19 Inflammatory Cytokine IL-1β in Human A549+ Lung Cells That Is Blocked with the MEK1/2 MAPK ERK1/2 Inhibitor
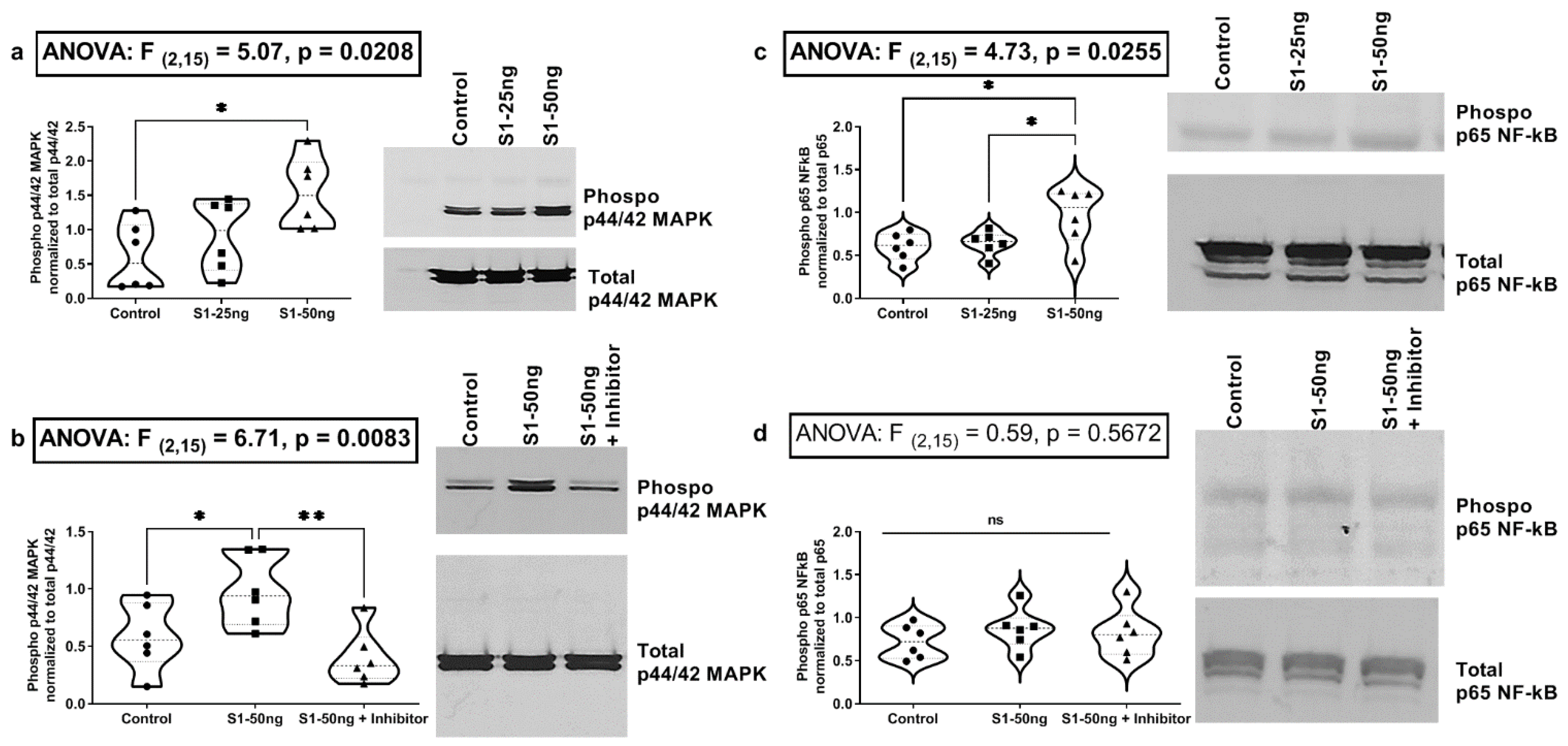
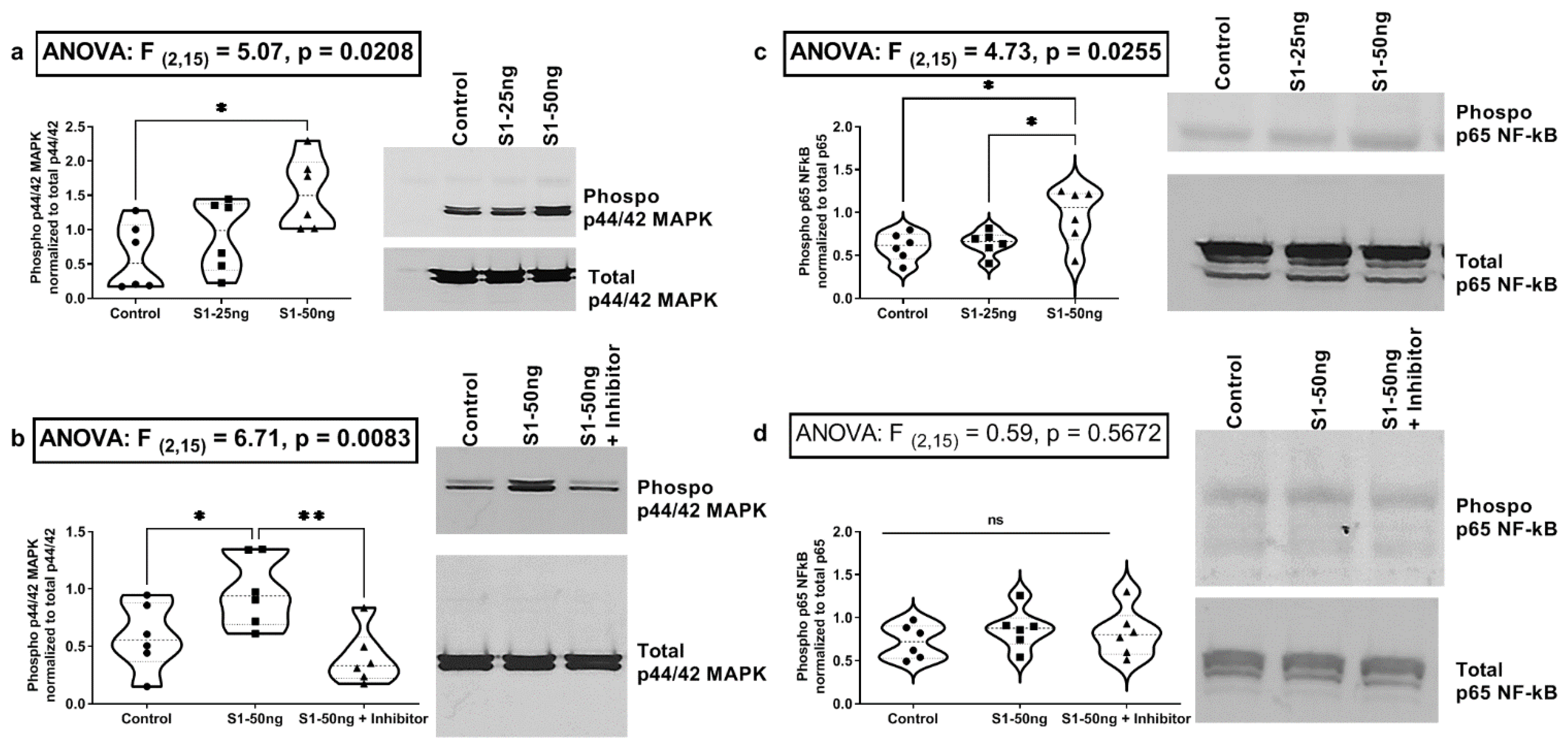
3.3. The SARS-CoV-2 S1 Spike Protein Activates the MAPK-ERK1/2 and the NF-kB p65 Signaling Pathways in Human A549+ Lung Cells
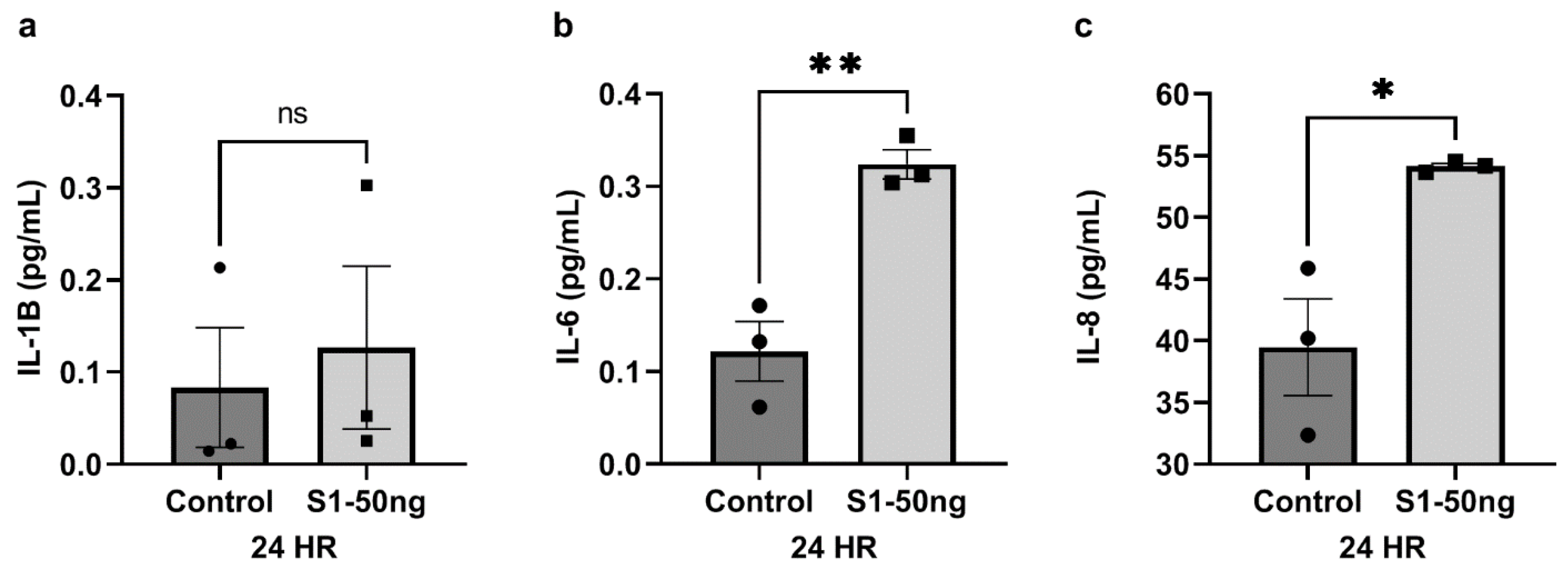
3.4. The SARS-CoV-2 S1 Spike Protein Stimulates Inflammatory Cytokines IL-6 and IL-8 Production in Caco-2 Human Intestinal Epithelial Cells
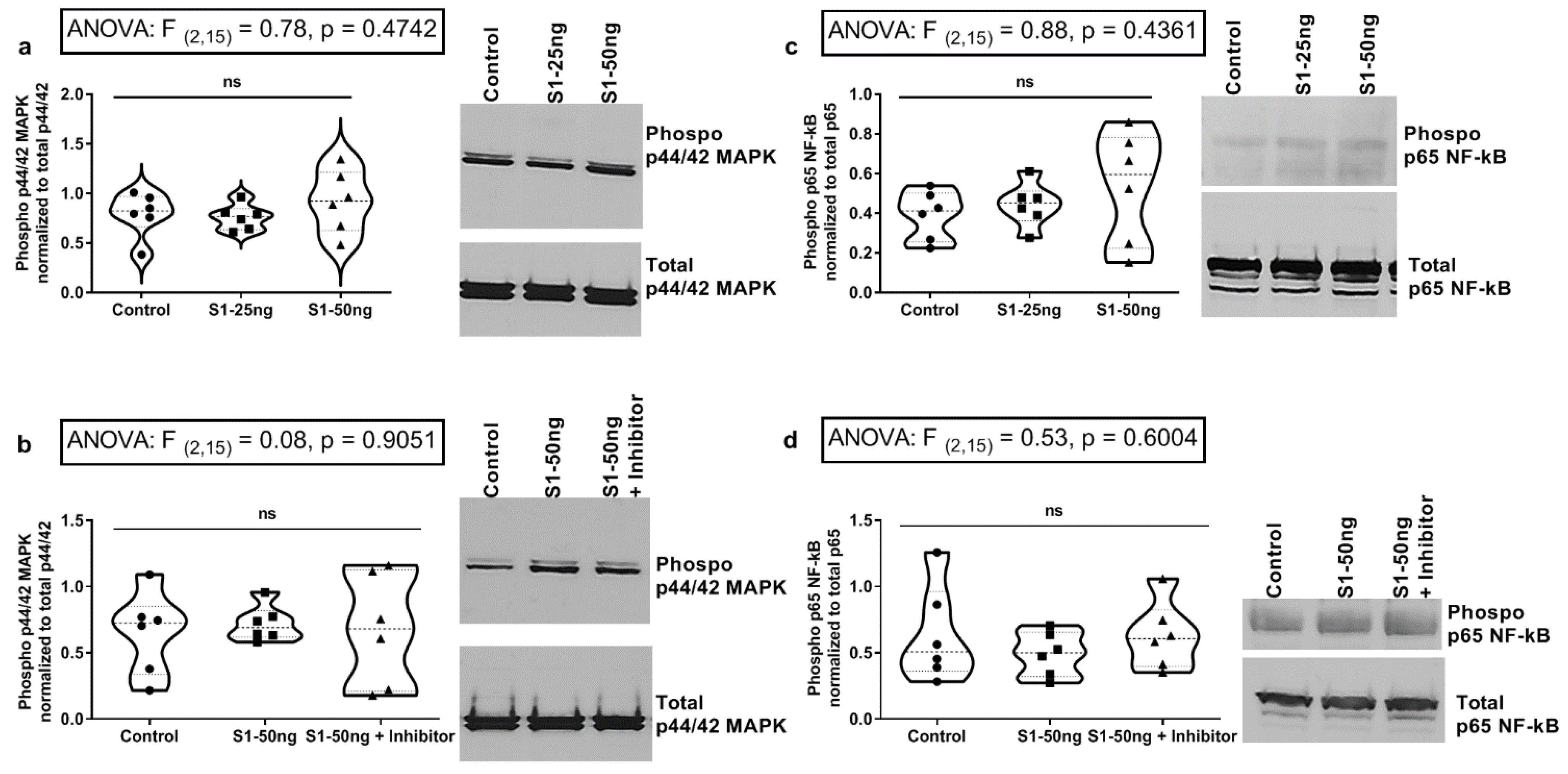
3.5. The SARS-CoV-2 S1 Spike Protein Does Not Stimulate MAPK p-ERK1/2 or NF-kB p-p65 Signaling Pathways in Human Caco-2 Intestinal Epithelial Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Worldometer. Coronavirus. Available online: https://www.worldometers.info/coronavirus/ (accessed on 3 March 2022).
- Moss, M.; Burnham, E.L. Chronic alcohol abuse, acute respiratory distress syndrome, and multiple organ dysfunction. Crit. Care Med. 2003, 31, S207–S212. [Google Scholar] [CrossRef]
- Dickson, R.P. The Lung Microbiome and ARDS. It Is Time to Broaden the Model. Am. J. Respir. Crit. Care Med. 2018, 197, 549–551. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Marx, W.; O’Neil, A.; Athan, E.; Walder, K.; Berk, M.; Olive, L.; Carvalho, A.F.; et al. The cytokine storms of COVID-19, H1N1 influenza, CRS and MAS compared. Can one sized treatment fit all? Cytokine 2021, 144, 155593. [Google Scholar] [CrossRef]
- Que, Y.; Hu, C.; Wan, K.; Hu, P.; Wang, R.; Luo, J.; Li, T.; Ping, R.; Hu, Q.; Sun, Y.; et al. Cytokine release syndrome in COVID-19: A major mechanism of morbidity and mortality. Int. Rev. Immunol. 2022, 41, 217–230. [Google Scholar] [CrossRef]
- Rahban, M.; Stanek, A.; Hooshmand, A.; Khamineh, Y.; Ahi, S.; Kazim, S.N.; Ahmad, F.; Muronetz, V.; Samy Abousenna, M.; Zolghadri, S.; et al. Infection of Human Cells by SARS-CoV-2 and Molecular Overview of Gastrointestinal, Neurological, and Hepatic Problems in COVID-19 Patients. J. Clin. Med. 2021, 10, 4802. [Google Scholar] [CrossRef]
- Kumar, A.; Narayan, R.K.; Prasoon, P.; Kumari, C.; Kaur, G.; Kumar, S.; Kulandhasamy, M.; Sesham, K.; Pareek, V.; Faiq, M.A.; et al. COVID-19 Mechanisms in the Human Body-What We Know So Far. Front. Immunol. 2021, 12, 693938. [Google Scholar] [CrossRef]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.C.; Klaser, K.; Antonelli, M.; Canas, L.S.; et al. Attributes and predictors of long COVID. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Ramakrishnan, R.K.; Kashour, T.; Hamid, Q.; Halwani, R.; Tleyjeh, I.M. Unraveling the Mystery Surrounding Post-Acute Sequelae of COVID-19. Front. Immunol. 2021, 12, 686029. [Google Scholar] [CrossRef]
- Letarov, A.V.; Babenko, V.V.; Kulikov, E.E. Free SARS-CoV-2 Spike Protein S1 Particles May Play a Role in the Pathogenesis of COVID-19 Infection. Biochem. Biokhimiia 2021, 86, 257–261. [Google Scholar] [CrossRef]
- Suzuki, Y.J.; Gychka, S.G. SARS-CoV-2 Spike Protein Elicits Cell Signaling in Human Host Cells: Implications for Possible Consequences of COVID-19 Vaccines. Vaccines 2021, 9, 36. [Google Scholar] [CrossRef]
- Theoharides, T.C. Could SARS-CoV-2 Spike Protein Be Responsible for Long-COVID Syndrome? Mol. Neurobiol. 2022, 59, 1850–1861. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; Zheng, Q.; Zhang, H.; Niu, Y.; Lou, Y.; Wang, H. The SARS-CoV-2 Spike Glycoprotein Biosynthesis, Structure, Function, and Antigenicity: Implications for the Design of Spike-Based Vaccine Immunogens. Front. Immunol. 2020, 11, 576622. [Google Scholar] [CrossRef]
- Martínez-Flores, D.; Zepeda-Cervantes, J.; Cruz-Reséndiz, A.; Aguirre-Sampieri, S.; Sampieri, A.; Vaca, L. SARS-CoV-2 Vaccines Based on the Spike Glycoprotein and Implications of New Viral Variants. Front. Immunol. 2021, 12, 701501. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Mendonça, L.; Yang, Y.; Gao, Y.; Shen, C.; Liu, J.; Ni, T.; Ju, B.; Liu, C.; Tang, X.; et al. The Architecture of Inactivated SARS-CoV-2 with Postfusion Spikes Revealed by Cryo-EM and Cryo-ET. Structure 2020, 28, 1218–1224.e1214. [Google Scholar] [CrossRef]
- Bar-On, Y.M.; Flamholz, A.; Phillips, R.; Milo, R. SARS-CoV-2 (COVID-19) by the numbers. eLife 2020, 9, e57309. [Google Scholar] [CrossRef] [Green Version]
- Ogata, A.F.; Maley, A.M.; Wu, C.; Gilboa, T.; Norman, M.; Lazarovits, R.; Mao, C.P.; Newton, G.; Chang, M.; Nguyen, K.; et al. Ultra-Sensitive Serial Profiling of SARS-CoV-2 Antigens and Antibodies in Plasma to Understand Disease Progression in COVID-19 Patients with Severe Disease. Clin. Chem. 2020, 66, 1562–1572. [Google Scholar] [CrossRef]
- Ogata, A.F.; Cheng, C.A.; Desjardins, M.; Senussi, Y.; Sherman, A.C.; Powell, M.; Novack, L.; Von, S.; Li, X.; Baden, L.R.; et al. Circulating SARS-CoV-2 Vaccine Antigen Detected in the Plasma of mRNA-1273 Vaccine Recipients. Clin. Infect. Dis. 2021, 74, 715–718. [Google Scholar] [CrossRef]
- Suzuki, Y.J. The viral protein fragment theory of COVID-19 pathogenesis. Med. Hypotheses 2020, 144, 110267. [Google Scholar] [CrossRef]
- Ning, Q.; Wu, D.; Wang, X.; Xi, D.; Chen, T.; Chen, G.; Wang, H.; Lu, H.; Wang, M.; Zhu, L.; et al. The mechanism underlying extrapulmonary complications of the coronavirus disease 2019 and its therapeutic implication. Signal. Transduct. Target. Ther. 2022, 7, 57. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.F.; Yuen, T.T.; Shuai, H.; Yuan, S.; Wang, Y.; Hu, B.; Yip, C.C.; Tsang, J.O.; Huang, X.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe 2020, 1, e14–e23. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Schreiber, A.; Viemann, D.; Schöning, J.; Schloer, S.; Mecate Zambrano, A.; Brunotte, L.; Faist, A.; Schöfbänker, M.; Hrincius, E.; Hoffmann, H.; et al. The MEK1/2-inhibitor ATR-002 efficiently blocks SARS-CoV-2 propagation and alleviates pro-inflammatory cytokine/chemokine responses. Cell Mol. Life Sci. 2022, 79, 65. [Google Scholar] [CrossRef]
- Polak, S.B.; Van Gool, I.C.; Cohen, D.; von der Thüsen, J.H.; van Paassen, J. A systematic review of pathological findings in COVID-19: A pathophysiological timeline and possible mechanisms of disease progression. Mod. Pathol. 2020, 33, 2128–2138. [Google Scholar] [CrossRef]
- Ramani, R.; Laplante, J.M.; Church, T.M.; Farrell, G.M.; Lamson, D.M.; St George, K. CACO-2 cells: A continuous cell line with sensitive and broad-spectrum utility for respiratory virus culture. J. Virol. Methods 2021, 293, 114120. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, A.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Cinatl, J., Jr.; Hoever, G.; Morgenstern, B.; Preiser, W.; Vogel, J.U.; Hofmann, W.K.; Bauer, G.; Michaelis, M.; Rabenau, H.F.; Doerr, H.W. Infection of cultured intestinal epithelial cells with severe acute respiratory syndrome coronavirus. Cell Mol. Life Sci. 2004, 61, 2100–2112. [Google Scholar] [CrossRef]
- Ozer, E.A.; Simons, L.M.; Adewumi, O.M.; Fowotade, A.A.; Omoruyi, E.C.; Adeniji, J.A.; Olayinka, O.A.; Dean, T.J.; Zayas, J.; Bhimalli, P.P.; et al. Multiple expansions of globally uncommon SARS-CoV-2 lineages in Nigeria. Nat. Commun. 2022, 13, 688. [Google Scholar] [CrossRef]
- Mamede, J.I.; Sitbon, M.; Battini, J.L.; Courgnaud, V. Heterogeneous susceptibility of circulating SIV isolate capsids to HIV-interacting factors. Retrovirology 2013, 10, 77. [Google Scholar] [CrossRef]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef]
- Suzuki, Y.J.; Nikolaienko, S.I.; Dibrova, V.A.; Dibrova, Y.V.; Vasylyk, V.M.; Novikov, M.Y.; Shults, N.V.; Gychka, S.G. SARS-CoV-2 spike protein-mediated cell signaling in lung vascular cells. Vasc. Pharmacol. 2021, 137, 106823. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.L.; Solopov, P.A.; Sharlow, E.R.; Lazo, J.S.; Marik, P.E.; Catravas, J.D. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Κ18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L477–L484. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, C.B.; Shaikh, M.; Bishehsari, F.; Swanson, G.; Voigt, R.M.; Dodiya, H.; Wilkinson, P.; Samelco, B.; Song, S.; Keshavarzian, A. Alcohol Feeding in Mice Promotes Colonic Hyperpermeability and Changes in Colonic Organoid Stem Cell Fate. Alcohol. Clin. Exp. Res. 2017, 41, 2100–2113. [Google Scholar] [CrossRef]
- Giron, L.B.; Dweep, H.; Yin, X.; Wang, H.; Damra, M.; Goldman, A.R.; Gorman, N.; Palmer, C.S.; Tang, H.Y.; Shaikh, M.W.; et al. Plasma Markers of Disrupted Gut Permeability in Severe COVID-19 Patients. Front. Immunol. 2021, 12, 686240. [Google Scholar] [CrossRef]
- Cory, T.J.; Emmons, R.S.; Yarbro, J.R.; Davis, K.L.; Pence, B.D. Metformin Suppresses Monocyte Immunometabolic Activation by SARS-CoV-2 Spike Protein Subunit 1. Front. Immunol. 2021, 12, 733921. [Google Scholar] [CrossRef]
- Paidi, R.K.; Jana, M.; Mishra, R.K.; Dutta, D.; Pahan, K. Selective Inhibition of the Interaction between SARS-CoV-2 Spike S1 and ACE2 by SPIDAR Peptide Induces Anti-Inflammatory Therapeutic Responses. J. Immunol. 2021, 207, 2521–2533. [Google Scholar] [CrossRef]
- Low, J.S.; Jerak, J.; Tortorici, M.A.; McCallum, M.; Pinto, D.; Cassotta, A.; Foglierini, M.; Mele, F.; Abdelnabi, R.; Weynand, B.; et al. ACE2-binding exposes the SARS-CoV-2 fusion peptide to broadly neutralizing coronavirus antibodies. Science 2022, 377, 735–742. [Google Scholar] [CrossRef]
- Kaminska, B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—From molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta 2005, 1754, 253–262. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef] [PubMed]
- Ombrello, M.J.; Schulert, G.S. COVID-19 and cytokine storm syndrome: Are there lessons from macrophage activation syndrome? Transl. Res. 2021, 232, 19. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, J.T.; Alexander, S.; Christoph, K.; Matthias, Z.; Julia, F.; Marie-Christine, A.; Jakob, J.M.; Jessica, G.; Sandra, W.; de Silva, U.S.; et al. The SARS-CoV-2 spike protein primes inflammasome-mediated interleukin-1-beta secretion in COVID-19 patient-derived macrophages. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Barilli, A.; Visigalli, R.; Ferrari, F.; Bianchi, M.G.; Dall’Asta, V.; Rotoli, B.M. Immune-Mediated Inflammatory Responses of Alveolar Epithelial Cells: Implications for COVID-19 Lung Pathology. Biomedicines 2022, 10, 618. [Google Scholar] [CrossRef]
- Vora, S.M.; Lieberman, J.; Wu, H. Inflammasome activation at the crux of severe COVID-19. Nat. Rev. Immunol. 2021, 21, 694–703. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- Khani, E.; Shahrabi, M.; Rezaei, H.; Pourkarim, F.; Afsharirad, H.; Solduzian, M. Current evidence on the use of anakinra in COVID-19. Int. Immunopharmacol. 2022, 111, 109075. [Google Scholar] [CrossRef]
- Paniri, A.; Akhavan-Niaki, H. Emerging role of IL-6 and NLRP3 inflammasome as potential therapeutic targets to combat COVID-19: Role of lncRNAs in cytokine storm modulation. Life Sci. 2020, 257, 118114. [Google Scholar] [CrossRef]
- Rahman, M.; Irmler, M.; Keshavan, S.; Introna, M.; Beckers, J.; Palmberg, L.; Johanson, G.; Ganguly, K.; Upadhyay, S. Differential Effect of SARS-CoV-2 Spike Glycoprotein 1 on Human Bronchial and Alveolar Lung Mucosa Models: Implications for Pathogenicity. Viruses 2021, 13, 2537. [Google Scholar] [CrossRef]
- Olajide, O.A.; Iwuanyanwu, V.U.; Lepiarz-Raba, I.; Al-Hindawi, A.A. Induction of Exaggerated Cytokine Production in Human Peripheral Blood Mononuclear Cells by a Recombinant SARS-CoV-2 Spike Glycoprotein S1 and Its Inhibition by Dexamethasone. Inflammation 2021, 44, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Olajide, O.A.; Iwuanyanwu, V.U.; Adegbola, O.D.; Al-Hindawi, A.A. SARS-CoV-2 Spike Glycoprotein S1 Induces Neuroinflammation in BV-2 Microglia. Mol. Neurobiol. 2022, 59, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Corpetti, C.; Del Re, A.; Seguella, L.; Palenca, I.; Rurgo, S.; De Conno, B.; Pesce, M.; Sarnelli, G.; Esposito, G. Cannabidiol inhibits SARS-Cov-2 spike (S) protein-induced cytotoxicity and inflammation through a PPARγ-dependent TLR4/NLRP3/Caspase-1 signaling suppression in Caco-2 cell line. Phytother. Res. PTR 2021, 35, 6893–6903. [Google Scholar] [CrossRef] [PubMed]
- Wurtz, N.; Penant, G.; Jardot, P.; Duclos, N.; La Scola, B. Culture of SARS-CoV-2 in a panel of laboratory cell lines, permissivity, and differences in growth profile. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2021, 40, 477–484. [Google Scholar] [CrossRef]
- de Souza, G.A.P.; Le Bideau, M.; Boschi, C.; Ferreira, L.; Wurtz, N.; Devaux, C.; Colson, P.; La Scola, B. Emerging SARS-CoV-2 Genotypes Show Different Replication Patterns in Human Pulmonary and Intestinal Epithelial Cells. Viruses 2021, 14, 23. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Li, Y.; Wang, Z.; Ma, F.; Luo, R.; Xu, X.; Zhou, G.; Wang, J.; Niu, J.; et al. Platelets mediate inflammatory monocyte activation by SARS-CoV-2 spike protein. J. Clin. Investig. 2022, 132, e150101. [Google Scholar] [CrossRef]
- Paidi, R.K.; Jana, M.; Raha, S.; McKay, M.; Sheinin, M.; Mishra, R.K.; Pahan, K. Eugenol, a Component of Holy Basil (Tulsi) and Common Spice Clove, Inhibits the Interaction Between SARS-CoV-2 Spike S1 and ACE2 to Induce Therapeutic Responses. J. Neuroimmune Pharmacol. 2021, 16, 743–755. [Google Scholar] [CrossRef]
- Sharma, V.K.; Prateeksha; Singh, S.P.; Singh, B.N.; Rao, C.V.; Barik, S.K. Nanocurcumin Potently Inhibits SARS-CoV-2 Spike Protein-Induced Cytokine Storm by Deactivation of MAPK/NF-κB Signaling in Epithelial Cells. ACS Appl. Bio Mater. 2022, 5, 483–491. [Google Scholar] [CrossRef]
- Hoter, A.; Naim, H.Y. Biochemical Characterization of SARS-CoV-2 Spike RBD Mutations and Their Impact on ACE2 Receptor Binding. Front. Mol. BioSci. 2022, 9, 893843. [Google Scholar] [CrossRef]
- Kircheis, R.; Planz, O. Could a Lower Toll-like Receptor (TLR) and NF-κB Activation Due to a Changed Charge Distribution in the Spike Protein Be the Reason for the Lower Pathogenicity of Omicron? Int. J. Mol. Sci. 2022, 23, 5966. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Ao, Z.; Ouyang, M.J.; Olukitibi, T.A.; Yao, X. SARS-CoV-2 Delta spike protein enhances the viral fusogenicity and inflammatory cytokine production. iScience 2022, 25, 104759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N.; et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature 2021, 592, 122–127. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Woo, H.G. Omicron: A Heavily Mutated SARS-CoV-2 Variant Exhibits Stronger Binding to ACE2 and Potently Escapes Approved COVID-19 Therapeutic Antibodies. Front. Immunol. 2021, 12, 830527. [Google Scholar] [CrossRef] [PubMed]
- He, C.; He, X.; Yang, J.; Lei, H.; Hong, W.; Song, X.; Yang, L.; Li, J.; Wang, W.; Shen, G.; et al. Spike protein of SARS-CoV-2 Omicron (B.1.1.529) variant have a reduced ability to induce the immune response. Signal. Transduct Target. Ther. 2022, 7, 119. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.H.; Zhang, S.; Porritt, R.A.; Noval Rivas, M.; Paschold, L.; Willscher, E.; Binder, M.; Arditi, M.; Bahar, I. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 25254–25262. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.K.; Francisco, E.B.; Yogendra, R.; Long, E.; Pise, A.; Rodrigues, H.; Hall, E.; Herrera, M.; Parikh, P.; Guevara-Coto, J.; et al. Persistence of SARS CoV-2 S1 Protein in CD16+ Monocytes in Post-Acute Sequelae of COVID-19 (PASC) up to 15 Months Post-Infection. Front. Immunol. 2021, 12, 746021. [Google Scholar] [CrossRef]
- Files, J.K.; Sarkar, S.; Fram, T.R.; Boppana, S.; Sterrett, S.; Qin, K.; Bansal, A.; Long, D.M.; Sabbaj, S.; Kobie, J.J.; et al. Duration of post-COVID-19 symptoms is associated with sustained SARS-CoV-2-specific immune responses. JCI Insight. 2021, 6, e151544. [Google Scholar] [CrossRef]
- Frank, M.G.; Nguyen, K.H.; Ball, J.B.; Hopkins, S.; Kelley, T.; Baratta, M.V.; Fleshner, M.; Maier, S.F. SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: Evidence of PAMP-like properties. Brain Behav. Immun. 2022, 100, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Swank, Z.; Senussi, Y.; Alter, G.; Walt, D.R. Persistent circulating SARS-CoV-2 spike is associated with post-acute COVID-19 sequelae. medRxiv 2022. Preprint. [Google Scholar] [CrossRef]
- Raghavan, S.; Kenchappa, D.B.; Leo, M.D. SARS-CoV-2 Spike Protein Induces Degradation of Junctional Proteins That Maintain Endothelial Barrier Integrity. Front. Cardiovasc. Med. 2021, 8, 687783. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forsyth, C.B.; Zhang, L.; Bhushan, A.; Swanson, B.; Zhang, L.; Mamede, J.I.; Voigt, R.M.; Shaikh, M.; Engen, P.A.; Keshavarzian, A. The SARS-CoV-2 S1 Spike Protein Promotes MAPK and NF-kB Activation in Human Lung Cells and Inflammatory Cytokine Production in Human Lung and Intestinal Epithelial Cells. Microorganisms 2022, 10, 1996. https://doi.org/10.3390/microorganisms10101996
Forsyth CB, Zhang L, Bhushan A, Swanson B, Zhang L, Mamede JI, Voigt RM, Shaikh M, Engen PA, Keshavarzian A. The SARS-CoV-2 S1 Spike Protein Promotes MAPK and NF-kB Activation in Human Lung Cells and Inflammatory Cytokine Production in Human Lung and Intestinal Epithelial Cells. Microorganisms. 2022; 10(10):1996. https://doi.org/10.3390/microorganisms10101996
Chicago/Turabian StyleForsyth, Christopher B., Lijuan Zhang, Abhinav Bhushan, Barbara Swanson, Li Zhang, João I. Mamede, Robin M. Voigt, Maliha Shaikh, Phillip A. Engen, and Ali Keshavarzian. 2022. "The SARS-CoV-2 S1 Spike Protein Promotes MAPK and NF-kB Activation in Human Lung Cells and Inflammatory Cytokine Production in Human Lung and Intestinal Epithelial Cells" Microorganisms 10, no. 10: 1996. https://doi.org/10.3390/microorganisms10101996
APA StyleForsyth, C. B., Zhang, L., Bhushan, A., Swanson, B., Zhang, L., Mamede, J. I., Voigt, R. M., Shaikh, M., Engen, P. A., & Keshavarzian, A. (2022). The SARS-CoV-2 S1 Spike Protein Promotes MAPK and NF-kB Activation in Human Lung Cells and Inflammatory Cytokine Production in Human Lung and Intestinal Epithelial Cells. Microorganisms, 10(10), 1996. https://doi.org/10.3390/microorganisms10101996