The COVID-19 Pandemic during the Time of the Diabetes Pandemic: Likely Fraternal Twins?

 ,
,
Abstract
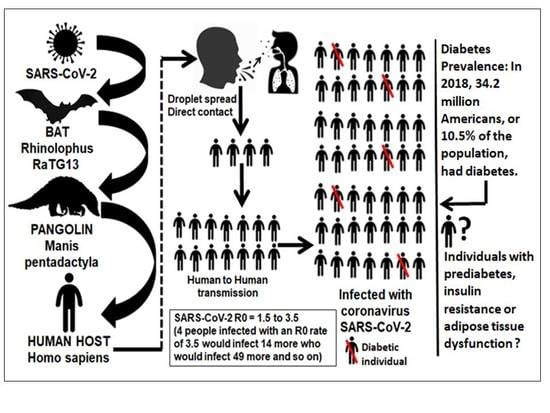
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Viral Infections and Immunometabolic Disease
3. At the Crossroad of COVID-19 and Diabetes Epidemiology
4. Obesity Meets COVID-19
5. Mirror Images: Immunoinflammatory Pathobiology of COVID-19 and Diabetes
6. Chronic Low-Grade Subclinical Systemic Inflammation in Prediabetes and Adipose Tissue Dysfunction Meets COVID-19
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fauci, A.S.; Lane, H.C.; Redfield, R.R. Covid-19-Navigating the Uncharted. N. Engl. J. Med. 2020, 382, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Bio-Med. Atenei Parm. 2020, 91, 157–160. [Google Scholar]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef]
- Yang, J.K.; Feng, Y.; Yuan, M.Y.; Yuan, S.Y.; Fu, H.J.; Wu, B.Y.; Sun, G.Z.; Yang, G.R.; Zhang, X.L.; Wang, L.; et al. Plasma glucose levels and diabetes are independent predictors for mortality and morbidity in patients with SARS. Diabet. Med. 2006, 23, 623–628. [Google Scholar] [CrossRef]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Guo, W.; Li, M.; Dong, Y.; Zhou, H.; Zhang, Z.; Tian, C.; Qin, R.; Wang, H.; Shen, Y.; Du, K.; et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab. Res. Rev. 2020, e3319. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef]
- Pal, R.; Bhadada, S.K. COVID-19 and non-communicable diseases. Postgrad. Med. J. 2020. [Google Scholar] [CrossRef]
- Knapp, S. Diabetes and infection: Is there a link?—A mini-review. Gerontology 2013, 59, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, A.; Cassani, G.; Puggioni, A.; Rossi, A.; Colombo, A.; Onodera, T.; Ferrannini, E. The diabetes pandemic and associated infections: Suggestions for clinical microbiology. Rev. Med. Microbiol. 2019, 30, 1–17. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Dalan, R.; Hopkins, D.; Mingrone, G.; Boehm, B.O. Endocrine and metabolic link to coronavirus infection. Nat. Rev. Endocrinol. 2020, 16, 297–298. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Calvo, T. Enterovirus infection and type 1 diabetes: Unraveling the crime scene. Clin. Exp. Immunol. 2019, 195, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Serfaty, L. Metabolic Manifestations of Hepatitis C Virus: Diabetes Mellitus, Dyslipidemia. Clin. Liver Dis. 2017, 21, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Soto, C. Prion-Like Protein Aggregates and Type 2 Diabetes. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, K.; Morris, J.; Bridson, T.; Govan, B.; Rush, C.; Ketheesan, N. Immunological mechanisms contributing to the double burden of diabetes and intracellular bacterial infections. Immunology 2015, 144, 171–185. [Google Scholar] [CrossRef]
- Gonzalez Plaza, J.J.; Hulak, N.; Kausova, G.; Zhumadilov, Z.; Akilzhanova, A. Role of metabolism during viral infections, and crosstalk with the innate immune system. Intractable Rare Dis. Res. 2016, 5, 90–96. [Google Scholar] [CrossRef]
- Zhou, T.; Hu, Z.; Yang, S.; Sun, L.; Yu, Z.; Wang, G. Role of Adaptive and Innate Immunity in Type 2 Diabetes Mellitus. J. Diabetes Res. 2018, 2018, 7457269. [Google Scholar] [CrossRef]
- Udler, M.S. Type 2 Diabetes: Multiple Genes, Multiple Diseases. Curr. Diabetes Rep. 2019, 19, 55. [Google Scholar] [CrossRef]
- Kulcsar, K.A.; Coleman, C.M.; Beck, S.E.; Frieman, M.B. Comorbid diabetes results in immune dysregulation and enhanced disease severity following MERS-CoV infection. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Allard, R.; Leclerc, P.; Tremblay, C.; Tannenbaum, T.N. Diabetes and the severity of pandemic influenza A (H1N1) infection. Diabetes Care 2010, 33, 1491–1493. [Google Scholar] [CrossRef] [PubMed]
- Bode, B.; Garrett, V.; Messler, J.; McFarland, R.; Crowe, J.; Booth, R.; Klonoff, D.C. Glycemic Characteristics and Clinical Outcomes of COVID-19 Patients Hospitalized in the United States. J. Diabetes Sci. Technol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetol. 2010, 47, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Redondo, M.J.; Steck, A.K.; Pugliese, A. Genetics of type 1 diabetes. Curr. Opin. Genet. Del. 2018, 19, 346–353. [Google Scholar] [CrossRef]
- Johnson, M.B.; Cerosaletti, K.; Flanagan, S.E.; Buckner, J.H. Genetic Mechanisms Highlight Shared Pathways for the Pathogenesis of Polygenic Type 1 Diabetes and Monogenic Autoimmune Diabetes. Curr. Diabetes Rep. 2019, 19, 20. [Google Scholar] [CrossRef]
- Varo, N.; Libby, P.; Nuzzo, R.; Italiano, J.; Doria, A.; Schonbeck, U. Elevated release of sCD40L from platelets of diabetic patients by thrombin, glucose and advanced glycation end products. Diabetes Vasc. Dis. Res. 2005, 2, 81–87. [Google Scholar] [CrossRef]
- Teijaro, J.R. Type I interferons in viral control and immune regulation. Curr. Opin. Virol. 2016, 16, 31–40. [Google Scholar] [CrossRef]
- Wang, Q.; Fang, P.; He, R.; Li, M.; Yu, H.; Zhou, L.; Yi, Y.; Wang, F.; Rong, Y.; Zhang, Y.; et al. O-GlcNAc transferase promotes influenza A virus–induced cytokine storm by targeting interferon regulatory factor–5. Sci. Adv. 2020, 6, eaaz7086. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Weerarathna, T.; McCarthy, J.S.; Davis, T.M. Common infections in diabetes: Pathogenesis, management and relationship to glycaemic control. Diabetes Metab. Res. Rev. 2007, 23, 3–13. [Google Scholar] [CrossRef]
- Kao, W.H.; Hsueh, W.C.; Rainwater, D.L.; O’Leary, D.H.; Imumorin, I.G.; Stern, M.P.; Mitchell, B.D. Family history of type 2 diabetes is associated with increased carotid artery intimal-medial thickness in Mexican Americans. Diabetes Care 2005, 28, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Luzi, L.; Radaelli, M.G. Influenza and obesity: Its odd relationship and the lessons for COVID-19 pandemic. Acta Diabetol. 2020, 57, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Zhang, J.J.; Dong, X.; Cao, Y.Y.; Yuan, Y.D.; Yang, Y.B.; Yan, Y.Q.; Akdis, C.A.; Gao, Y.D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities in the novel Wuhan coronavirus (COVID-19) infection: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef]
- Zhou, B.; Lu, Y.; Hajifathalian, K.; Bentham, J.; Di Cesare, M.; Danaei, G.; Lo, W.C. Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity 2020. [Google Scholar] [CrossRef]
- Lighter, J.; Phillips, M.; Hochman, S.; Sterling, S.; Johnson, D.; Francois, F.; Stachel, A. Obesity in patients younger than 60 years is a risk factor for Covid-19 hospital admission. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dietz, W.; Santos-Burgoa, C. Obesity and its Implications for COVID-19 Mortality. Obesity 2020. [Google Scholar] [CrossRef] [PubMed]
- Venkata, C.; Sampathkumar, P.; Afessa, B. Hospitalized patients with 2009 H1N1 influenza infection: The Mayo Clinic experience. Mayo Clin. Proc. 2010, 85, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Louie, J.K.; Acosta, M.; Winter, K.; Jean, C.; Gavali, S.; Schechter, R.; Vugia, D.; Harriman, K.; Matyas, B.; Glaser, C.A.; et al. Factors associated with death or hospitalization due to pandemic 2009 influenza A(H1N1) infection in California. JAMA 2009, 302, 1896–1902. [Google Scholar] [CrossRef]
- Thompson, D.L.; Jungk, J.; Hancock, E.; Smelser, C.; Landen, M.; Nichols, M.; Selvage, D.; Baumbach, J.; Sewell, M. Risk factors for 2009 pandemic influenza A (H1N1)-related hospitalization and death among racial/ethnic groups in New Mexico. Am. J. Public Health 2011, 101, 1776–1784. [Google Scholar] [CrossRef]
- Cheung, B.M.; Li, C. Diabetes and hypertension: Is there a common metabolic pathway? Curr. Atheroscler. Rep. 2012, 14, 160–166. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Pal, R.; Bhansali, A. COVID-19, diabetes mellitus and ACE2: The conundrum. Diabetes Res. Clin. Pract. 2020, 162, 108132. [Google Scholar] [CrossRef]
- Rodriguez-Ayala, E.; Gallegos-Cabrales, E.C.; Gonzalez-Lopez, L.; Laviada-Molina, H.A.; Salinas-Osornio, R.A.; Nava-Gonzalez, E.J.; Leal-Berumen, I.; Escudero-Lourdes, C.; Escalante-Araiza, F.; Buenfil-Rello, F.A.; et al. Towards precision medicine: Defining and characterizing adipose tissue dysfunction to identify early immunometabolic risk in symptom-free adults from the GEMM family study. Adipocyte 2020, 9, 153–169. [Google Scholar] [CrossRef]
- Bastarrachea, R.A.; Laviada-Molina, H.A.; Nava-Gonzalez, E.J.; Leal-Berumen, I.; Escudero-Lourdes, C.; Escalante-Araiza, F.; Peschard, V.G.; Veloz-Garza, R.A.; Haack, K.; Martinez-Hernandez, A.; et al. Deep Multi-OMICs and Multi-Tissue Characterization in a Pre- and Postprandial State in Human Volunteers: The GEMM Family Study Research Design. Genes 2018, 9, 532. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Apovian, C. Macrophage functions in lean and obese adipose tissue. Metabolism 2017, 72, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeck, G.; Catalan, V.; Rodriguez, A.; Ramirez, B.; Becerril, S.; Salvador, J.; Colina, I.; Gomez-Ambrosi, J. Adiponectin-leptin Ratio is a Functional Biomarker of Adipose Tissue Inflammation. Nutrients 2019, 11, 454. [Google Scholar] [CrossRef]
- Trayhurn, P. Hypoxia and adipocyte physiology: Implications for adipose tissue dysfunction in obesity. Annu. Rev. 2014, 34, 207–236. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Harrison, S.A.; Belfort-Aguilar, R.; Hardies, L.J.; Balas, B.; Schenker, S.; Cusi, K. Importance of changes in adipose tissue insulin resistance to histological response during thiazolidinedione treatment of patients with nonalcoholic steatohepatitis. Hepatology 2009, 50, 1087–1093. [Google Scholar] [CrossRef]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef]
- Wen, J.; Cai, X.; Zhang, J.; Jiang, J.; Li, W.; Liu, G.; Wang, M.; Gaisano, H.Y.; Pan, Y.; He, Y. Relation of adipose tissue insulin resistance to prediabetes. Endocrine 2020, 68, 93–102. [Google Scholar] [CrossRef]
- Michalakis, K.; Ilias, I. SARS-CoV-2 infection and obesity: Common inflammatory and metabolic aspects. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 469–471. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020. [Google Scholar] [CrossRef]
- Iacobini, C.; Pugliese, G.; Blasetti Fantauzzi, C.; Federici, M.; Menini, S. Metabolically healthy versus metabolically unhealthy obesity. Metabolism 2019, 92, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, C.M.; Jones, S.A.; Yang, J.; Rajagopalan, H.; O’Donnell, L.F.; Chernyak, Y.; Tobin, K.; Cerfolio, R.J.; Fritz, F.; Horwitz, L.I. Factors associated with hospitalization and critical illness among 4,103 patients with Covid-19 disease in New York City. MedRXiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.04.08.20057794v1 (accessed on 13 May 2020). [CrossRef]
- Hussain, M.; Jabeen, N.; Raza, F.; Shabbir, S.; Baig, A.A.; Amanullah, A.; Aziz, B. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Conti, P.; Younes, A. Coronavirus COV-19/SARS-CoV-2 affects women less than men: Clinical response to viral infection. J. Biol. Regul. Homeost. Agents 2020, 34, 339–343. [Google Scholar]
- Boettler, T.; Newsome, P.N.; Mondelli, M.U.; Maticic, M.; Cordero, E.; Cornberg, M.; Berg, T. Care of patients with liver disease during the COVID-19 pandemic: EASL-ESCMID position paper. JHEP Rep. 2020, 2, 100113. [Google Scholar] [CrossRef]
- Vardavas, C.I.; Nikitara, K. COVID-19 and smoking: A systematic review of the evidence. Tob. Induc. Dis. 2020, 18, 20. [Google Scholar] [CrossRef]
- Despres, J.P. Body fat distribution and risk of cardiovascular disease: An update. Circulation 2012, 126, 1301–1313. [Google Scholar] [CrossRef]
- Reddy, P.; Lent-Schochet, D.; Ramakrishnan, N.; McLaughlin, M.; Jialal, I. Metabolic syndrome is an inflammatory disorder: A conspiracy between adipose tissue and phagocytes. Clin. Chim. Acta 2019, 496, 35–44. [Google Scholar] [CrossRef]
- Coppell, K.J.; Hall, R.M.; Downie, M.; Fraser, S.K.; Garrett, M.; Jefferies, C.A.; Kenealy, T.W.; Milne, R.E.; Orr-Walker, B.J.; Paul, R.G.; et al. Diabetes and COVID-19-the meeting of two pandemics: What are the concerns? N. Z. Med. J. 2020, 133, 85–87. [Google Scholar]
- Drucker, D.J. Coronavirus Infections and Type 2 Diabetes-Shared Pathways with Therapeutic Implications. Endocr. Rev. 2020, 41. [Google Scholar] [CrossRef] [PubMed]
- Nakhleh, A.; Shehadeh, N. Interactions between antihyperglycemic drugs and the renin-angiotensin system: Putative roles in COVID-19. A mini-review. Diabetes Metab. Syndr. 2020, 14, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Lipska, K.J.; De Rekeneire, N.; Van Ness, P.H.; Johnson, K.C.; Kanaya, A.; Koster, A.; Strotmeyer, E.S.; Goodpaster, B.H.; Harris, T.; Gill, T.M.; et al. Identifying dysglycemic states in older adults: Implications of the emerging use of hemoglobin A1c. J. Clin. Endocrinol. Metab. 2010, 95, 5289–5295. [Google Scholar] [CrossRef] [PubMed]
- Mechanick, J.I.; Garber, A.J.; Grunberger, G.; Handelsman, Y.; Garvey, W.T. Dysglycemia-based chronic disease: An american association of clinical endocrinologists position statement. Endocr. Pract. 2018, 24, 995–1011. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cole, S.A.; Laviada-Molina, H.A.; Serres-Perales, J.M.; Rodriguez-Ayala, E.; Bastarrachea, R.A. The COVID-19 Pandemic during the Time of the Diabetes Pandemic: Likely Fraternal Twins? Pathogens 2020, 9, 389. https://doi.org/10.3390/pathogens9050389
Cole SA, Laviada-Molina HA, Serres-Perales JM, Rodriguez-Ayala E, Bastarrachea RA. The COVID-19 Pandemic during the Time of the Diabetes Pandemic: Likely Fraternal Twins? Pathogens. 2020; 9(5):389. https://doi.org/10.3390/pathogens9050389
Chicago/Turabian StyleCole, Shelley A., Hugo A. Laviada-Molina, Jeannette M. Serres-Perales, Ernesto Rodriguez-Ayala, and Raul A. Bastarrachea. 2020. "The COVID-19 Pandemic during the Time of the Diabetes Pandemic: Likely Fraternal Twins?" Pathogens 9, no. 5: 389. https://doi.org/10.3390/pathogens9050389
APA StyleCole, S. A., Laviada-Molina, H. A., Serres-Perales, J. M., Rodriguez-Ayala, E., & Bastarrachea, R. A. (2020). The COVID-19 Pandemic during the Time of the Diabetes Pandemic: Likely Fraternal Twins? Pathogens, 9(5), 389. https://doi.org/10.3390/pathogens9050389