Comparative Genomic Analysis Provides Insights into the Phylogeny, Resistome, Virulome, and Host Adaptation in the Genus Ewingella

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Genomic Features of E. americana B6-1
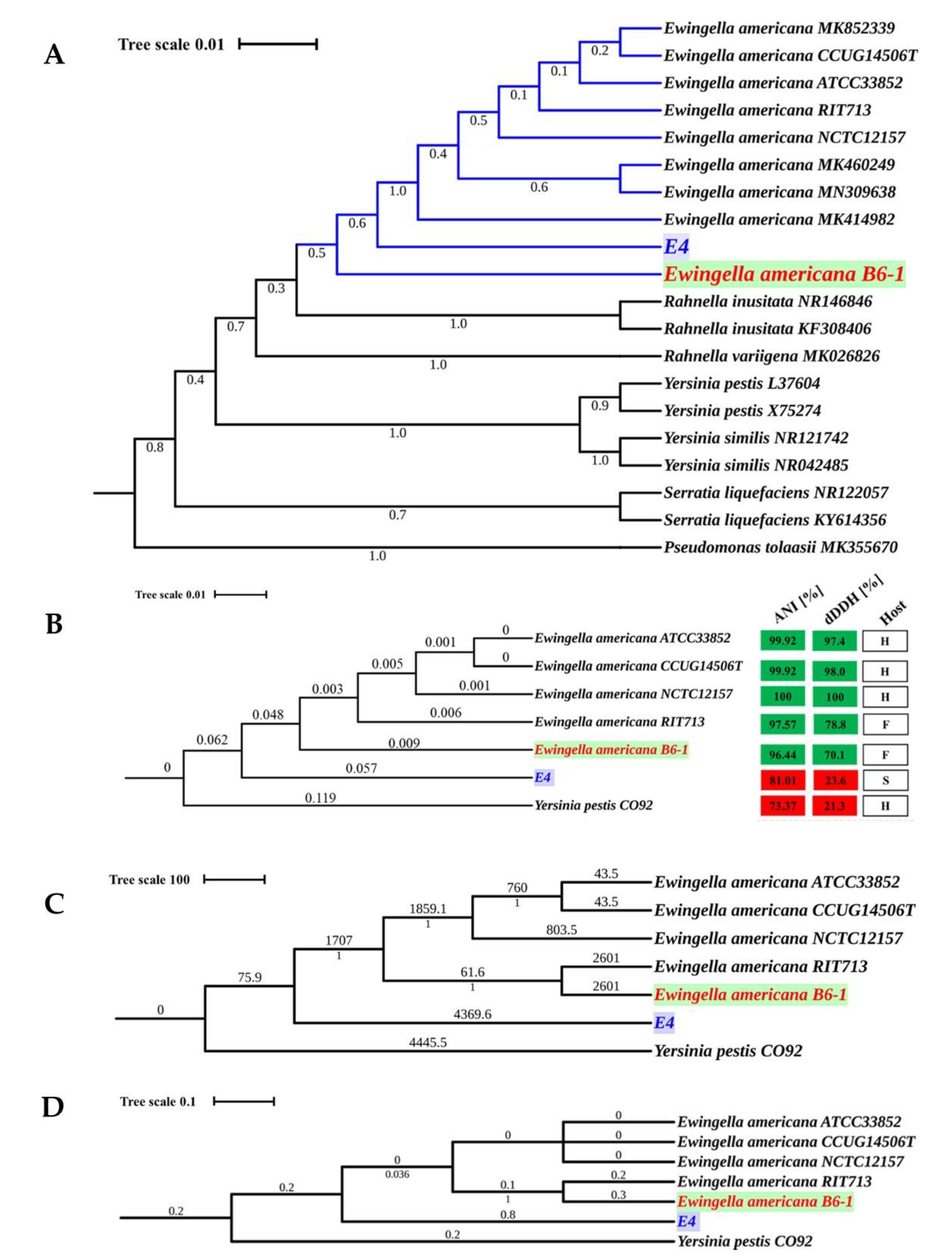
2.2. Average Nucleotide Identity Calculations and Phylogenetic Analyses
2.3. Orthology and Pan-Genome Analysis
2.4. Resistome and Antimicrobial Susceptibility Profile of B6-1
2.5. Mobile Genetic Elements (MGE)
2.6. CRISPR-CAS System, Restriction Modification System, and Toxin-Antitoxin System
2.7. Pathogenicity and Virulence Factors
2.8. Stress Response
2.9. Annotation of Carbohydrate-Active Enzymes (CAZymes)
2.10. Secondary Metabolites and Bacteriocins
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Characterization
4.2. DNA Extraction, Genome Sequencing, and Annotation
4.3. Average Nucleotide Identity and Phylogenetic Analyses
4.4. Orthology and Pan-Genome Analyses
4.5. Resistome and Antimicrobial Susceptibility Profile of B6-1
4.6. Mobile Genetic Elements (MGE) and Bacterial Defense
4.7. Pathogenicity, and Virulence Factors
4.8. CAZymes and Secondary Metabolites
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| B6-1 | Ewingella americana B6-1 |
| ATCC | Ewingella americana ATCC 33852 |
| CCUG | Ewingella americana CCUG 14506T |
| NCTC | Ewingella americana NCTC12157 |
| RIT713 | Ewingella americana RIT713 |
| E4 | E4 |
| E. americana | Ewingella americana |
| ANI | Average Nucleotide Identity |
| CAZyme | Carbohydrate-active enzymes |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| dDDH | digital DNA-DNA hybridization |
| GC | Guanine-cytosine |
| MGE | Mobile genetic elements |
| NCBI | National Center for Biotechnology Information |
| SMs | Secondary metabolites |
| SNP | Single-nucleotide polymorphism |
| R-M system | Restriction-modification system |
| TA system | Toxin-antitoxin system |
References
- Adeolu, M.; Alnajar, S.; Naushad, S.; Gupta, R.S. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: Proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar] [CrossRef] [PubMed]
- Grimont, P.A.D.; Farmer, J.J.; Grimont, F.; Asbury, M.A.; Brenner, D.J.; Deval, C. Ewingella americana gen.nov., sp. nov., a new Enterobacteriaceae isolated from clinical specimens. Annales de l’Institut Pasteur Microbiologie 1983, 134, 39–52. [Google Scholar] [CrossRef]
- Esposito, S.; Miconi, F.; Molinari, D.; Savarese, E.; Celi, F.; Marchese, L.; Valloscuro, S.; Miconi, G.; Principi, N. What is the role of Ewingella americana in humans? A case report in a healthy 4-year-old girl. BMC Infect. Dis. 2019, 19, 386. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.E.; Fanning, G.R.; Brenner, D.J. Isolation of Ewingella americana from mollusks. Curr. Microbiol. 1995, 31, 287–290. [Google Scholar] [CrossRef]
- Hamilton-Miller, J.; Shah, S. Identity and antibiotic susceptibility of enterobacterial flora of Lettuce vegetables. Int. J. Antimicrob. Agents 2001, 18, 81–83. [Google Scholar] [CrossRef]
- Wei, M.; Zhao, Z.B.; Liu, H.Y.; Fu, C.H.; Yu, L.J. First Report of Ewingella americana Causing Bacterial Shot Hole on Anoectochilus roxburghii in China. Plant Dis. 2020. [Google Scholar] [CrossRef]
- Helps, C.; Harbour, D.; Corry, J. PCR-based 16S ribosomal DNA detection technique for Clostridium estertheticum causing spoilage in vacuum-packed chill-stored beef. Int. J. Food Microbiol. 1999, 52, 57–65. [Google Scholar] [CrossRef]
- Lyon, W.J.; Milliet, J.B. Microbial Flora Associated with Louisiana Processed Frozen and Fresh Nutria (Myocastor coypus) Carcasses. J. Food Sci. 2000, 65, 1041–1045. [Google Scholar] [CrossRef]
- Inglis, P.W.; Burden, J.L.; Peberdy, J.F. Evidence for the association of the enteric bacterium Ewingella americana with internal stipe necrosis of Agaricus bisporus. Microbiology 1996, 142, 3253–3260. [Google Scholar] [CrossRef][Green Version]
- Liu, Z.H.; Sossah, F.L.; Li, Y.; Fu, Y.P. First Report of Ewingella americana Causing Bacterial Brown Rot Disease on Cultivated Needle Mushroom (Flammulina velutipes) in China. Plant Dis. 2018, 102, 2633. [Google Scholar] [CrossRef]
- Inglis, P.W.; Peberdy, J.F. Isolation of Ewingella americana from the Cultivated Mushroom, Agaricus bisporus. Curr. Microbiol. 1996, 33, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.R.; Pay, J.; Braithwaite, M. Isolation, identification and ecology of Ewingella americana (the causal agent of internal stipe necrosis) from cultivated mushrooms in New Zealand. Australas. Plant Pathol. 2007, 36, 424–428. [Google Scholar] [CrossRef]
- Hassan, S.; Amer, S.; Mittal, C.; Sharma, R. Ewingella americana: An emerging true pathogen. Case Rep. Infect. Dis. 2012, 2012, 2. [Google Scholar] [CrossRef]
- Reyes, J.E.; Venturini, M.E.; Oria, R.; Blanco, D. Prevalence of Ewingella americana in retail fresh cultivated mushrooms (Agaricus bisporus, Lentinula edodes and Pleurotus ostreatus) in Zaragoza (Spain). FEMS Microbiol. Ecol. 2004, 47, 291–296. [Google Scholar] [CrossRef]
- Bukhari, S.; Hussain, W.A.; Fatani, M.; Ashshi, A. Multi-drug resistant Ewingella americana. Saudi Med. J. 2008, 29, 1051–1053. [Google Scholar] [CrossRef] [PubMed]
- Madbouly, A.K.; Elshatoury, E.; Abouzeid, M. Etiology of stipe necrosis of cultivated mushrooms (Agaricus bisporus) in Egypt. Phytopathol. Mediterr. 2014, 53, 124–129. [Google Scholar] [CrossRef]
- Buermans, H.P.J.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1932–1941. [Google Scholar] [CrossRef]
- Yin, Z.; Yuan, C.; Du, Y.; Yang, P.; Qian, C.; Wei, Y.; Zhang, S.; Huang, D.; Liu, B. Comparative genomic analysis of the Hafnia genus reveals an explicit evolutionary relationship between the species alvei and paralvei and provides insights into pathogenicity. BMC Genom. 2019, 20, 768. [Google Scholar] [CrossRef]
- Urbaniak, C.; van Dam, P.; Zaborin, A.; Zaborina, O.; Gilbert, J.A.; Torok, T.; Wang, C.C.C.; Venkateswaran, K. Genomic Characterization and Virulence Potential of Two Fusarium oxysporum Isolates Cultured from the International Space Station. mSystems 2019, 4, e00345-18. [Google Scholar] [CrossRef]
- Johnson, T.J.; Nolan, L.K. Pathogenomics of the Virulence Plasmids of Escherichia coli. Microbiol. Mol. Biol. Rev. 2009, 73, 750–774. [Google Scholar] [CrossRef]
- Leopold, S.R.; Goering, R.V.; Witten, A.; Harmsen, D.; Mellmann, A. Bacterial Whole-Genome Sequencing Revisited: Portable, Scalable, and Standardized Analysis for Typing and Detection of Virulence and Antibiotic Resistance Genes. J. Clin. Microbiol. 2014, 52, 2365–2370. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef]
- Pound, M.W.; Tart, S.B.; Okoye, O. Multidrug-Resistant Ewingella americana: A Case Report and Review of the Literature. Ann. Pharmacother. 2007, 41, 2066–2070. [Google Scholar] [CrossRef]
- Naito, M.; Pawlowska, T.E. The role of mobile genetic elements in evolutionary longevity of heritable endobacteria. Mob. Genet. Elem. 2016, 6, e1136375. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.; da Costa, M.; Rooney, A.; Yi, H.; Xu, X.-W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef]
- Venter, S.N.; Palmer, M.; Beukes, C.W.; Chan, W.-Y.; Shin, G.; van Zyl, E.; Seale, T.; Coutinho, T.A.; Steenkamp, E.T. Practically delineating bacterial species with genealogical concordance. Antonie van Leeuwenhoek 2017, 110, 1311–1325. [Google Scholar] [CrossRef]
- Jensen, R.A. Orthologs and paralogs—We need to get it right. Genom. Biol. 2001, 2. [Google Scholar] [CrossRef]
- Segerman, B. The genetic integrity of bacterial species: The core genome and the accessory genome, two different stories. Front. Cell. Infect. Microbiol. 2012, 2. [Google Scholar] [CrossRef]
- Cotty, P.J.; Bayman, P.; Egel, D.S.; Elias, K.S. Agriculture, aflatoxins and Aspergillus. In The Genus Aspergillus; Powell, K.A., Renwick, A., Peberdy, J.F., Eds.; Springer: New York, NY, USA, 1994. [Google Scholar]
- Lackner, G.; Partida-Martinez, L.P.; Hertweck, C. Endofungal bacteria as producers of mycotoxins. Trends Microbiol. 2009, 17, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K. Cultivation of Mushrooms in Plastic Bottles and Small Bags. In Edible and Medicinal Mushrooms; Diego, C.Z., Pardo-Giménez, A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 309–338. [Google Scholar] [CrossRef]
- Chhotaray, C.; Wang, S.; Tan, Y.; Ali, A.; Shehroz, M.; Fang, C.; Liu, Y.; Lu, Z.; Cai, X.; Hameed, H.M.A.; et al. Comparative Analysis of Whole-Genome and Methylome Profiles of a Smooth and a Rough Mycobacterium abscessus Clinical Strain. G3 (Bethesda) 2020, 10, 13–22. [Google Scholar] [CrossRef]
- Lehtinen, S.; Blanquart, F.; Lipsitch, M.; Fraser, C.; The Maela Pneumococcal Collaboration. On the evolutionary ecology of multidrug resistance in bacteria. PLoS Pathog. 2019, 15, e1007763. [Google Scholar] [CrossRef]
- Amador, P.; Fernandes, R.; Prudêncio, C.; Duarte, I. Prevalence of Antibiotic Resistance Genes in Multidrug-Resistant Enterobacteriaceae on Portuguese Livestock Manure. Antibiotics 2019, 8, 23. [Google Scholar] [CrossRef]
- Nikaido, H. Multidrug Resistance in Bacteria. Ann. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef]
- Aminov, R.I. Horizontal gene exchange in environmental microbiota. Front. Microbiol. 2011, 2, 158. [Google Scholar] [CrossRef]
- Van der Beek, S.L.; Zorzoli, A.; Çanak, E.; Chapman, R.N.; Lucas, K.; Meyer, B.H.; Evangelopoulos, D.; de Carvalho, L.P.S.; Boons, G.-J.; Dorfmueller, H.C.; et al. Streptococcal dTDP-L-rhamnose biosynthesis enzymes: Functional characterization and lead compound identification. Mol. Microbiol. 2019, 111, 951–964. [Google Scholar] [CrossRef]
- Kati, C.; Bibashi, E.; Kokolina, E.; Sofianou, D. Case of peritonitis caused by Ewingella americana in a patient undergoing continuous ambulatory peritoneal dialysis. J. Clin. Microbiol. 1999, 37, 3733–3734. [Google Scholar] [CrossRef]
- Repizo, G.D.; Espariz, M.; Blancato, V.S.; Suárez, C.A.; Esteban, L.; Magni, C. Genomic comparative analysis of the environmental Enterococcus mundtii against enterococcal representative species. BMC Genom. 2014, 15, 489. [Google Scholar] [CrossRef]
- Gomez, A.; Ladire, M.; Marcille, F.; Nardi, M.; Fons, M. Characterization of ISRgn1, a Novel Insertion Sequence of the IS3 Family Isolated from a Bacteriocin-Negative Mutant of Ruminococcus gnavus E1. Appl. Environ. Microbiol. 2002, 68, 4136–4139. [Google Scholar] [CrossRef]
- Nicolas, E.; Oger, C.A.; Nguyen, N.; Lambin, M.; Draime, A.; Leterme, S.C.; Chandler, M.; Hallet, B.F.J. Unlocking Tn3-family transposase activity in vitro unveils an asymetric pathway for transposome assembly. Proc. Natl. Acad. Sci. USA 2017, 114, E669–E678. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [PubMed]
- Smillie, C.; Garcillán-Barcia, M.P.; Francia, M.V.; Rocha, E.P.C.; de la Cruz, F. Mobility of Plasmids. Microbiol. Mol. Biol. Rev. 2010, 74, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 2018, 359, eaar4120. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, H.; Ortega-Polo, R.; Zaheer, R.; Goji, N.; Amoako, K.K.; Brown, R.S.; Majury, A.; Liss, S.N.; McAllister, T.A. Comparative genomics of multidrug-resistant Enterococcus spp. isolated from wastewater treatment plants. BMC Microbiol. 2020, 20, 20. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Pawluk, A.; Maxwell, K.L.; Davidson, A.R. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 2013, 493, 429–432. [Google Scholar] [CrossRef]
- Vasu, K.; Nagaraja, V. Diverse Functions of Restriction-Modification Systems in Addition to Cellular Defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef]
- Negri, A.; Jąkalski, M.; Szczuka, A.; Pryszcz, L.P.; Mruk, I. Transcriptome analyses of cells carrying the Type II Csp231I restriction-modification system reveal cross-talk between two unrelated transcription factors: C protein and the Rac prophage repressor. Nucleic Acids Res. 2019, 47, 9542–9556. [Google Scholar] [CrossRef]
- Donahue, J.P.; Peek, R.M., Jr. Restriction and Modification Systems. In Helicobacter Pylori: Physiology and Genetics; Mobley, H.L.T., Mendz, G.L., Hazell, S.L., Eds.; ASM Press: Washington, DC, USA, 2001. [Google Scholar]
- Wang, X.; Wood, T.K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011, 77, 5577–5583. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Park, J.-H.; Inouye, M. Toxin-Antitoxin Systems in Bacteria and Archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef]
- Shao, Y.; Harrison, E.M.; Bi, D.; Tai, C.; He, X.; Ou, H.-Y.; Rajakumar, K.; Deng, Z. TADB: A web-based resource for Type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011, 39, D606–D611. [Google Scholar] [CrossRef]
- Wen, Y.; Behiels, E.; Devreese, B. Toxin–Antitoxin systems: Their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 2014, 70, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Broxterman, S.E.; Schols, H.A. Interactions between pectin and cellulose in primary plant cell walls. Carbohydr. Polym. 2018, 192, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, I.R.; Brouillet, S.; Soulié, M.-C.; Gribaldo, S.; Sirven, C.; Charron, N.; Boccara, M.; Choquer, M. Genome-wide analyses of chitin synthases identify horizontal gene transfers towards bacteria and allow a robust and unifying classification into fungi. BMC Evol. Biol. 2016, 16, 252. [Google Scholar] [CrossRef]
- Killiny, N.; Martinez, R.H.; Dumenyo, C.K.; Cooksey, D.A.; Almeida, R.P.P. The Exopolysaccharide of Xylella fastidiosa Is Essential for Biofilm Formation, Plant Virulence, and Vector Transmission. Mol. Plant-Microbe Interact. 2013, 26, 1044–1053. [Google Scholar] [CrossRef]
- Looi, H.K.; Toh, Y.F.; Yew, S.M.; Na, S.L.; Tan, Y.-C.; Chong, P.-S.; Khoo, J.-S.; Yee, W.-Y.; Ng, K.P.; Kuan, C.S. Genomic insight into pathogenicity of dematiaceous fungus Corynespora cassiicola. PeerJ 2017, 5, e2841. [Google Scholar] [CrossRef]
- Sangwan, N.S.; Jadaun, J.S.; Tripathi, S.; Mishra, B.; Narnoliya, L.K.; Sangwan, R.S. Chapter 9—Plant Metabolic Engineering. In Omics Technologies and Bio-Engineering; Barh, D., Azevedo, V., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 143–175. [Google Scholar] [CrossRef]
- Shankar-Sinha, S.; Valencia, G.A.; Janes, B.K.; Rosenberg, J.K.; Whitfield, C.; Bender, R.A.; Standiford, T.J.; Younger, J.G. The Klebsiella pneumoniae O antigen contributes to bacteremia and lethality during murine pneumonia. Infect. Immun. 2004, 72, 1423–1430. [Google Scholar] [CrossRef]
- Abreu, A.G.; Barbosa, A.S. How Escherichia coli Circumvent Complement-Mediated Killing. Front. Immunol. 2017, 8, 452. [Google Scholar] [CrossRef]
- Albelda-Berenguer, M.; Monachon, M.; Joseph, E. Chapter Five—Siderophores: From natural roles to potential applications. In Advances in Applied Microbiology; Gadd, G.M., Sariaslani, S., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 106, pp. 193–225. [Google Scholar]
- Telford, J.R.; Raymond, K.N. Amonabactin: A family of novel siderophores from a pathogenic bacterium. JBIC J. Biol. Inorg. Chem. 1997, 2, 750–761. [Google Scholar] [CrossRef]
- Grammbitter, G.L.C.; Schmalhofer, M.; Karimi, K.; Shi, Y.-M.; Schöner, T.A.; Tobias, N.J.; Morgner, N.; Groll, M.; Bode, H.B. An Uncommon Type II PKS Catalyzes Biosynthesis of Aryl Polyene Pigments. J. Am. Chem. Soc. 2019, 141, 16615–16623. [Google Scholar] [CrossRef]
- Schöner, T.A.; Gassel, S.; Osawa, A.; Tobias, N.J.; Okuno, Y.; Sakakibara, Y.; Shindo, K.; Sandmann, G.; Bode, H.B. Aryl Polyenes, a Highly Abundant Class of Bacterial Natural Products, Are Functionally Related to Antioxidative Carotenoids. ChemBioChem 2016, 17, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 2010, 8, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ghoul, M.; Mitri, S. The Ecology and Evolution of Microbial Competition. Trends Microbiol. 2016, 24, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Zeth, K.; Römer, C.; Patzer, S.I.; Braun, V. Crystal Structure of Colicin M, a Novel Phosphatase Specifically Imported by Escherichia coli. J. Biol. Chem. 2008, 283, 25324–25331. [Google Scholar] [CrossRef]
- Chikindas, M.L.; Weeks, R.; Drider, D.; Chistyakov, V.A.; Dicks, L.M. Functions and emerging applications of bacteriocins. Curr. Opin. Biotechnol. 2018, 49, 23–28. [Google Scholar] [CrossRef]
- Yang, S.-C.; Lin, C.-H.; Sung, C.T.; Fang, J.-Y. Antibacterial activities of bacteriocins: Application in foods and pharmaceuticals. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Whole-Genome Sequence of Ewingella americana ATCC 33852. Available online: https://www.ncbi.nlm.nih.gov/genome/11412?genome_assembly_id=204696 (accessed on 8 January 2020).
- Draft Genome Sequence of Ewingella americana CCUG 14506T. Available online: https://www.ncbi.nlm.nih.gov/genome/11412?genome_assembly_id=694515 (accessed on 8 January 2020).
- Genome Sequence of Ewingella americana NCTC12157. Available online: https://www.ncbi.nlm.nih.gov/genome/11412?genome_assembly_id=392147 (accessed on 8 January 2020).
- Wong, N.H.; Rosato, A.J.; Rose, Y.M.; Penix, T.S.; Fung, J.B.; Vanitski, A.L.; Goossen, C.J.; Bradshaw, S.G.; Lopp, S.M.; Pennington, A.D.; et al. Isolation and Whole-Genome Sequencing of 12 Mushroom-Associated Bacterial Strains: An Inquiry-Based Laboratory Exercise in a Genomics Course at the Rochester Institute of Technology. Microbiol. Resour. Announc. 2020, 9, e01457-19. [Google Scholar] [CrossRef]
- Altshuler, I.; Hamel, J.; Turney, S.; Magnuson, E.; Levesque, R.; Greer, C.W.; Whyte, L.G. Species interactions and distinct microbial communities in high Arctic permafrost affected cryosols are associated with the CH4 and CO2 gas fluxes. Environ. Microbiol. 2019, 21, 3711–3727. [Google Scholar] [CrossRef]
- Mergaert, J.; Verdonck, L.; Kersters, K. Transfer of Erwinia ananas (synonym, Erwinia uredovora) and Erwinia stewartii to the Genus Pantoea emend. as Pantoea ananas (Serrano 1928) comb. nov. and Pantoea stewartii (Smith 1898) comb. nov., Respectively, and Description of Pantoea stewartii subsp. indologenes subsp. nov. Int. J. Syst. Evol. Microbiol. 1993, 43, 162–173. [Google Scholar] [CrossRef]
- Ivanovic, N.; Minic, R.; Djuricic, I.; Radojevic Skodric, S.; Zivkovic, I.; Sobajic, S.; Djordjevic, B. Active Lactobacillus rhamnosus LA68 or Lactobacillus plantarum WCFS1 administration positively influences liver fatty acid composition in mice on a HFD regime. Food Funct. 2016, 7, 2840–2848. [Google Scholar] [CrossRef]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.; Abrams, N.; Jackson, J.; Jacobs, A.; Kiryutin, B.; Koonin, E.; Krylov, D.; Mazumder, R.; Mekhedov, S.; Nikolskaya, A.; et al. The COG Database: An Updated Version Includes Eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.; Auch, A.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Richter, M.; Rossello-Mora, R.; Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.; Roos, D. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Chiou, C.-S.; Chen, C.-C. PGAdb-builder: A web service tool for creating pan-genome allele database for molecular fine typing. Sci. Rep. 2016, 6, 36213. [Google Scholar] [CrossRef]
- Kaas, R.S.; Leekitcharoenphon, P.; Aarestrup, F.M.; Lund, O. Solving the Problem of Comparing Whole Bacterial Genomes across Different Sequencing Platforms. PLoS ONE 2014, 9, e104984. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2016, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Hudzicki, J. Kirby-bauer disk diffusion susceptibility test protocol. Am. Soc. Microbiol. Protoc. 2009. Available online: https://www.asm.org/getattachment/2594ce26-bd44-47f6-8287-0657aa9185ad/Kirby-Bauer-Disk-Diffusion-Susceptibility-Test-Protocol-pdf.pdf (accessed on 8 January 2020).
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Approved Standard; 25th Informational Supplement; CLSI Document M100-S25; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015. [Google Scholar]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Moura, A.; Soares, M.; Pereira, C.; Leitão, N.; Henriques, I.; Correia, A. INTEGRALL: A database and search engine for integrons, integrases and gene cassettes. Bioinformatics (Oxf. Engl.) 2009, 25, 1096–1098. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Dandan, Z.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2018, 47. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, S.; Larsen, M.; Aarestrup, F.; Lund, O. PathogenFinder—Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Abby, S.; Néron, B.; Ménager, H.; Touchon, M.; Rocha, E. MacSyFinder: A Program to Mine Genomes for Molecular Systems with an Application to CRISPR-Cas Systems. PLoS ONE 2014, 9, e110726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Van Heel, A.J.; de Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic Class. | Antimicrobial Agent | * Susceptibility (Average Diameter/mm) |
|---|---|---|
| β-Lactams | Ampicillin | R (11.10) |
| Cephalosporins | Aztreonam | S (23.34) |
| Cefazolin | R (11.78) | |
| Cefixime | I (17.69) | |
| Ceftriaxone | S (21.51) | |
| Fluoroquinolone | Ciprofloxacin | S (23.75) |
| Ofloxacin | S (23.33) | |
| Aminoglycosides | Streptomycin | S (14.87) |
| Gentamicin | S (17.85) | |
| Kanamycin | S (19.67) | |
| Large ring lactone | Erythromycin | I (14.05) |
| Tetracycline | Tetracycline | R (12.64) |
| Rifamycin | Rifampicin | R (8.54) |
| Lincosamide | Clindamycin | R (0.00) |
| Sugar peptide | Vancomycin | R (0.00) |
| Novobiocin | R (0.00) |
| Strain | Integron (In1) | Insertion Sequence | Plasmid | GI | CRISPR Spacers | R-M System | TA System | ||
|---|---|---|---|---|---|---|---|---|---|
| Type I | Type II | Type IV | |||||||
| B6-1 | 4 | 2 | 2 | 19 | 4 | I, II, IV | 2 | 22 | 3 |
| ATCC | 4 | 4 | 0 | 20 | 5 | I, II | 0 | 29 | 1 |
| CCUG | 4 | 4 | 0 | 23 | 4 | I, II | 0 | 26 | 1 |
| NCTC | 4 | 4 | 0 | 24 | 4 | I, II | 0 | 30 | 1 |
| RIT713 | 5 | 2 | 0 | 23 | 2 | I, II, IV | 1 | 30 | 4 |
| Strain Name | Region | Length (kb) | Completeness | No. of CDS | GC% | Predominant Phage |
|---|---|---|---|---|---|---|
| B6-1 | 1 | 43.9 | Intact | 52 | 51.81 | Entero_mEp390_NC_019721 |
| 2 | 17.9 | Intact | 25 | 52.35 | Erwini_ENT90_NC_019932 | |
| ATCC | 3 | 51.8 | Intact | 48 | 50.54 | Entero_mEp460_NC_019716 |
| CCUG | 1 | 35.8 | Intact | 43 | 51.88 | Haemop_HP1_NC_001697 |
| 1 | 44.5 | Intact | 51 | 50.42 | Entero_mEp460_NC_019716 | |
| NCTC | 1 | 24.8 | Questionable | 37 | 50.50 | Entero_mEp460_NC_019716 |
| 2 | 33.6 | Intact | 24 | 53.04 | Entero_N15_NC_001901 | |
| 1 | 35.6 | Intact | 45 | 51.93 | Aeromo_phiO18P_NC_009542 | |
| 2 | 18.1 | Intact | 24 | 54.42 | Erwini_ENT90_NC_019932 | |
| RIT713 | 1 | 47.0 | Intact | 83 | 49.9 | Edward_GF_2_NC_026611 |
| 1 | 35.6 | Intact | 45 | 51.93 | Aeromo_phiO18P_NC_009542 | |
| 2 | 18.1 | Intact | 24 | 54.42 | Erwini_ENT90_NC_019932 |
| Strain | Host | Predicted Pathogenicity Score | Probability of Being a Human Pathogen | Pathogenic Families Matched | Being a Human Pathogen |
|---|---|---|---|---|---|
| B6-1 | F. filiformis | 46.24 | 0.60 | 24 | Yes |
| RIT713 | Craterellus sp. | 53.60 | 0.64 | 24 | Yes |
| ATCC | Human | 46.21 | 0.61 | 24 | Yes |
| CCUG | Human | 48.41 | 0.62 | 24 | Yes |
| NCTC | Human | 54.71 | 0.63 | 25 | Yes |
| Yersinia pestis CO92 | Human | 6174.52 | 0.91 | 1569 | Yes |
| CAZYme | E. americana Strains | ||||
|---|---|---|---|---|---|
| Module | B6-1 | ATCC | CCUG | NCTC | RIT713 |
| AA | 6 | 6 | 6 | 6 | 7 |
| CBM | 13 | 13 | 13 | 12 | 14 |
| CE | 16 | 15 | 16 | 15 | 15 |
| GH | 70 | 68 | 68 | 73 | 65 |
| GT | 53 | 52 | 52 | 60 | 48 |
| PL | 2 | 2 | 2 | 2 | 2 |
| Total | 160 | 156 | 157 | 168 | 151 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Sheng, H.; Okorley, B.A.; Li, Y.; Sossah, F.L. Comparative Genomic Analysis Provides Insights into the Phylogeny, Resistome, Virulome, and Host Adaptation in the Genus Ewingella. Pathogens 2020, 9, 330. https://doi.org/10.3390/pathogens9050330
Liu Z, Sheng H, Okorley BA, Li Y, Sossah FL. Comparative Genomic Analysis Provides Insights into the Phylogeny, Resistome, Virulome, and Host Adaptation in the Genus Ewingella. Pathogens. 2020; 9(5):330. https://doi.org/10.3390/pathogens9050330
Chicago/Turabian StyleLiu, Zhenghui, Hongyan Sheng, Benjamin Azu Okorley, Yu Li, and Frederick Leo Sossah. 2020. "Comparative Genomic Analysis Provides Insights into the Phylogeny, Resistome, Virulome, and Host Adaptation in the Genus Ewingella" Pathogens 9, no. 5: 330. https://doi.org/10.3390/pathogens9050330
APA StyleLiu, Z., Sheng, H., Okorley, B. A., Li, Y., & Sossah, F. L. (2020). Comparative Genomic Analysis Provides Insights into the Phylogeny, Resistome, Virulome, and Host Adaptation in the Genus Ewingella. Pathogens, 9(5), 330. https://doi.org/10.3390/pathogens9050330