Resistance of Tick Gut Microbiome to Anti-Tick Vaccines, Pathogen Infection and Antimicrobial Peptides




Abstract
1. Introduction
2. Methods
2.1. Original Data Sets
- i)
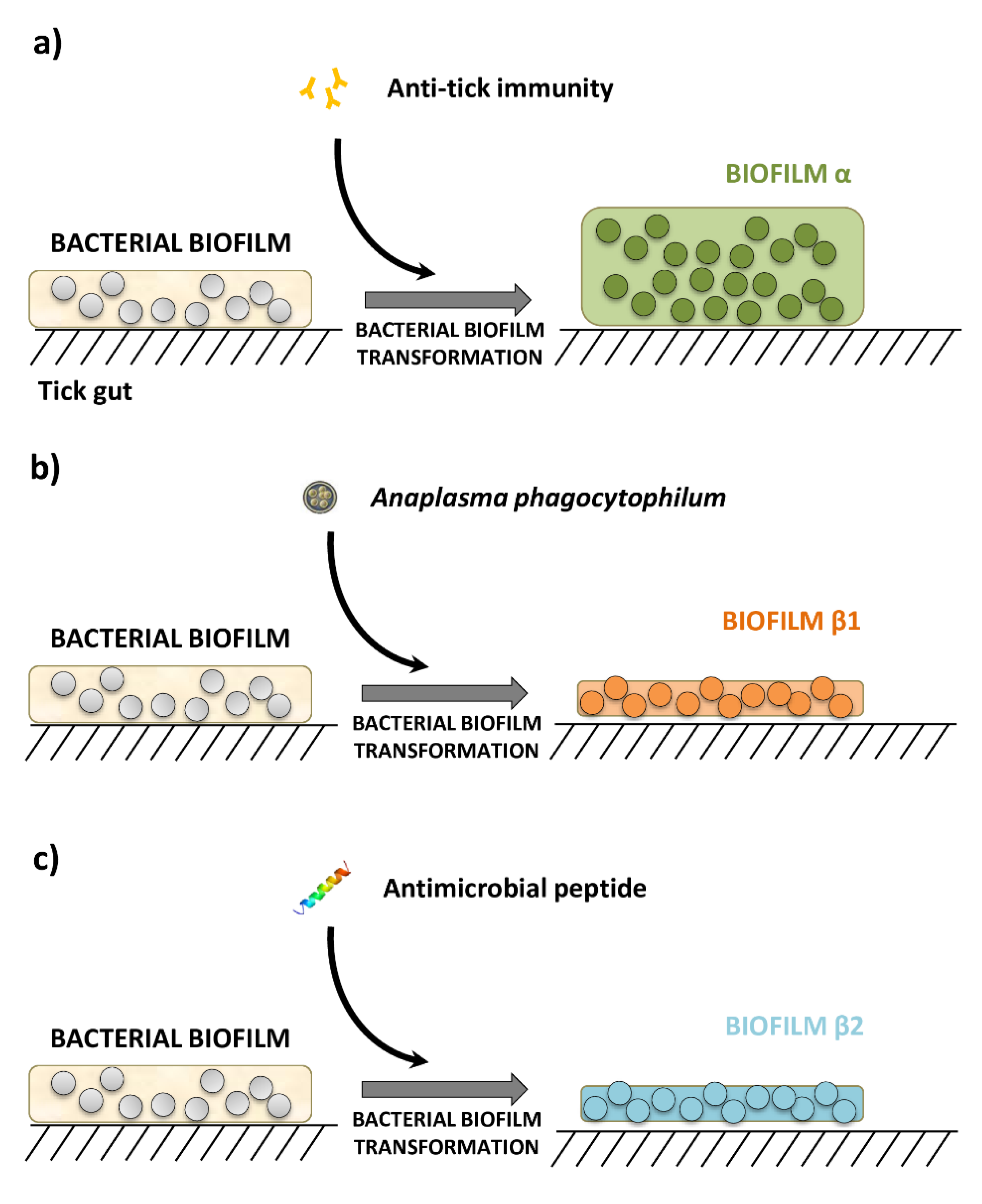
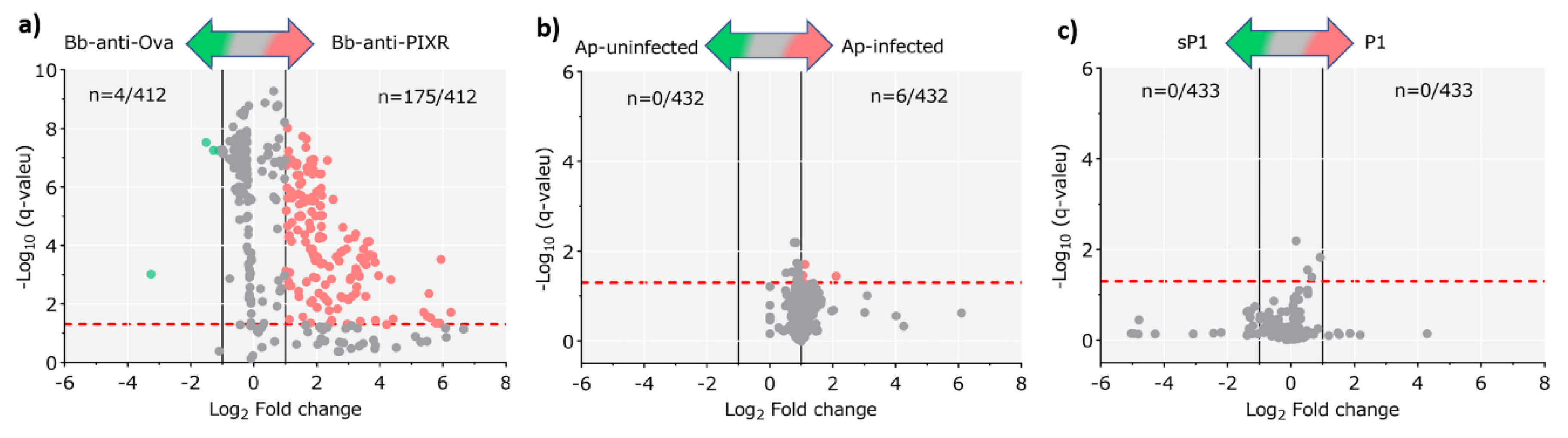
- The study by Narasimhan et al. [4] reported that the infection of larvae of I. scapularis with B. burgdorferi increased the expression of the tick protein PIXR, which facilitates B. burgdorferi infection and molting of larvae. The composition of the microbiota of tick larvae fed on C3H/HeJ mice immunized with recombinant PIXR and infected with B. burgdorferi was compared with that of ticks fed on C3H/HeJ mice immunized with Ovalbumin (OVA, an antigen not found in ticks) and infected with B. burgdorferi. The authors also showed that anti-tick immunity specific to PIXR induces the formation of dense bacterial biofilms in the tick gut. These two 16S datasets are hereinafter referred to as ‘Bb-anti-PIXR’ (n = 16) and ‘Bb-anti-Ova’ (n = 24). Comparisons were used to represent the microbiota composition in response to anti-tick immunity as disturbing factor;
- ii)
- The report by Abraham et al. [3] studied the changes in gut microbiota composition and biofilms of nymphs of I. scapularis fed on A. phagocytophilum-infected or not infected C3H/HeJ mice. These two 16S datasets are hereinafter referred to as ‘Ap-infected’ (n = 20) and ‘Ap-uninfected’ (n = 10). Comparisons between them were used to assess the microbiota composition in response to the disturbing factor A. phagocytophilum infection;
- iii)
- In addition, Abraham et al. [3] showed that A. phagocytophilum induces ticks to express an anti-freeze glycoprotein (IAFGP), an antimicrobial protein with the ability to alter microbiota composition [3] inducing the formation of scattered and diffused bacterial biofilms [9]. A derivative peptide of IAFGP, P1 (PARKARAATAATAATAATAATAAT) affect the tick gut microbial community compared with a scrambled P1 (sP1) peptide (AATAATATAAARRAAAAPTTAKTT) [3]. These two 16S datasets are hereinafter referred to as ‘P1’ (n = 16) and ‘sP1’ (n = 12) and represents an example of microbiota composition in response to antimicrobial peptides.
2.2. Processing of Original Raw Sequences
2.3. Prediction of Functional Traits in Tick Microbiome
2.4. Taxonomic and Functional Co-Occurrence Networks and Network Resistance Analysis
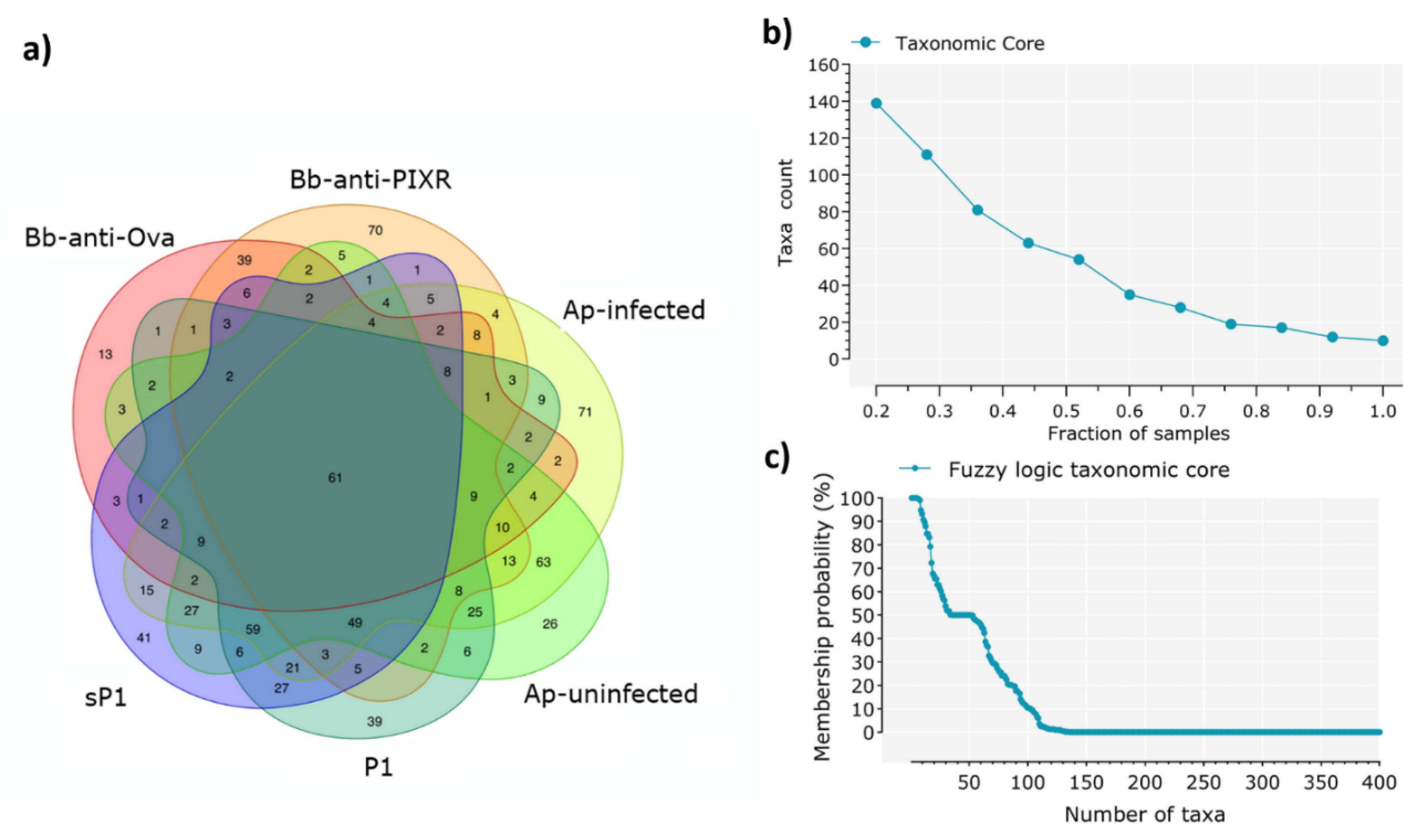
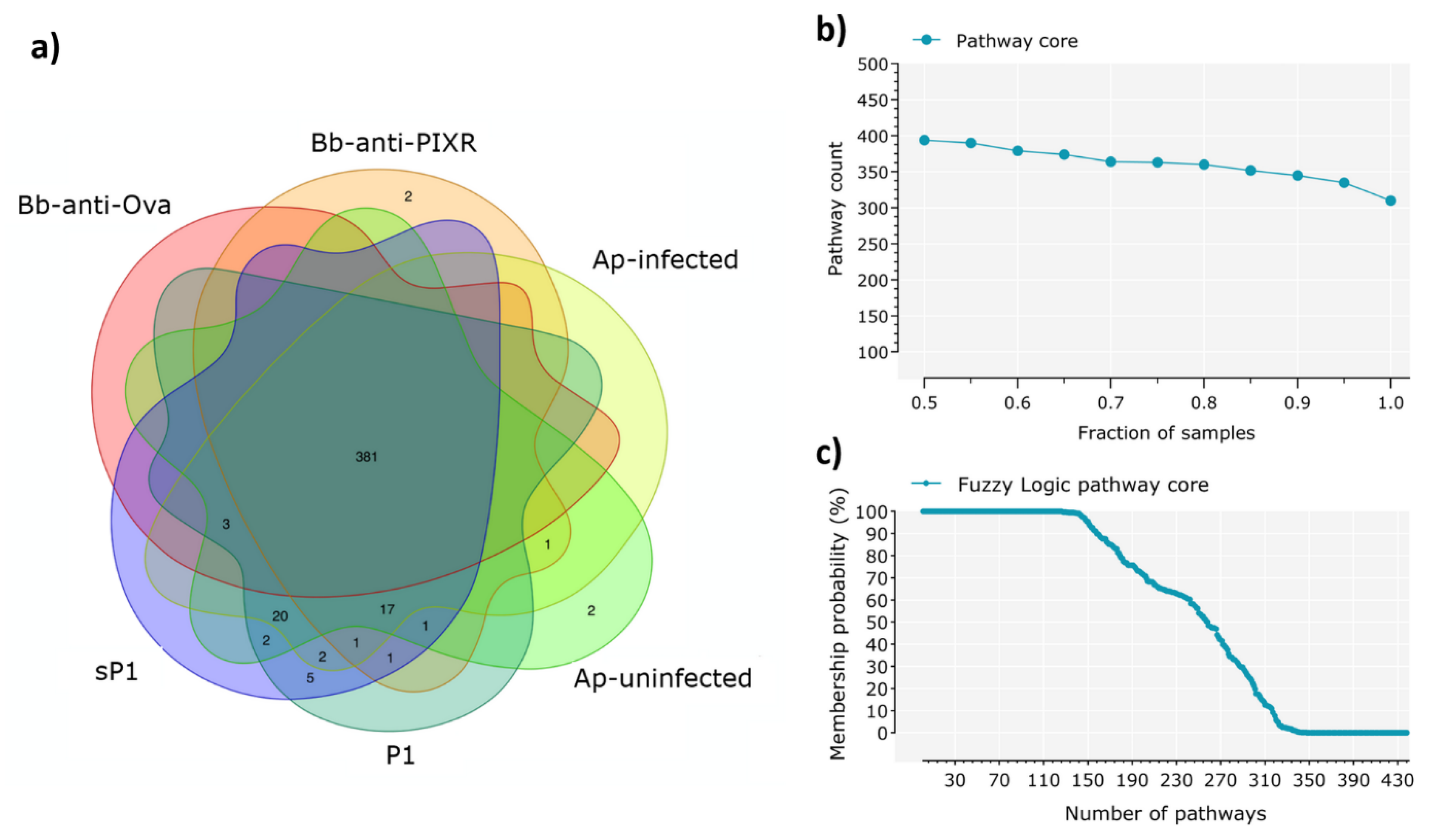
2.5. Identification of the Core Microbiome
3. Results
3.1. Effect of Biological Disturbance on Taxonomic and Functional Profiles of Tick Microbiome
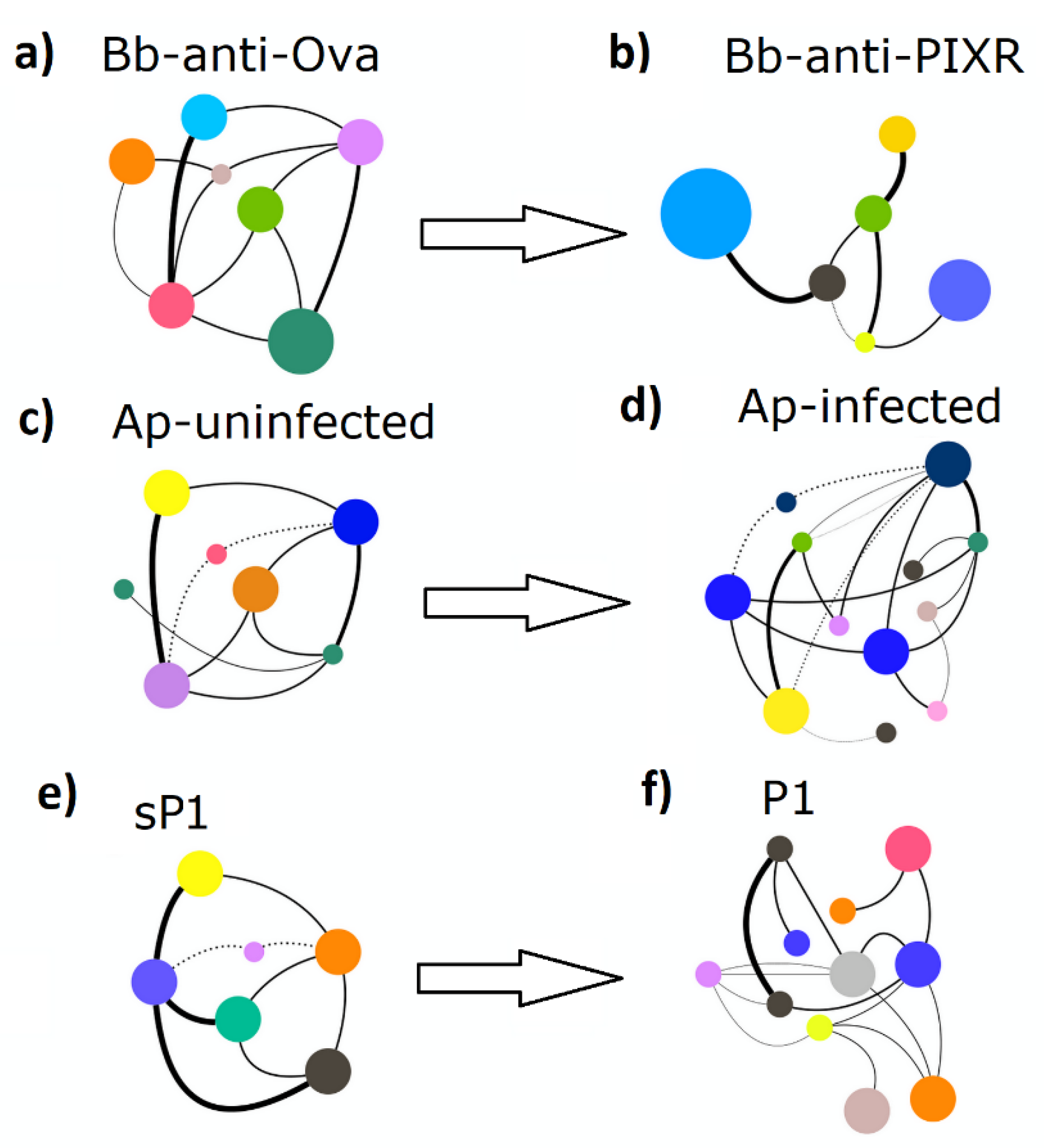
3.2. Disturbance Reshapes Co-Ocurrence Networks and Reduces Networks Tolerance to Taxa Extinction
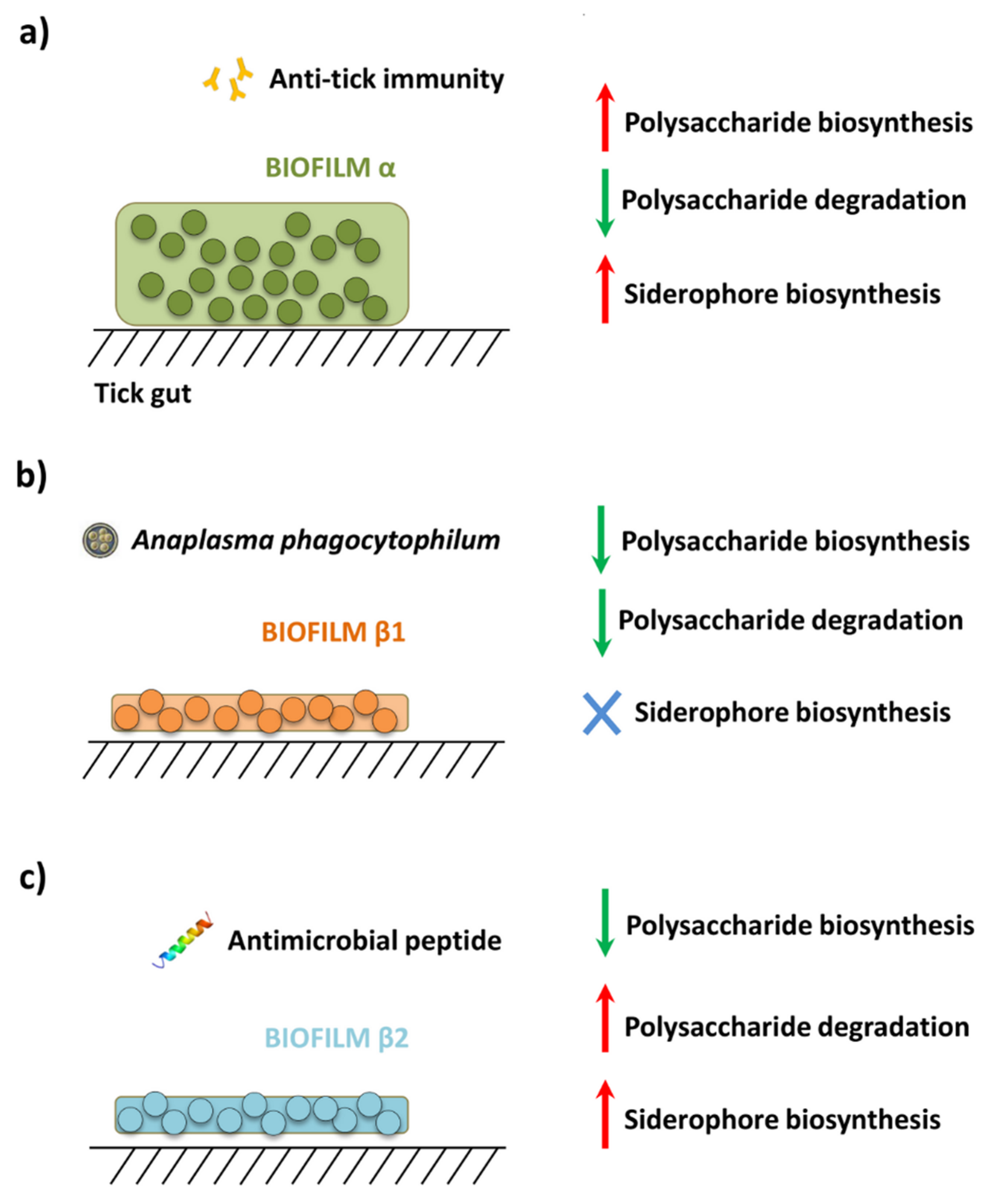
3.3. Anti-Tick Immunity Increases the Representation of Biofilm Formation Pathways in Tick Microbiome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De la Fuente, J.; Antunes, S.; Bonnet, S.; Cabezas-cruz, A.; Domingos, A.G.; Estrada-peña, A.; Johnson, N.; Kocan, K.M.; Mansfield, K.L.; Nijhof, A.M.; et al. Tick-pathogen interactions and vector competence: identification of molecular drivers for tick-borne diseases. Front. Cell. Infect. Microbiol. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Cabezas-Cruz, A.; Pollet, T.; Estrada-Peña, A.; Allain, E.; Bonnet, S.I.; Moutailler, S. Handling the microbial complexity associated to ticks. In Ticks and Tick-Borne Pathogens; Abubakar, D.M., Ed.; TechOpen: London, UK, 2018; pp. 1–36. [Google Scholar]
- Abraham, N.M.; Liu, L.; Lyon, B.; Yadav, A.K.; Narasimhan, S. Pathogen-mediated manipulation of arthropod microbiota to promote infection. PNAS 2017, 114, 781–790. [Google Scholar] [CrossRef]
- Narasimhan, S.; Schuijt, T.J.; Abraham, N.M.; Rajeevan, N.; Coumou, J.; Graham, M.; Robson, A.; Wu, M.; Daffre, S.; Hovius, J.W.; et al. Modulation of the tick gut milieu by a secreted tick protein favors Borrelia burgdorferi colonization. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Narasimhan, S.; Rajeevan, N.; Liu, L.; Zhao, Y.O.; Heisig, J.; Pan, J.; Eppler-epstein, R.; Deponte, K.; Fish, D.; Fikrig, E. Gut Microbiota of the Tick Vector Ixodes scapularis Modulate Colonization of the Lyme Disease Spirochete. Cell Host Microbe 2014, 15, 58–71. [Google Scholar] [CrossRef]
- Purdy, A.E.; Watnick, P.I. Spatially selective colonization of the arthropod intestine through activation of Vibrio cholerae biofilm formation. Proc. Natl. Acad. Sci. USA 2011, 108, 19737–19742. [Google Scholar] [CrossRef]
- Kim, J.K.; Kwon, J.Y.; Kim, S.K.; Han, S.H.; Won, Y.J.; Lee, J.H.; Kim, C.H.; Fukatsu, T.; Lee, B.L. Purine biosynthesis, biofilm formation, and persistence of an insect-microbe gut symbiosis. Appl. Environ. Microbiol. 2014, 80, 4374–4382. [Google Scholar] [CrossRef]
- Maltz, M.A.; Weiss, B.L.; O’neill, M.; Wu, Y.; Aksoy, S. OmpA-mediated biofilm formation is essential for the commensal bacterium Sodalis glossinidius to colonize the tsetse fly gut. Appl. Environ. Microbiol. 2012, 78, 7760–7768. [Google Scholar] [CrossRef]
- Heisig, M.; Abraham, N.M.; Liu, L.; Neelakanta, G.; Mattessich, S.; Sultana, H.; Shang, Z.; Ansari, J.M.; Killiam, C.; Walker, W.; et al. Antivirulence properties of an antifreeze protein. Cell Rep. 2014, 9, 417–424. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Langille, C.M.T.C.H.M.G.I. PICRUSt2: An improved and extensible approach for metagenome inference Gavin. bioRxiv 2019, 1–42. [Google Scholar] [CrossRef]
- Shade, A.; Peter, H.; Allison, S.D.; Baho, D.L.; Berga, M.; Bürgmann, H.; Huber, D.H.; Langenheder, S.; Lennon, J.T.; Martiny, J.B.H.; et al. Fundamentals of microbial community resistance and resilience. Front. Microbiol. 2012, 3, 1–19. [Google Scholar] [CrossRef]
- Allison, S.D.; Martiny, J.B.H. Resistance, resilience, and redundancy in microbial communities. Light Evol. 2009, 2, 149–166. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2011, 39, 2010–2012. [Google Scholar] [CrossRef]
- Blankenberg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A.; Team, G. Manipulation of FASTQ data with galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef]
- Bolyen, E.; Dillon, M.; Bokulich, N.; Abnet, C.; Al-Ghalith, G.; Alexander, H.; Alm, E.; Arumugam, M.; Asnicar, F.; Bai, Y.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2 - Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Werner, J.J.; Koren, O.; Hugenholtz, P.; Desantis, T.Z.; Walters, W.A.; Caporaso, J.G.; Angenent, L.T.; Knight, R.; Ley, R.E. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2012, 6, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Morton, J.T.; Sanders, J.; Quinn, R.A.; Mcdonald, D.; Gonzalez, A.; Vázquez-baeza, Y.; Navas-molina, J.A.; Song, S.J.; Metcalf, J.L.; Hyde, E.R.; et al. Balance trees reveal microbial niche differentiation. mSystems 2017, 2, 1–11. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, 1–11. [Google Scholar] [CrossRef]
- Newman, M.E.J. Modularity and community structure in networks. PNAS 2006, 19, 8577–8582. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Jacomy, M. Gephi: An Open source software for exploring and manipulating networks. In Proceedings of the Third International Conference on Weblogs and Social Media, ICWSM 2009, San Jose, CA, USA, 17–20 May 2009; pp. 4–6. [Google Scholar]
- Lhomme, S. NetSwan: Network strengths and weaknesses analysis. R Pack Version. 2015, pp. 1–8. Available online: https://rdrr.io/cran/NetSwan/ (accessed on 16 April 2020).
- Yazdanbakhsh, O.; Dick, S. A systematic review of complex fuzzy sets and logic. Fuzzy Sets Syst. 2018, 338, 1–22. [Google Scholar] [CrossRef]
- Esteban, J.; García-Coca, M. Mycobacterium biofilms. Front. Microbiol. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Zumsteg, A.; Urwyler, S.K.; Glaubitz, J. Characterizing bacterial communities in paper production—troublemakers revealed. Microbiologyopen 2017, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Palmer, J.; Shah, N.; Valm, A.; Paster, B.; Dewhirst, F.; Inui, T.; Cisara, J.O. Interbacterial adhesion networks within early oral biofilms of single human hosts. Appl. Environ. Microbiol. 2017, 83, 1–16. [Google Scholar]
- Leathers, T.D.; Bischoff, K.M. Biofilm formation by strains of Leuconostoc citreum and L. mesenteroides. Biotechnol. Lett. 2011, 33, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.L.; Harkins, D.M.; Torralba, M.; Gomez, A.; Highlander, S.K.; Jones, M.B.; Leong, P.; Saffery, R.; Bockmann, M.; Kuelbs, C.; et al. Supragingival plaque microbiome ecology and functional potential in the context of health and disease. MBio 2018, 9, 1–21. [Google Scholar] [CrossRef]
- Girvan, M.; Newman, M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef]
- Limoli, D.H.; Jones, C.J.; Wozniak, D.J. Bacterial extracellular polysaccharides in biofilm formation and function. Microbiol. Spectr. 2015, 3, 1–19. [Google Scholar] [CrossRef]
- Brackman, G.; Breyne, K.; De Rycke, R.; Vermote, A.; Van Nieuwerburgh, F.; Meyer, E.; Van Calenbergh, S.; Coenye, T. The quorum sensing inhibitor hamamelitannin increases antibiotic susceptibility of Staphylococcus aureus biofilms by affecting Peptidoglycan biosynthesis and eDNA Release. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Hathroubi, S.; Hancock, M.A.; Bosse, J.T.; Langford, P.R.; Tremblay, Y.D.N.; Labrie, J.; Jacques, M. Surface polysaccharide mutants reveal that absence of o antigen reduces biofilm formation of Actinobacillus pleuropneumoniae. Infect. Immun. 2016, 84, 127–137. [Google Scholar] [CrossRef] [PubMed]
- May, T.; Okabe, S. Enterobactin is required for biofilm development in reduced-genome Escherichia coli. Environ. Microbiol. 2011, 13, 3149–3162. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.; Buckling, A. Siderophore production and biofilm formation as linked social traits. ISME J. 2009, 3, 632–634. [Google Scholar] [CrossRef]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling interactions in the microbiome: A network perspective. Trends Microbiol. 2017, 25, 217–228. [Google Scholar] [CrossRef]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 1–14. [Google Scholar] [CrossRef]
- Narasimhan, S.; Fikrig, E. Tick microbiome: The force within. Trends Parasitol. 2015, 31, 315–323. [Google Scholar] [CrossRef]
- Chicana, B.; Couper, L.I.; Kwan, J.Y.; Tahiraj, E.; Swei, A. Comparative Microbiome Profiles of Sympatric Tick Species from the Far-Western United States. Insects 2019, 10, 353. [Google Scholar] [CrossRef]
- Gall, C.A.; Scoles, G.A.; Magori, K.; Mason, K.L.; Brayton, K.A. Laboratory colonization stabilizes the naturally dynamic microbiome composition of field collected Dermacentor andersoni ticks. Microbiome 2017, 5, 133. [Google Scholar] [CrossRef]
- Bonnet, S.I.; Binetruy, F.; Hernández-jarguín, A.M.; Duron, O. The tick microbiome: why non-pathogenic microorganisms matter in tick biology and pathogen transmission. Front. Cell. Infect. Microbiol. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Agler, M.T.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.T.; Weigel, D.; Kemen, E.M. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016, 14, 1–31. [Google Scholar] [CrossRef]
- Obregón, D.; Bard, E.; Abrial, D.; Estrada-Peña, A.; Cabezas-Cruz, A. sex-specific linkages between taxonomic and functional profiles of tick gut microbiomes. Front. Cell. Infect. Microbiol. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Díaz-Sánchez, S.; Estrada-Peña, A.; Cabezas-Cruz, A.; de la Fuente, J. evolutionary insights into the tick hologenome. Trends Parasitol. 2019, 35, 725–737. [Google Scholar] [CrossRef]
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef]
- Gardner, M.R.; Ashby, W.R. Connectance of large dynamic (Cybernetic) systems: Critical values for stability. Nature 1970, 228, 784. [Google Scholar] [CrossRef]
- Zimmerli, W.; Low, P.D.; Waldvogel, F.A. Bacterial extracellular polysaccharides in biofilm formation and function. J. Clin. Invest. 1984, 73, 1191–1200. [Google Scholar] [CrossRef]
- Ricciardi, B.F.; Muthukrishnan, G.; Masters, E.; Ninomiya, M.; Lee, C.C.; Schwarz, E.M. Staphylococcus aureus evasion of host immunity in the setting of prosthetic joint infection: biofilm and beyond. Curr. Rev. Musculoskelet. Med. 2018, 11, 389–400. [Google Scholar] [CrossRef]
- González, J.F.; Hahn, M.M.; Gunn, J.S. Chronic biofilm-based infections: Skewing of the immune response. Pathog. Dis. 2018, 76, 1–7. [Google Scholar] [CrossRef]
- Scherr, T.D.; Heim, C.E.; Morrison, J.M.; Kielian, T. Hiding in plain sight: Interplay between staphylococcal biofilms and host immunity. Front. Immunol. 2014, 5, 1–7. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Toole, G.O.; Kaplan, H.B.; Kolter, R. Biofilm formation as microbial development. Annu. Rev. Microbiol. 2000, 54, 49–79. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Topological Features | Anti-Tick Immunity | A. phagocytophilum Infection | Antimicrobial Peptide | |||
|---|---|---|---|---|---|---|
| Bb-anti-OVA | Bb-anti-PIXR | Ap-uninfected | Ap-infected | sP1 | P1 | |
| Nodes a | 204 | 329 | 416 | 506 | 378 | 364 |
| Edges b | 2522 | 6974 | 11,170 | 43,389 | 7976 | 12,787 |
| Modules c | 8 | 8 | 6 | 10 | 8 | 12 |
| Network diameter d | 3 | 5 | 4 | 9 | 4 | 5 |
| Average degree e | 24.73 | 26.82 | 53.71 | 17.15 | 42.20 | 26.29 |
| Weighted degree f | 4.38 | 10.25 | 23.52 | 6.51 | 18.04 | 10.58 |
| Clustering coefficient g | 0.393 (8313) | 0.581 (34,613) | 0.528 (113,571) | 0.376 (12,102) | 0.521 (61,799) | 0.490 (22,053) |
| Topological Features | Anti-Tick Immunity | A. phagocytophilum Infection | Antimicrobial Peptide | |||
|---|---|---|---|---|---|---|
| Bb-anti-OVA | Bb-anti-PIXR | Ap-uninfected | Ap-infected | sP1 | P1 | |
| Nodes a | 392 | 404 | 427 | 429 | 425 | 427 |
| Edges b | 19,284 | 44,362 | 32,654 | 28,067 | 93,162 | 66,514 |
| Modules c | 13 | 3 | 5 | 9 | 3 | 3 |
| Network diameter d | 6 | 4 | 4 | 4 | 4 | 4 |
| Average degree (D) e | 98.14 | 124.15 | 152.95 | 130.86 | 125.41 | 103.75 |
| Weighted degree (WD) f | 74.04 | 86.51 | 103.47 | 85.022 | 82.62 | 67.91 |
| Clustering coefficient g | 0.783 (777,010) | 0.742 (1,087,589) | 0.768 (1,603,192) | 0.757 (1,302,711) | 0.730 (986,125) | 0.663 (689,790) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estrada-Peña, A.; Cabezas-Cruz, A.; Obregón, D. Resistance of Tick Gut Microbiome to Anti-Tick Vaccines, Pathogen Infection and Antimicrobial Peptides. Pathogens 2020, 9, 309. https://doi.org/10.3390/pathogens9040309
Estrada-Peña A, Cabezas-Cruz A, Obregón D. Resistance of Tick Gut Microbiome to Anti-Tick Vaccines, Pathogen Infection and Antimicrobial Peptides. Pathogens. 2020; 9(4):309. https://doi.org/10.3390/pathogens9040309
Chicago/Turabian StyleEstrada-Peña, Agustín, Alejandro Cabezas-Cruz, and Dasiel Obregón. 2020. "Resistance of Tick Gut Microbiome to Anti-Tick Vaccines, Pathogen Infection and Antimicrobial Peptides" Pathogens 9, no. 4: 309. https://doi.org/10.3390/pathogens9040309
APA StyleEstrada-Peña, A., Cabezas-Cruz, A., & Obregón, D. (2020). Resistance of Tick Gut Microbiome to Anti-Tick Vaccines, Pathogen Infection and Antimicrobial Peptides. Pathogens, 9(4), 309. https://doi.org/10.3390/pathogens9040309