Free to Circulate: An Update on the Epidemiological Dynamics of Porcine Circovirus 2 (PCV-2) in Italy Reveals the Role of Local Spreading, Wild Populations, and Foreign Countries

 , , ,
, , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
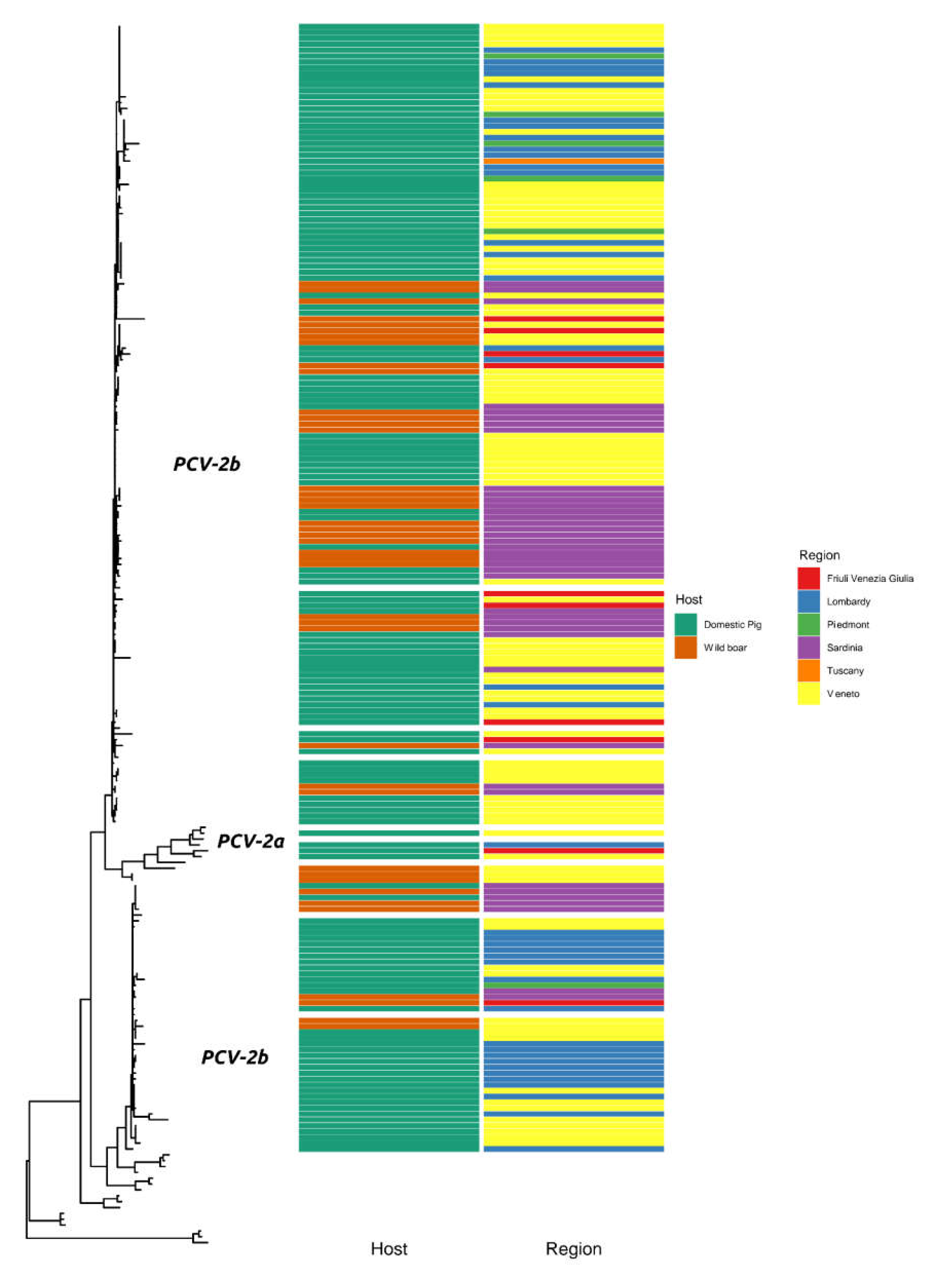
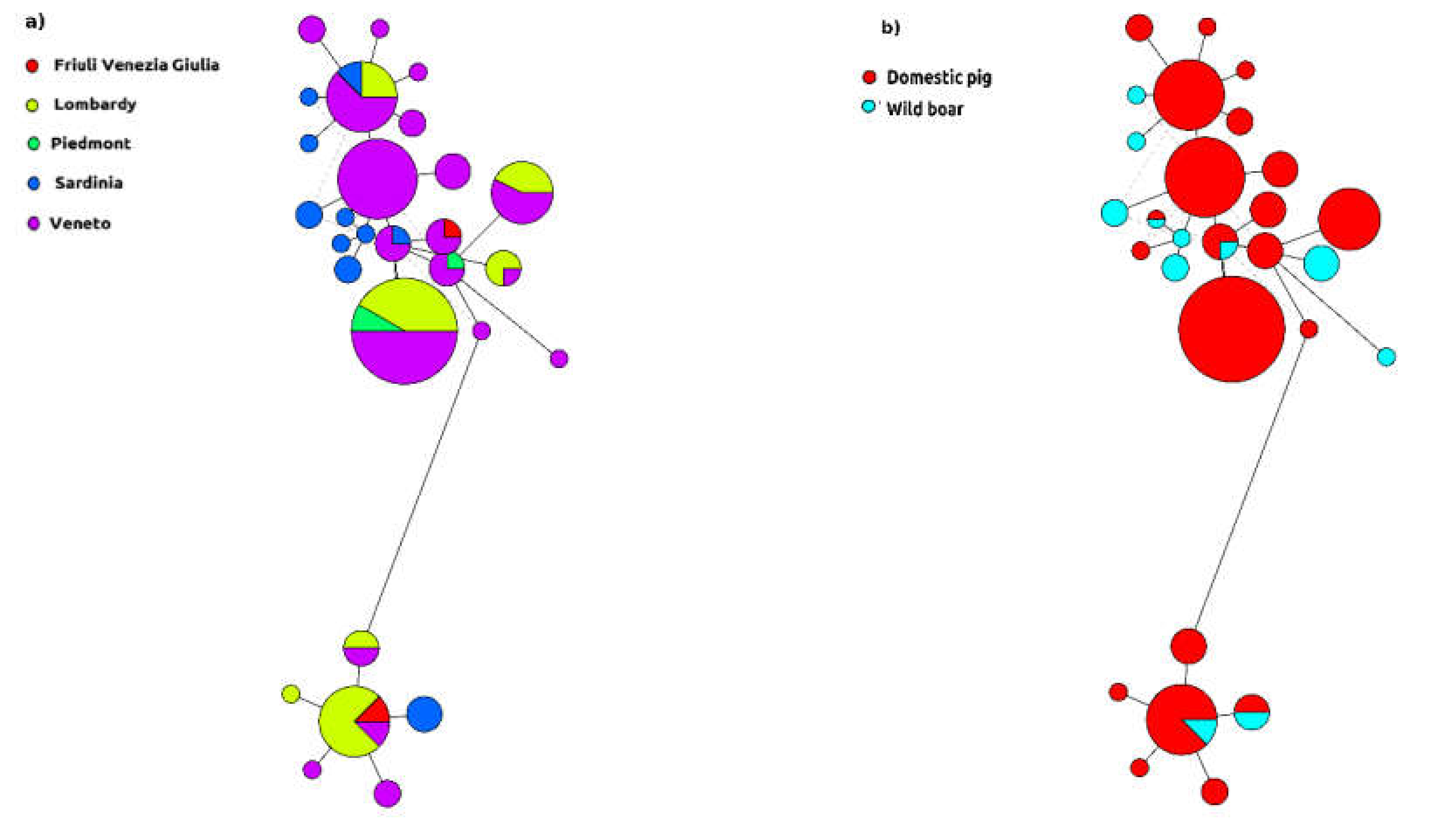
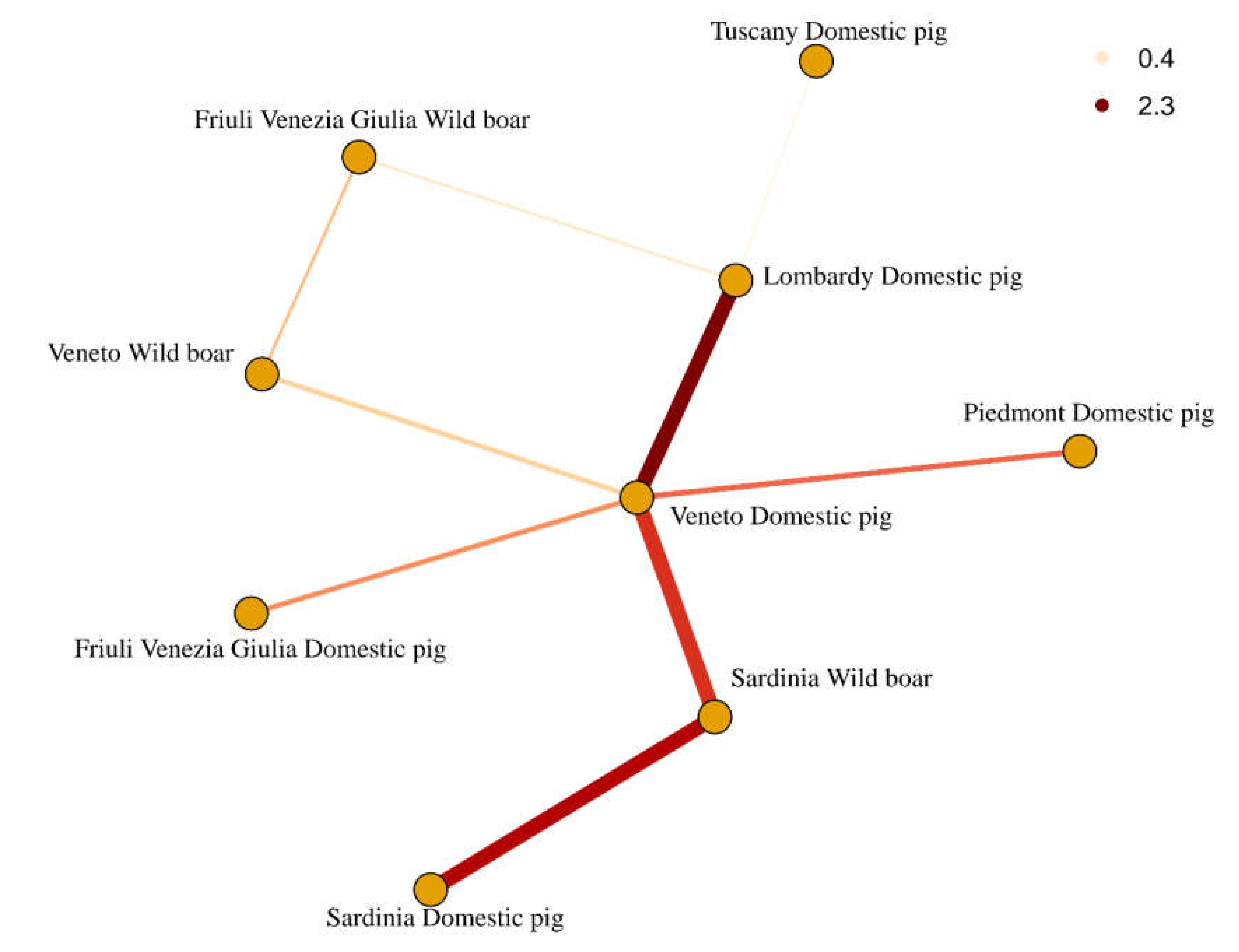
2. Results
- -
- strain 9607 originated from a recombination event between PCV-2d (region 0–350) and PCV-2a (region 351–699);
- -
- strain 16860 between PCV-2b (region 0–105 and 270–699) and PCV-2d (region 106–269);
- -
- strains 9473 and 13719 between PCV-2d (region 0–312) and PCV-2b (region 313–699).
3. Discussion
4. Material and Methods
4.1. Italian Samples
4.2. Sequence Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Segalés, J.; Kekarainen, T.; Cortey, M. The natural history of porcine circovirus type 2: From an inoffensive virus to a devastating swine disease? Vet. Microbiol. 2013, 165, 13–20. [Google Scholar] [CrossRef]
- Allan, G.M.; McNeilly, F.; Kennedy, S.; Daft, B.; Clarke, E.G.; Ellis, J.A.; Haines, D.M.; Meehan, B.M.; Adair, B.M. Isolation of porcine circovirus-like viruses from pigs with a wasting disease in the USA and Europe. J. Vet. Diagnostic Investig. 1998, 10, 3–10. [Google Scholar] [CrossRef]
- Harding, J.C.S.; Clark, E.T.G.; Strokappe, J.H.; Willson, P.I.; Ellis, J. A Postweaning multisystemic wasting syndrome: Epidemiology and clinical presentation. Swine Heal. Prod. 1998, 6, 249–254. [Google Scholar]
- Grau-Roma, L.; Fraile, L.; Segalés, J. Recent advances in the epidemiology, diagnosis and control of diseases caused by porcine circovirus type 2. Vet. J. 2011, 187, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Segalés, J. Porcine circovirus 2 (PCV-2) genotype update and proposal of a new genotyping methodology. PLoS ONE 2018, 13, e0208585. [Google Scholar] [CrossRef] [PubMed]
- Segalés, J. Porcine circovirus type 2 (PCV2) infections: Clinical signs, pathology and laboratory diagnosis. Virus Res. 2012, 164, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Young, M.G.; Cunningham, G.L.; Sanford, S.E. Circovirus vaccination in pigs with subclinical porcine circovirus type 2 infection complicated by ileitis. J. Swine Heal. Prod. 2011, 19, 175–180. [Google Scholar]
- Jacobsen, B.; Krueger, L.; Seeliger, F.; Bruegmann, M.; Segalés, J.; Baumgaertner, W. Retrospective study on the occurrence of porcine circovirus 2 infection and associated entities in Northern Germany. Vet. Microbiol. 2009, 138, 27–33. [Google Scholar] [CrossRef]
- Franzo, G.; Cortey, M.; Segalés, J.; Hughes, J.; Drigo, M. Phylodynamic analysis of porcine circovirus type 2 reveals global waves of emerging genotypes and the circulation of recombinant forms. Mol. Phylogenet. Evol. 2016, 100, 269–280. [Google Scholar] [CrossRef]
- Zhai, S.-L.; Lu, S.-S.; Wei, W.-K.; Lv, D.-H.; Wen, X.-H.; Zhai, Q.; Chen, Q.-L.; Sun, Y.-W.; Xi, Y. Reservoirs of Porcine Circoviruses: A Mini Review. Front. Vet. Sci. 2019, 6, 319. [Google Scholar] [CrossRef]
- Barbosa, C.N.; Martins, N.R.S.; Freitas, T.R.P.; Lobato, Z.I.P. Serological Survey of Porcine circovirus-2 in Captive Wild Boars (Sus scrofa) from Registered Farms of South and South-east Regions of Brazil. Transbound. Emerg. Dis. 2016, 63, e278–e280. [Google Scholar] [CrossRef] [PubMed]
- Hälli, O.; Ala-Kurikka, E.; Nokireki, T.; Skrzypczak, T.; Raunio-Saarnisto, M.; Peltoniemi, O.A.T.; Heinonen, M. Prevalence of and risk factors associated with viral and bacterial pathogens in farmed European wild boar. Vet. J. 2012, 194, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Petrini, S.; Barocci, S.; Gavaudan, S.; Villa, R.; Briscolini, S.; Sabbatini, M.; Mattozzi, C.; Barchiesi, F.; Salamida, S.; Ferrari, M.; et al. Detection of porcine circovirus type 2 (PCV2) from wild boars in central Italy. Eur. J. Wildl. Res. 2009, 55, 465–469. [Google Scholar] [CrossRef]
- Turcitu, M.A.; Wellenberg, G.J.; Barboi, G.; Codreanu, M.D.; Vuta, V.B.; Nicolae, S.; Barbuceanu, F.; Coste, H.; Cioranu, R. Genetic diversity of porcine circovirus type 2 (PCV2) in the Romanian wild boar population. Res. Vet. Sci. 2011, 91, e103–e106. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Guo, K.; Zhang, Y. Current understanding of genomic DNA of porcine circovirus type 2. Virus Genes 2014, 49, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.-Y.; Choi, Y.-C.; Park, I.-B.; Lee, C.-H.; Kang, S.-J.; Chun, T. The ORF5 protein of porcine circovirus type 2 enhances viral replication by dampening type I interferon expression in porcine epithelial cells. Vet. Microbiol. 2018, 226, 50–58. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Cao, J.; Zhou, N.; Jin, Y.; Wu, J.; Zhou, J. Identification and Functional Analysis of the Novel ORF4 Protein Encoded by Porcine Circovirus Type 2. J. Virol. 2013, 87, 1420–1429. [Google Scholar] [CrossRef]
- Li, D.; Wang, J.; Xu, S.; Cai, S.; Ao, C.; Fang, L.; Xiao, S.; Chen, H.; Jiang, Y. Identification and functional analysis of the novel ORF6 protein of porcine circovirus type 2 in vitro. Vet. Res. Commun. 2018, 42, 1–10. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Y.; Chen, I.; Lau, J.; He, F.; Lau, A.; Wang, Z.; Karuppannan, A.K.; Kwang, J. The ORF3 Protein of Porcine Circovirus Type 2 Interacts with Porcine Ubiquitin E3 Ligase Pirh2 and Facilitates p53 Expression in Viral Infection. J. Virol. 2007, 81, 9560–9567. [Google Scholar] [CrossRef]
- Xiao, C.T.; Harmon, K.M.; Halbur, P.G.; Opriessnig, T. PCV2d-2 is the predominant type of PCV2 DNA in pig samples collected in the U.S. during 2014–2016. Vet. Microbiol. 2016, 197, 72–77. [Google Scholar] [CrossRef]
- Cortey, M.; Pileri, E.; Sibila, M.; Pujols, J.; Balasch, M.; Plana, J.; Segalés, J. Genotypic shift of porcine circovirus type 2 from PCV-2a to PCV-2b in Spain from 1985 to 2008. Vet. J. 2011, 187, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Dupont, K.; Nielsen, E.O.; Bækbo, P.; Larsen, L.E. Genomic analysis of PCV2 isolates from Danish archives and a current PMWS case-control study supports a shift in genotypes with time. Vet. Microbiol. 2008, 128, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Guo, X.; Ge, X.; Wang, Z.; Chen, Y.; Cha, Z.; Yang, H. Genetic variation analysis of Chinese strains of porcine circovirus type 2. Virus Res. 2009, 145, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Cortey, M.; Segalés, J.; Hughes, J.; Drigo, M. Phylodynamic analysis of porcine circovirus type 2: Methodological approach and datasets. DATA Br. 2016, 8, 549–552. [Google Scholar] [CrossRef]
- Franzo, G.; Tucciarone, C.M.; Dotto, G.; Gigli, A.; Ceglie, L.; Drigo, M. International trades, local spread and viral evolution: The case of porcine circovirus type 2 (PCV2) strains heterogeneity in Italy. Infect. Genet. Evol. 2015, 32, 409–415. [Google Scholar] [CrossRef]
- Dei Giudici, S.; Lo Presti, A.; Bonelli, P.; Angioi, P.P.; Sanna, G.; Zinellu, S.; Balzano, F.; Salis, F.; Ciccozzi, M.; Oggiano, A. Phylogenetic analysis of porcine circovirus type 2 in Sardinia, Italy, shows genotype 2d circulation among domestic pigs and wild boars. Infect. Genet. Evol. 2019, 71, 189–196. [Google Scholar] [CrossRef]
- Mur, L.; Atzeni, M.; Martínez-López, B.; Feliziani, F.; Rolesu, S.; Sanchez-Vizcaino, J.M. Thirty-Five-Year Presence of African Swine Fever in Sardinia: History, Evolution and Risk Factors for Disease Maintenance. Transbound. Emerg. Dis. 2016, 63, e165–e177. [Google Scholar] [CrossRef]
- Kwon, T.; Lee, D.U.; Yoo, S.J.; Je, S.H.; Shin, J.Y.; Lyoo, Y.S. Genotypic diversity of porcine circovirus type 2 (PCV2) and genotype shift to PCV2d in Korean pig population. Virus Res. 2017, 228, 24–29. [Google Scholar] [CrossRef]
- Guo, L.; Fu, Y.; Wang, Y.; Lu, Y.; Wei, Y.; Tang, Q.; Fan, P.; Liu, J.; Zhang, L.; Zhang, F.; et al. A porcine circovirus type 2 (PCV2) mutant with 234 amino acids in Capsid protein showed more virulence in vivo, compared with classical PCV2a/b strain. PLoS ONE 2012, 7, e41463. [Google Scholar] [CrossRef]
- Opriessnig, T.; Gerber, P.F.; Xiao, C.T.; Halbur, P.G.; Matzinger, S.R.; Meng, X.J. Commercial PCV2a-based vaccines are effective in protecting naturally PCV2b-infected finisher pigs against experimental challenge with a 2012 mutant PCV2. Vaccine 2014, 32, 4342–4348. [Google Scholar] [CrossRef]
- Park, K.H.; Oh, T.; Yang, S.; Cho, H.; Kang, I.; Chae, C. Evaluation of a porcine circovirus type 2a (PCV2a) vaccine efficacy against experimental PCV2a, PCV2b, and PCV2d challenge. Vet. Microbiol. 2019, 231, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Huang, L.; Bussalleu, E.; Lefebvre, D.J.; Fort, M.; Van Doorsselaere, J.; Nauwynck, H.J. Antigenic subtyping and epitopes’ competition analysis of porcine circovirus type 2 using monoclonal antibodies. Vet. Microbiol. 2012, 157, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Wang, N.; Zhu, Z.; Wang, Z.; Wang, A.; Deng, Z.; Yang, Y. In silico analyses of antigenicity and surface structure variation of an emerging porcine circovirus genotype 2b mutant, prevalent in Southern China from 2013 to 2015. J. Gen. Virol. 2016, 97, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Tucciarone, C.M.; Cecchinato, M.; Drigo, M. Porcine circovirus type 2 (PCV2) evolution before and after the vaccination introduction: A large scale epidemiological study. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Jeong, J.; Park, C.; Choi, K.; Chae, C. Comparison of three commercial one-dose porcine circovirus type 2 (PCV2) vaccines in a herd with concurrent circulation of PCV2b and mutant PCV2b. Vet. Microbiol. 2015, 177, 43–52. [Google Scholar] [CrossRef]
- Karuppannan, A.K.; Opriessnig, T. Porcine circovirus type 2 (PCV2) vaccines in the context of current molecular epidemiology. Viruses 2017, 9, 99. [Google Scholar] [CrossRef]
- Opriessnig, T.; Xiao, C.T.; Halbur, P.G.; Gerber, P.F.; Matzinger, S.R.; Meng, X.J. A commercial porcine circovirus (PCV) type 2a-based vaccine reduces PCV2d viremia and shedding and prevents PCV2d transmission to naïve pigs under experimental conditions. Vaccine 2017, 35, 248–254. [Google Scholar] [CrossRef]
- Opriessnig, T.; O’Neill, K.; Gerber, P.F.; de Castro, A.M.M.G.; Gimenéz-Lirola, L.G.; Beach, N.M.; Zhou, L.; Meng, X.J.; Wang, C.; Halbur, P.G. A PCV2 vaccine based on genotype 2b is more effective than a 2a-based vaccine to protect against PCV2b or combined PCV2a/2b viremia in pigs with concurrent PCV2, PRRSV and PPV infection. Vaccine 2013, 31, 487–494. [Google Scholar] [CrossRef]
- Edlefsen, P.T. Leaky vaccines protect highly exposed recipients at a lower rate: Implications for vaccine efficacy estimation and sieve analysis. Comput. Math. Methods Med. 2014, 2014. [Google Scholar] [CrossRef]
- Franzo, G.; Dotto, G.; Cecchinato, M.; Pasotto, D.; Martini, M.; Drigo, M. Phylodynamic analysis of porcine reproductive and respiratory syndrome virus (PRRSV) in Italy: Action of selective pressures and interactions between different clades. Infect. Genet. Evol. 2015, 31, 149–157. [Google Scholar] [CrossRef]
- Franzo, G.; Massi, P.; Tucciarone, C.M.; Barbieri, I.; Tosi, G.; Fiorentini, L.; Ciccozzi, M.; Lavazza, A.; Cecchinato, M.; Moreno, A. Think globally, act locally: Phylodynamic reconstruction of infectious bronchitis virus (IBV) QX genotype (GI-19 lineage) reveals different population dynamics and spreading patterns when evaluated on different epidemiological scales. PLoS ONE 2017, 12, e0184401. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Lett, J.-M.; Varsani, A.; Martin, D.P. Widely Conserved Recombination Patterns among Single-Stranded DNA Viruses. J. Virol. 2009, 83, 2697–2707. [Google Scholar] [CrossRef] [PubMed]
- Correa-Fiz, F.; Franzo, G.; Llorens, A.; Segalés, J.; Kekarainen, T. Porcine circovirus 2 (PCV-2) genetic variability under natural infection scenario reveals a complex network of viral quasispecies. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.; Segalés, J.; Höfle, U.; Balasch, M.; Plana-Durán, J.; Domingo, M.; Gortázar, C. Epidemiological study on porcine circovirus type 2 (PCV2) infection in the European wild boar (Sus scrofa). Vet. Res. 2004, 35, 243–253. [Google Scholar] [CrossRef]
- Franzo, G.; Tucciarone, C.M.; Drigo, M.; Cecchinato, M.; Martini, M.; Mondin, A.; Menandro, M.L. First report of wild boar susceptibility to Porcine circovirus type 3: High prevalence in the Colli Euganei Regional Park (Italy) in the absence of clinical signs. Transbound. Emerg. Dis. 2018, 65, 957–962. [Google Scholar] [CrossRef]
- Franzo, G.; Grassi, L.; Tucciarone, C.M.; Drigo, M.; Martini, M.; Pasotto, D.; Mondin, A.; Menandro, M.L. A wild circulation: High presence of Porcine circovirus 3 in different mammalian wild hosts and ticks. Transbound. Emerg. Dis. 2019, 66, 1548–1557. [Google Scholar] [CrossRef]
- Opriessnig, T.; Yu, S.; Gallup, J.M.; Evans, R.B.; Fenaux, M.; Pallares, F.; Thacker, E.L.; Brockus, C.W.; Ackermann, M.R.; Thomas, P.; et al. Effect of vaccination with selective bacterins on conventional pigs infected with type 2 porcine circovirus. Vet. Pathol. 2003, 40, 521–529. [Google Scholar] [CrossRef]
- Standley, K. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability.(outlines version 7). Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E. Pegas: An R package for population genetics with an integrated-modular approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzo, G.; Tinello, S.; Grassi, L.; Tucciarone, C.M.; Legnardi, M.; Cecchinato, M.; Dotto, G.; Mondin, A.; Martini, M.; Pasotto, D.; et al. Free to Circulate: An Update on the Epidemiological Dynamics of Porcine Circovirus 2 (PCV-2) in Italy Reveals the Role of Local Spreading, Wild Populations, and Foreign Countries. Pathogens 2020, 9, 221. https://doi.org/10.3390/pathogens9030221
Franzo G, Tinello S, Grassi L, Tucciarone CM, Legnardi M, Cecchinato M, Dotto G, Mondin A, Martini M, Pasotto D, et al. Free to Circulate: An Update on the Epidemiological Dynamics of Porcine Circovirus 2 (PCV-2) in Italy Reveals the Role of Local Spreading, Wild Populations, and Foreign Countries. Pathogens. 2020; 9(3):221. https://doi.org/10.3390/pathogens9030221
Chicago/Turabian StyleFranzo, Giovanni, Susanna Tinello, Laura Grassi, Claudia Maria Tucciarone, Matteo Legnardi, Mattia Cecchinato, Giorgia Dotto, Alessandra Mondin, Marco Martini, Daniela Pasotto, and et al. 2020. "Free to Circulate: An Update on the Epidemiological Dynamics of Porcine Circovirus 2 (PCV-2) in Italy Reveals the Role of Local Spreading, Wild Populations, and Foreign Countries" Pathogens 9, no. 3: 221. https://doi.org/10.3390/pathogens9030221
APA StyleFranzo, G., Tinello, S., Grassi, L., Tucciarone, C. M., Legnardi, M., Cecchinato, M., Dotto, G., Mondin, A., Martini, M., Pasotto, D., Menandro, M. L., & Drigo, M. (2020). Free to Circulate: An Update on the Epidemiological Dynamics of Porcine Circovirus 2 (PCV-2) in Italy Reveals the Role of Local Spreading, Wild Populations, and Foreign Countries. Pathogens, 9(3), 221. https://doi.org/10.3390/pathogens9030221