A Quantitative Live-Cell Superresolution Imaging Framework for Measuring the Mobility of Single Molecules at Sites of Virus Assembly



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
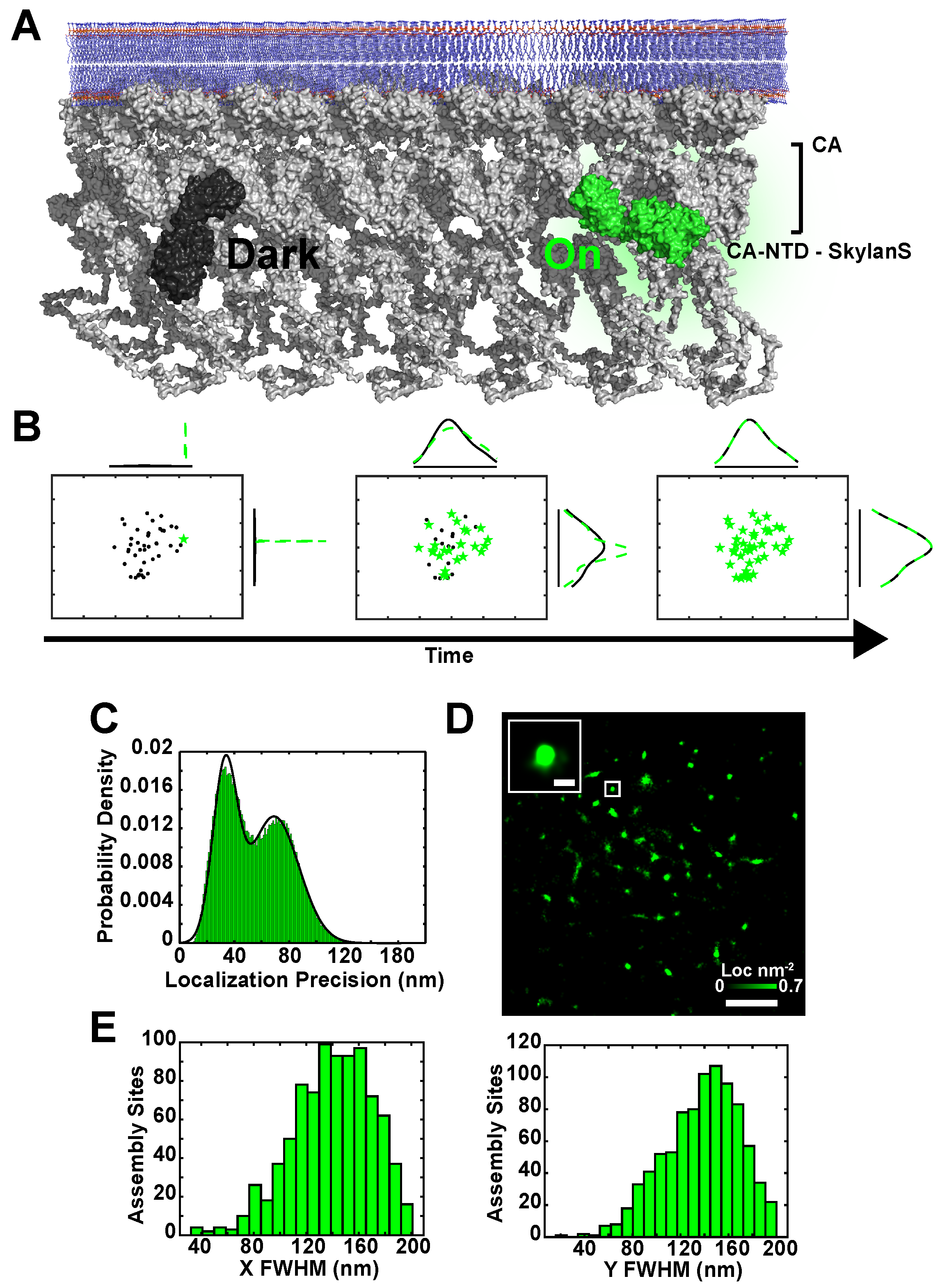
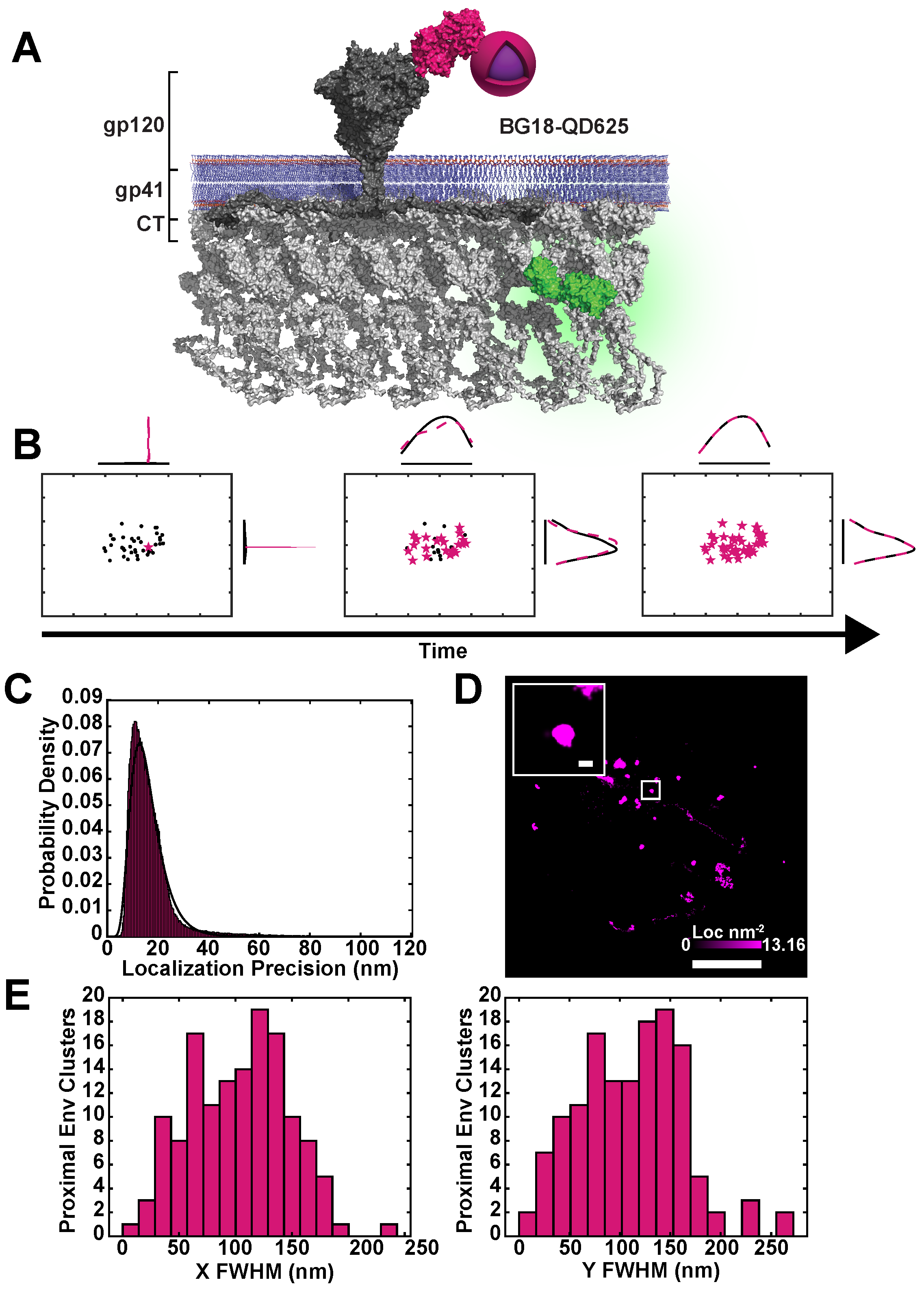
2.1. Simultaneous Superresolution Reconstruction and Tracking of HIV-1 Gag and Env
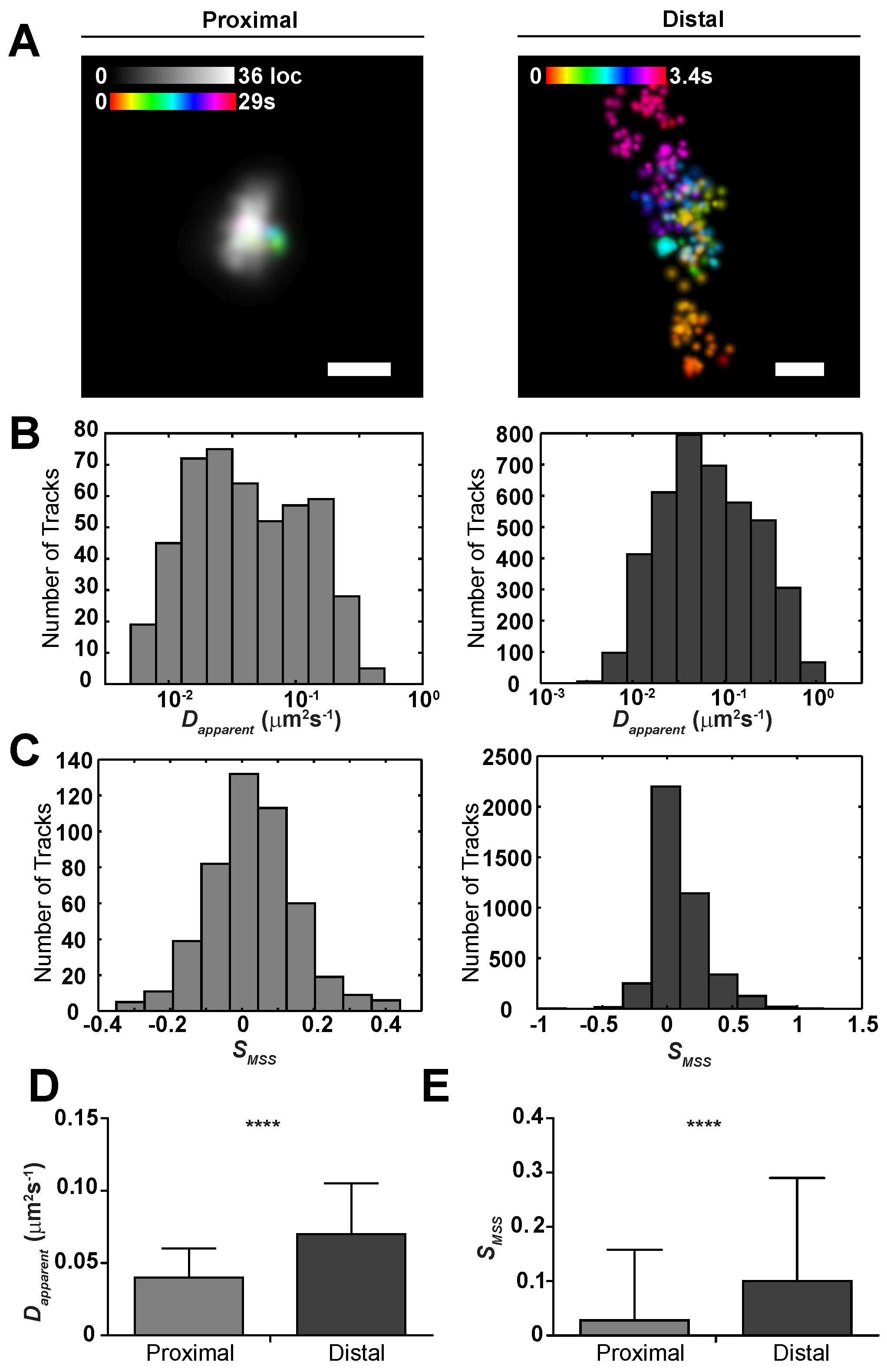
2.2. Sub-Viral Quantification of Env Diffusion
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Generation of CA-Skylan-S Expressing Viruses
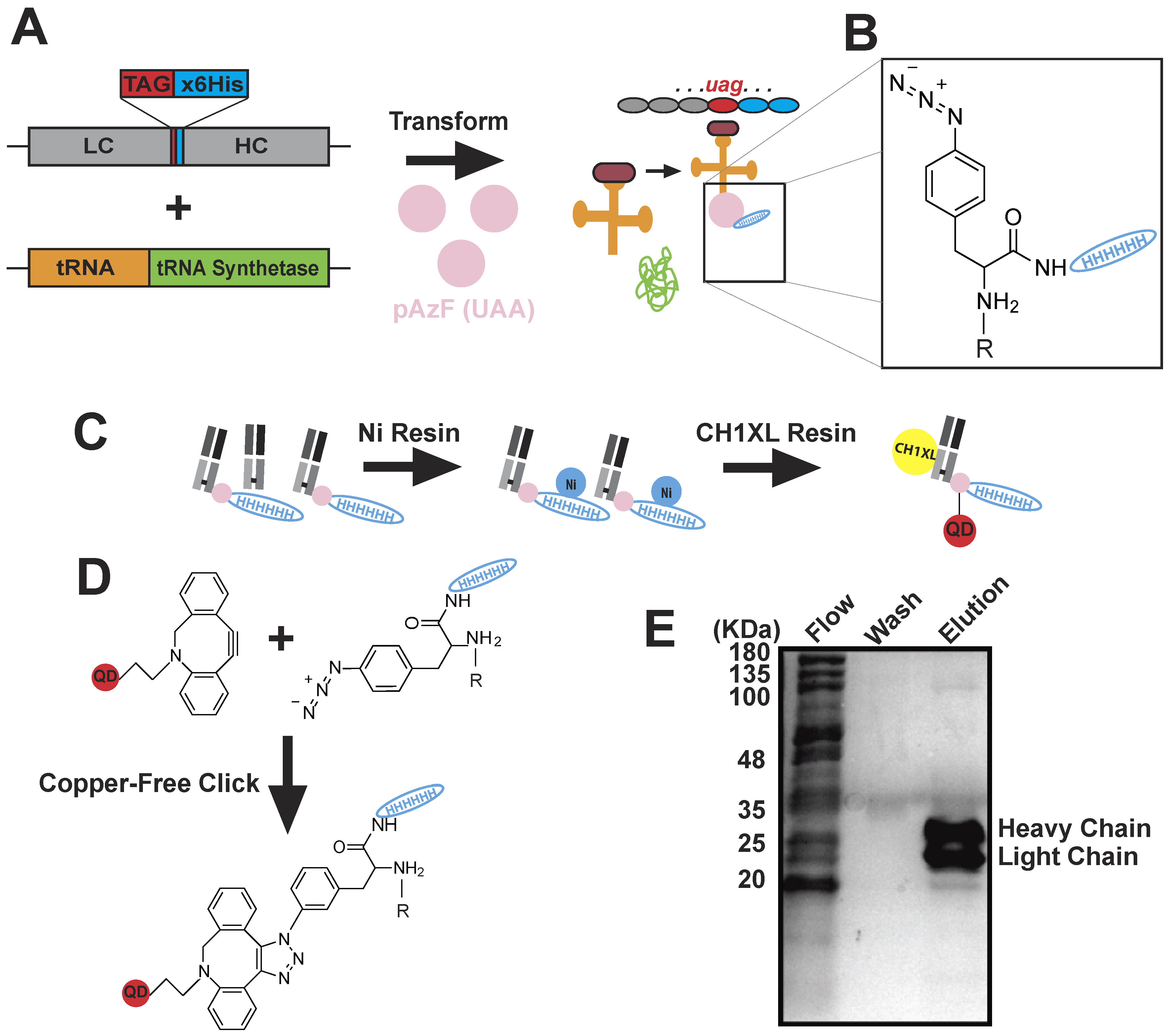
4.3. Monovalent BG18-QD625 Production
4.4. Sample Preparation and Imaging Conditions
4.5. Image Processing and Analysis
4.6. Monte Carlo Simulations of Single Molecule Localization and HIV-1 Assembly Site Reconstruction
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Env | Envelope |
| MA | Matrix |
| CA | Capsid |
| SPT | Single Particle Tracking |
| MSD | Mean Squared Displacement |
| Slope of the Moment Scaling Spectrum | |
| TIRF-M | Total Internal Reflection Fluorescence Microscopy |
References
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hell, S.W. Far-field optical nanoscopy. Science 2007, 316, 1153–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef] [Green Version]
- Buttler, C.A.; Pezeshkian, N.; Fernandez, M.V.; Aaron, J.; Norman, S.; Freed, E.O.; Van Engelenburg, S.B. Single molecule fate of HIV-1 envelope reveals late-stage viral lattice incorporation. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lara-Tejero, M.; Bewersdorf, J.; Galán, J.E. Visualization and characterization of individual type III protein secretion machines in live bacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 6098–6103. [Google Scholar] [CrossRef] [Green Version]
- Han, J.J.; Kunde, Y.; Hong-Geller, E.; Werner, J.H. Actin restructuring during Salmonella typhimurium infection investigated by confocal and super-resolution microscopy. J. Biomed. Opt. 2014, 19, 016011. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Chen, L.; Aguilar, H.C.; Chou, K.C. A stochastic assembly model for Nipah virus revealed by super-resolution microscopy. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gray, R.D.; Beerli, C.; Pereira, P.M.; Scherer, K.M.; Samolej, J.; Bleck, C.K.E.; Mercer, J.; Henriques, R. VirusMapper: open-source nanoscale mapping of viral architecture through super-resolution microscopy. Sci. Rep. 2016, 6, 29132. [Google Scholar] [CrossRef] [Green Version]
- Horsington, J.; Lynn, H.; Turnbull, L.; Cheng, D.; Braet, F.; Diefenbach, R.J.; Whitchurch, C.B.; Karupiah, G.; Newsome, T.P. A36-dependent actin filament nucleation promotes release of vaccinia virus. PLoS Pathog. 2013, 9, e1003239. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.T.; Gould, T.J.; Gudheti, M.V.; Maas, S.A.; Mills, K.D.; Zimmerberg, J. Dynamic clustered distribution of hemagglutinin resolved at 40 nm in living cell membranes discriminates between raft theories. Proc. Natl. Acad. Sci. USA 2007, 104, 17370–17375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, R.F.; Albecka, A.; Van De Linde, S.; Rees, E.J.; Crump, C.M.; Kaminski, C.F. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floderer, C.; Masson, J.B.; Boilley, E.; Georgeault, S.; Merida, P.; El Beheiry, M.; Dahan, M.; Roingeard, P.; Sibarita, J.B.; Favard, C.; et al. Single molecule localisation microscopy reveals how HIV-1 Gag proteins sense membrane virus assembly sites in living host CD4 T cells. Sci. Rep. 2018, 8, 1–15. [Google Scholar]
- Lehmann, M.; Rocha, S.; Mangeat, B.; Blanchet, F.; Uji-i, H.; Hofkens, J.; Piguet, V. Quantitative multicolor super-resolution microscopy reveals tetherin HIV-1 interaction. PLoS Pathog. 2011, 7, e1002456. [Google Scholar] [CrossRef] [Green Version]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Müller, B.; Hell, S.W.; Kräusslich, H.G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.F.; Rossy, J.; Owen, D.M.; Mak, J.; Gaus, K. HIV taken by STORM: Super-resolution fluorescence microscopy of a viral infection. Virol. J. 2012, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Gunzenhäuser, J.; Olivier, N.; Pengo, T.; Manley, S. Quantitative super-resolution imaging reveals protein stoichiometry and nanoscale morphology of assembling HIV-Gag virions. Nano Lett. 2012, 12, 4705–4710. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.H.; Chan, J.; Lambelé, M.; Thali, M. Clustering and mobility of HIV-1 Env at viral assembly sites predict its propensity to induce cell-cell fusion. J. Virol. 2013, 87, 7516–7525. [Google Scholar] [CrossRef] [Green Version]
- Muranyi, W.; Malkusch, S.; Müller, B.; Heilemann, M.; Kräusslich, H.G. Super-Resolution Microscopy Reveals Specific Recruitment of HIV-1 Envelope Proteins to Viral Assembly Sites Dependent on the Envelope C-Terminal Tail. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef]
- Van Engelenburg, S.B.; Shtengel, G.; Sengupta, P.; Waki, K.; Jarnik, M.; Ablan, S.D.; Freed, E.O.; Hess, H.F.; Lippincott-Schwartz, J. Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science 2014, 343, 653–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrix, J.; Baumgärtel, V.; Schrimpf, W.; Ivanchenko, S.; Digman, M.A.; Gratton, E.; Kräusslich, H.G.; Müller, B.; Lamb, D.C. Live-cell observation of cytosolic HIV-1 assembly onset reveals RNA-interacting Gag oligomers. J. Cell Biol. 2015, 210, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Boutant, E.; Bonzi, J.; Anton, H.; Nasim, M.B.; Cathagne, R.; Réal, E.; Dujardin, D.; Carl, P.; Didier, P.; Paillart, J.C.; et al. Zinc Fingers in HIV-1 Gag Precursor Are Not Equivalent for gRNA Recruitment at the Plasma Membrane. Biophys. J. 2020, 119, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, K.H.; Berk, S.; Grigsby, I.F.; Chen, Y.; Mansky, L.M.; Mueller, J.D. Interrelationship between cytoplasmic retroviral Gag concentration and Gag–membrane association. J. Mol. Biol. 2014, 426, 1611–1624. [Google Scholar] [CrossRef] [Green Version]
- Sakin, V.; Hanne, J.; Dunder, J.; Anders-Össwein, M.; Laketa, V.; Nikić, I.; Kräusslich, H.G.; Lemke, E.A.; Müller, B. A Versatile Tool for Live-Cell Imaging and Super-Resolution Nanoscopy Studies of HIV-1 Env Distribution and Mobility. Cell Chem. Biol. 2017, 24, 635.e5–645.e5. [Google Scholar] [CrossRef] [Green Version]
- Chojnacki, J.; Waithe, D.; Carravilla, P.; Huarte, N.; Galiani, S.; Enderlein, J.; Eggeling, C. Envelope glycoprotein mobility on HIV-1 particles depends on the virus maturation state. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Pezeshkian, N.; Groves, N.; van Engelenburg, S. Single-molecule imaging of HIV-1 envelope glycoprotein dynamics and Gag lattice association exposes determinants responsible for virus incorporation. Proc. Natl. Acad. Sci. USA 2019, 116. [Google Scholar] [CrossRef] [Green Version]
- Inavalli, V.V.; Lenz, M.O.; Butler, C.; Angibaud, J.; Compans, B.; Levet, F.; Tønnesen, J.; Rossier, O.; Giannone, G.; Thoumine, O.; et al. A super-resolution platform for correlative live single-molecule imaging and STED microscopy. Nat. Methods 2019, 16, 1263–1268. [Google Scholar] [CrossRef]
- Saad, J.S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R.H.; Summers, M.F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11364–11369. [Google Scholar] [CrossRef] [Green Version]
- Alfadhli, A.; Still, A.; Barklis, E. Analysis of Human Immunodeficiency Virus Type 1 Matrix Binding to Membranes and Nucleic Acids. J. Virol. 2009, 83, 12196–12203. [Google Scholar] [CrossRef] [Green Version]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. HIV-1 Gag targeting to the plasma membrane. October 2004, 101, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Hill, C.P.; Sundquist, W.I.; Finch, J.T. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 2000, 407, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Gamble, T.R.; Yoo, S.; Vajdos, F.F.; von Schwedler, U.K.; Worthylake, D.K.; Wang, H.; McCutcheon, J.P.; Sundquist, W.I.; Hill, C.P. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 1997, 278, 849–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouvenet, N.; Bieniasz, P.D.; Simon, S.M. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 2008, 454, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Ivanchenko, S.; Godinez, W.J.; Lampe, M.; Kräusslich, H.G.; Eils, R.; Rohr, K.; Bräuchle, C.; Müller, B.; Lamb, D.C. Dynamics of HIV-1 assembly and release. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [Green Version]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef] [Green Version]
- Byland, R.; Vance, P.J.; Hoxie, J.A.; Marsh, M. A Conserved Dileucine Motif Mediates Clathrin and AP-2–dependent Endocytosis of the HIV-1 Envelope Protein. Mol. Biol. Cell 2007, 18, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Helma, J.; Schmidthals, K.; Lux, V.; Nüske, S.; Scholz, A.M.; Kräusslich, H.G.; Rothbauer, U.; Leonhardt, H. Direct and Dynamic Detection of HIV-1 in Living Cells. PLoS ONE 2012, 7, e50026. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, X.; Zeng, Z.; Zhang, M.; Sun, Y.; Xi, P.; Peng, J.; Xu, P. Development of a reversibly switchable fluorescent protein for super-resolution optical fluctuation imaging (SOFI). ACS Nano 2015, 9, 2659–2667. [Google Scholar] [CrossRef]
- Shroff, H.; Galbraith, C.G.; Galbraith, J.A.; Betzig, E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat. Methods 2008, 5, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Briggs, J.A.; Wilk, T.; Welker, R.; Kräusslich, H.G.; Fuller, S.D. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 2003, 22, 1707–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, N.T.; Wang, H.; Scharf, L.; Nogueira, L.; Horwitz, J.A.; Bar-On, Y.; Golijanin, J.; Sievers, S.A.; Sok, D.; Cai, H.; et al. Coexistence of potent HIV-1 broadly neutralizing antibodies and antibody-sensitive viruses in a viremic controller. Sci. Transl. Med. 2017, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, H.; Sheetz, M.P.; Elson, E.L. Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 1991, 60, 910–921. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R.; Manfroi, A.; Young, W. Strongly and weakly self-similar diffusion. Phys. Nonlinear Phenom. 2001, 154, 111–137. [Google Scholar] [CrossRef]
- Huang, F.; Hartwich, T.M.; Rivera-Molina, F.E.; Lin, Y.; Duim, W.C.; Long, J.J.; Uchil, P.D.; Myers, J.R.; Baird, M.A.; Mothes, W.; et al. Video-rate nanoscopy using sCMOS camera–specific single-molecule localization algorithms. Nat. Methods 2013, 10, 653–658. [Google Scholar] [CrossRef]
- Tremblay, M.; Rooke, R.; Geleziunas, R.; Wainberg, M.A.; Sullivan, A.K.; Tsoukas, C.; Gilmore, N.; Shematek, G. New cd4 (+) cell line susceptible to infection by hiv-1. J. Med. Virol. 1989, 28, 243–249. [Google Scholar] [CrossRef]
- Barnes, C.O.; Gristick, H.B.; Freund, N.T.; Escolano, A.; Lyubimov, A.Y.; Hartweger, H.; West, A.P.; Cohen, A.E.; Nussenzweig, M.C.; Bjorkman, P.J. Structural characterization of a highly-potent V3-glycan broadly neutralizing antibody bound to natively-glycosylated HIV-1 envelope. Nat. Commun. 2018, 9, 1251. [Google Scholar] [CrossRef] [Green Version]
- Barbas, C.F.; Hu, D.; Dunlop, N.; Sawyer, L.; Cababa, D.; Hendry, R.M.; Nara, P.L.; Burton, D.R. In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc. Natl. Acad. Sci. USA 1994, 91, 3809–3813. [Google Scholar] [CrossRef] [Green Version]
- Crocker, J.C.; Grier, D.G. Methods of digital video microscopy for colloidal studies. J. Colloid Interface Sci. 1996, 179, 298–310. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the probes and analysis code are available upon request from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groves, N.S.; Bruns, M.M.; van Engelenburg, S.B. A Quantitative Live-Cell Superresolution Imaging Framework for Measuring the Mobility of Single Molecules at Sites of Virus Assembly. Pathogens 2020, 9, 972. https://doi.org/10.3390/pathogens9110972
Groves NS, Bruns MM, van Engelenburg SB. A Quantitative Live-Cell Superresolution Imaging Framework for Measuring the Mobility of Single Molecules at Sites of Virus Assembly. Pathogens. 2020; 9(11):972. https://doi.org/10.3390/pathogens9110972
Chicago/Turabian StyleGroves, Nicholas S., Merissa M. Bruns, and Schuyler B. van Engelenburg. 2020. "A Quantitative Live-Cell Superresolution Imaging Framework for Measuring the Mobility of Single Molecules at Sites of Virus Assembly" Pathogens 9, no. 11: 972. https://doi.org/10.3390/pathogens9110972
APA StyleGroves, N. S., Bruns, M. M., & van Engelenburg, S. B. (2020). A Quantitative Live-Cell Superresolution Imaging Framework for Measuring the Mobility of Single Molecules at Sites of Virus Assembly. Pathogens, 9(11), 972. https://doi.org/10.3390/pathogens9110972