Multilocus Genotyping Reveals New Molecular Markers for Differentiating Distinct Genetic Lineages among “Candidatus Phytoplasma Solani” Strains Associated with Grapevine Bois Noir


 ,
,  , ,
, ,  ,
,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
2.1. CaPsol Identification
2.2. Characterization and Distribution of CaPsol tuf-Types
2.3. Possible Role of Protein Encoded by tufB Gene (EF-Tu) in Host Selection
2.4. Genetic Lineages in CaPsol Strains Determined by Multiple Gene Sequence Typing
2.5. RFLP Analyses: Prevalence of CaPsol Lineages
3. Materials and Methods
3.1. Sample Collection
3.2. CaPsol Molecular Identification
3.3. Molecular Characterization of CaPsol Strains through Multilocus Genotyping Analysis
3.4. Phylogenetic Analyses
3.5. Survey on CaPsol Genetic Lineages by Restriction Fragment Length Polymorphism Analyis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Quaglino, F.; Zhao, Y.; Casati, P.; Bulgari, D.; Bianco, P.A.; Wei, W.; Davis, R.E. ‘Candidatus Phytoplasma solani’, a novel taxon associated with stolbur and Bois Noir related diseases of plants. Int. J. Syst. Evol. Microbiol. 2013, 63, 2879–2894. [Google Scholar] [CrossRef] [PubMed]
- Romanazzi, G.; Murolo, S.; Feliziani, E. Effects of an innovative strategy to contain grapevine Bois noir: Field treatment with resistance inducers. Phytopathology 2013, 103, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Langer, M.; Maixner, M. Molecular characterisation of Grapevine yellows associated phytoplasmas of the stolbur-group based on RFLP-analysis of non-ribosomal DNA. Vitis 2004, 43, 191–200. [Google Scholar]
- Cvrković, T.; Jović, J.; Mitrović, M.; Krstić, Q.; Toševski, I. Experimental and molecular evidence of Reptalus panzeri as a natural vector of bois noir. Plant Pathol. 2014, 63, 42–53. [Google Scholar] [CrossRef]
- Mori, N.; Quaglino, F.; Tessari, F.; Pozzebon, A.; Bulgari, D.; Casati, P.; Bianco, P.A. Investigation on ‘bois noir’ epidemiology in north-eastern Italian vineyards through a multidisciplinary approach. Ann. Appl. Biol. 2015, 166, 75–89. [Google Scholar] [CrossRef]
- Kosovac, A.; Radonjić, S.; Hrnčić, S.; Krstić, O.; Toševski, I.; Jović, J. Molecular tracing of the transmission routes of bois noir in Mediterranean Vineyards of Montenegro and experimental evidence for the epidemiological role of Vitex agnus-castus (Lamiaceae) and associated Hyalesthes obsoletus (Cixiidae). Plant Pathol. 2016, 65, 285–298. [Google Scholar] [CrossRef]
- Kosovac, A.; Jakovljević, M.; Krstić, O.; Cvrković, T.; Mitrović, M.; Toševski, I.; Jović, J. Role of plant-specialized Hyalesthes obsoletus associated with Convolvulus arvensis and Crepis foetida in the transmission of ‘Candidatus Phytoplasma solani’-inflicted Bois noir disease of grapevine in Serbia. Eur. J. Plant Pathol. 2019, 153, 183–195. [Google Scholar] [CrossRef]
- Quaglino, F.; Sanna, F.; Moussa, A.; Faccincani, M.; Passera, A.; Casati, P.; Bianco, P.A.; Mori, N. Identification and ecology of alternative insect vectors of ‘Candidatus Phytoplasma solani’ to grapevine. Sci. Rep. 2019, 9, 19522. [Google Scholar] [CrossRef]
- Moussa, A.; Mori, N.; Faccincani, M.; Pavan, F.; Bianco, P.A.; Quaglino, F. Vitex agnus-castus cannot be used as trap plant for the vector Hyalesthes obsoletus to prevent infections by ‘Candidatus Phytoplasma solani’ in northern Italian vineyards: Experimental evidence. Ann. Appl. Biol. 2019, 175, 302–312. [Google Scholar] [CrossRef]
- Pierro, R.; Panattoni, A.; Passera, A.; Materazzi, A.; Luvisi, A.; Loni, A.; Ginanni, M.; Lucchi, A.; Bianco, P.A.; Quaglino, F. Proposal of a new Bois noir epidemiological pattern related to ‘Candidatus Phytoplasma solani’ strains characterized by a possible moderate virulence in Tuscany. Pathogens 2020, 9, 268. [Google Scholar] [CrossRef]
- Bianco, P.A.; Romanazzi, G.; Mori, N.; Myrie, W.; Bertaccini, A. Integrated management of phytoplasma diseases. In Transmission and Management of Phytoplasma—Associated Diseases. Phytoplasmas: Plant Pathogenic Bacteria–II; Bertaccini, A., Weintraub, G.P., Rao, G.P., Mori, N., Eds.; Springer Nature: Singapore, 2019; pp. 208–237. [Google Scholar]
- Murolo, S.; Romanazzi, G. In-vineyard population structure of ‘Candidatus Phytoplasma solani’ using multilocus sequence typing analysis. Infect. Gen. Evol. 2015, 31, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Quaglino, F.; Maghradze, D.; Casati, P.; Chkhaidze, N.; Lobjanidze, M.; Ravasio, A.; Passera, A.; Venturini, G.; Failla, O.; Bianco, P.A. Identification and characterization of new ‘Candidatus Phytoplasma solani’ strains associated with bois noir disease in Vitis vinifera L. cultivars showing a range of symptoms severity in Georgia, the Caucasus region. Plant Dis. 2016, 100, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Pierro, R.; Passera, A.; Panattoni, A.; Casati, P.; Luvisi, A.; Rizzo, D.; Bianco, P.A.; Quaglino, F.; Materazzi, A. Molecular typing of ‘Bois Noir’ phytoplasma strains in the Chianti Classico area (Tuscany, Central Italy) and their association with symptom severity in Vitis vinifera L. cv. Sangiovese. Phytopathology 2018, 108, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Pierro, R.; Passera, A.; Panattoni, A.; Rizzo, D.; Stefani, L.; Bartolini, L.; Casati, P.; Luvisi, A.; Quaglino, F.; Materazzi, A. Prevalence of a ‘Candidatus Phytoplasma solani’ strain, so far associated only with other hosts, in Bois Noir-affected grapevines within Tuscan vineyards. Ann. Appl. Biol. 2018, 173, 202–212. [Google Scholar] [CrossRef]
- Naser, S.M.; Vancanneyt, M.; Hoste, B.; Snauwaert, C.; Swings, J. Lactobacillus cypricasei Lawson et al. 2001 is a later heterotypic synonym of Lactobacillus acidipiscis Tanasupawat et al. 2000. Int. J. Syst. Evol. Microbiol. 2006, 56, 1681–1683. [Google Scholar] [CrossRef][Green Version]
- Mignard, S.; Flandrois, J.P. A seven-gene, multilocus, genus-wide approach to the phylogeny of mycobacteria using supertrees. Int. J. Syst. Evol. Microbiol. 2008, 58, 1432–1441. [Google Scholar] [CrossRef]
- Pascual, J.; Macián, M.C.; Arahal, D.R.; Garay, E.; Pujalte, M.J. Multilocus sequence analysis of the central clade of the genus Vibrio by using the 16S rRNA, recA, pyrH, rpoD, gyrB, rctB and toxR genes. Int. J. Syst. Evol. Microbiol. 2010, 60, 154–165. [Google Scholar] [CrossRef]
- Adkar-Purushothama, C.R.; Quaglino, F.; Casati, P.; Bianco, P.A. Molecular typing of Coorg black pepper yellows phytoplasma by multiple gene analysis. Ann. Appl. Biol. 2011, 159, 58–68. [Google Scholar] [CrossRef]
- Malembic-Maher, S.; Salar, P.; Filippin, L.; Carle, P.; Angelini, E.; Foissac, X. Genetic diversity of European phytoplasmas of the 16SrV taxonomic group and proposal of ‘Candidatus Phytoplasma rubi’. Int. J. Syst. Evol. Microbiol. 2011, 61, 2129–2134. [Google Scholar] [CrossRef]
- Lee, I.M.; Bottner-Parker, K.D.; Zhao, Y.; Bertaccini, A.; Davis, R.E. Differentiation and classification of phytoplasmas in the pigeon pea witches’-broom group (16SrIX): An update based on multiple gene sequence analysis. Int. J. Syst. Evol. Microbiol. 2012, 62, 2279–2285. [Google Scholar] [CrossRef]
- Durante, G.; Casati, P.; Clair, D.; Quaglino, F.; Bulgari, D.; Boudon-Padieu, E.; Bianco, P.A. Sequence analyses of S10-spc operon among 16SrV group phytoplasmas: Phylogenetic relationships and identification of discriminating single nucleotide polymorphisms. Ann. Appl. Biol. 2012, 161, 234–246. [Google Scholar] [CrossRef]
- Davis, R.E.; Zhao, Y.; Dally, E.L.; Lee, I.M.; Jomantiene, R.; Douglas, S.M. ‘Candidatus Phytoplasma pruni’, a novel taxon associated with X-disease of stone fruits, Prunus spp.: Multilocus characterization based on 16S rRNA, secY, and ribosomal protein genes. Int. J. Syst. Evol. Microbiol. 2013, 63, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Casati, P.; Quaglino, F.; Stern, A.R.; Tedeschi, R.; Alma, A.; Bianco, P.A. Multiple gene analyses reveal extensive genetic diversity among ‘Candidatus Phytoplasma mali’ populations. Ann. Appl. Biol. 2011, 158, 257–266. [Google Scholar] [CrossRef]
- Lee, I.-M.; Gundersen, D.E.; Hammond, R.W.; Davis, R.E. Use of mycoplasmalike organism (MLO) group-specific oligonucleotide primers for nested-PCR assays to detect mixed-MLO infections in a single host plant. Phytopathology 1994, 84, 559–566. [Google Scholar] [CrossRef]
- Oliveri, C.; Pacifico, D.; D’Urso, V.; La Rosa, R.; Marzachi, C.; Tessitori, M. Bois noir phytoplasma variability in a Mediterranean vineyard system: New plant host and putative vectors. Australas. Plant Pathol. 2015, 44, 235–244. [Google Scholar] [CrossRef]
- Kostadinovska, E.; Quaglino, F.; Mitrev, S.; Casati, P.; Bulgari, D.; Bianco, P.A. Multiple gene analyses identify distinct “bois noir” phytoplasma genotypes in the Republic of Macedonia. Phytopathol. Mediterr. 2014, 53, 491–501. [Google Scholar]
- Mori, N.; Pavan, F.; Reggiani, N.; Bacchiavini, M.; Mazzon, L.; Paltrinieri, S.; Bertaccini, A. Correlation of bois noir disease with nettle and vector abundance in northern Italy vineyards. J. Pest Sci. 2012, 85, 23–28. [Google Scholar] [CrossRef]
- Murolo, S.; Garbarino, M.; Mancini, V.; Romanazzi, G. Spatial pattern of Bois noir: Case study of a delicate balance between disease progression and recovery. Sci. Rep. 2020, 10, 9801. [Google Scholar] [CrossRef]
- Berg, K.A.; Dunlop, J.; Sanchez, T.; Silva, M.; Clarke, W.P. A conservative, single-amino acid substitution in the second cytoplasmic domain of the human serotonin2C receptor alters both ligand-dependent and -independent receptor signalling. J. Pharm. Exp. Therap. 2008, 324, 1084–1092. [Google Scholar] [CrossRef]
- Bruner, A.C.; Jung, S.; Abbott, A.G.; Powell, G.L. The naturally occurring high oleate oil character in some peanut varieties results from reduced oleoyl-PC desaturase activity from mutation of aspartate 150 to asparagine. Crop Sci. 2001, 4, 522–526. [Google Scholar] [CrossRef]
- Kawachi, T.; Sunaga, Y.; Ebato, M.; Hatanaka, T.; Harada, H. Repression of nitrate uptake by replacement of Asp105 by asparagine in AtNTR3.1 in Arabidopsis thaliana L. Plant Cell Physiol. 2006, 47, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Kavaliauskas, D.; Nissen, P.; Knudsen, C.R. The busiest of all ribosomal assistants: Elongation factor Tu. Biochemistry 2012, 51, 2642–2651. [Google Scholar] [CrossRef] [PubMed]
- Kunert, A.; Losse, J.; Gruszin, C.; Hiihn, M.; Kraendler, K.; Mikkat, S.; Volke, D.; Hoffmann, R.; Jokiranta, T.S.; Seeberger, H.; et al. Immune evasion of the human pathogen Pseudomonas aeruginosa: Elongation factor Tuf is a factor H and plasminogen binding protein. J. Immunol. 2007, 179, 2979–2988. [Google Scholar] [CrossRef] [PubMed]
- Archambaud, C.; Gouin, E.; Pizarro-Cerda, J.; Cossart, P.; Dussurget, O. Translation elongation factor EF-Tu is a target for Stp, a serine-threonine phosphatase involved in virulence of Listeria monocytogenes. Mol. Microbiol. 2005, 56, 383–396. [Google Scholar] [CrossRef]
- Aryan, A.; Brader, G.; Mörtel, J.; Pastar, M.; Riedle-Bauer, M. An abundant ‘Candidatus Phytoplasma solani’ tuf b strain is associated with grapevine, stinging nettle and Hyalesthes obsoletus. Eur. J. Plant Pathol. 2014, 140, 213–227. [Google Scholar] [CrossRef]
- Jamshidi, E.; Murolo, S.; Salehi, M.; Romanazzi, G. Sequence analysis of new tuf molecular types of ‘Candidatus Phytoplasma solani’ in Iranian vineyards. Pathogens 2020, 9, 508. [Google Scholar] [CrossRef]
- Goebel, W.; Chakraborty, T.; Kreft, J. Bacterial hemolysins as virulence factors. Ant. Van Leeuw. 1998, 54, 453–463. [Google Scholar] [CrossRef]
- Gambetta, G.A.; Matthews, M.A.; Syvanen, M. The Xylella fastidosa RTX operons: Evidence for the evolution of protein mosaics through novel genetic exchanges. BMC Genom. 2018, 19, 329. [Google Scholar] [CrossRef]
- Quaglino, F.; Casati, P.; Bianco, P.A. Distinct rpsC single nucleotide polymorphism lineages of Flavescence dorée subgroup 16SrV-D phytoplasma co-infect Vitis vinifera L. Folia Microbiol. 2010, 55, 251–257. [Google Scholar] [CrossRef]
- Mitrovic, J.; Kakizawa, S.; Duduk, B.; Oshima, K.; Namba, S.; Bertaccini, A. The groEL gene as an additional marker for finer differentiation of ‘Candidatus Phytoplasma asteris’-related strains. Ann. Appl. Biol. 2011, 159, 41–48. [Google Scholar] [CrossRef]
- Casati, P.; Jermini, M.; Quaglino, F.; Corbani, G.; Schaerer, S.; Passera, A.; Bianco, P.A.; Rigamonti, I.E. New insights on Flavescence dorée phytoplasma ecology in the vineyard agro-ecosystem in southern Switzerland. Ann. Appl. Biol. 2017, 171, 37–51. [Google Scholar] [CrossRef]
- Angelini, E.; Clair, D.; Borgo, M.; Bertaccini, A.; Boudon-Padieu, E. Flavescence dorée in France and Italy: Occurrence of closely related phytoplasma isolates and their near relationships to palatinate grapevine yellows and an alder yellows phytoplasma. Vitis 2001, 40, 79–86. [Google Scholar]
- Deng, S.; Hiruki, C. Genetic relatedness between two nonculturable mycoplasmalike organisms revealed by nucleic acid hybridization and polymerase chain reaction. Phytopathology 1991, 81, 1475–1479. [Google Scholar] [CrossRef]
- Quaglino, F.; Zhao, Y.; Bianco, P.A.; Wei, W.; Casati, P.; Durante, G.; Davis, R.E. New 16Sr subgroups and distinct single nucleotide polymorphism lineages among grapevine Bois noir phytoplasma populations. Ann. Appl. Biol. 2009, 154, 279–289. [Google Scholar] [CrossRef]
- Mitrović, J.; Siewert, C.; Duduk, B.; Hecht, J.; Mölling, K.; Broeker, F.; Beyerlein, P.; Büttner, C.; Bertaccini, A.; Kube, M. Generation and analysis of draft sequences of ‘stolbur’ phytoplasma from multiple displacement amplification templates. J. Mol. Microbiol. Biotechnol. 2014, 24, 1–11. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]





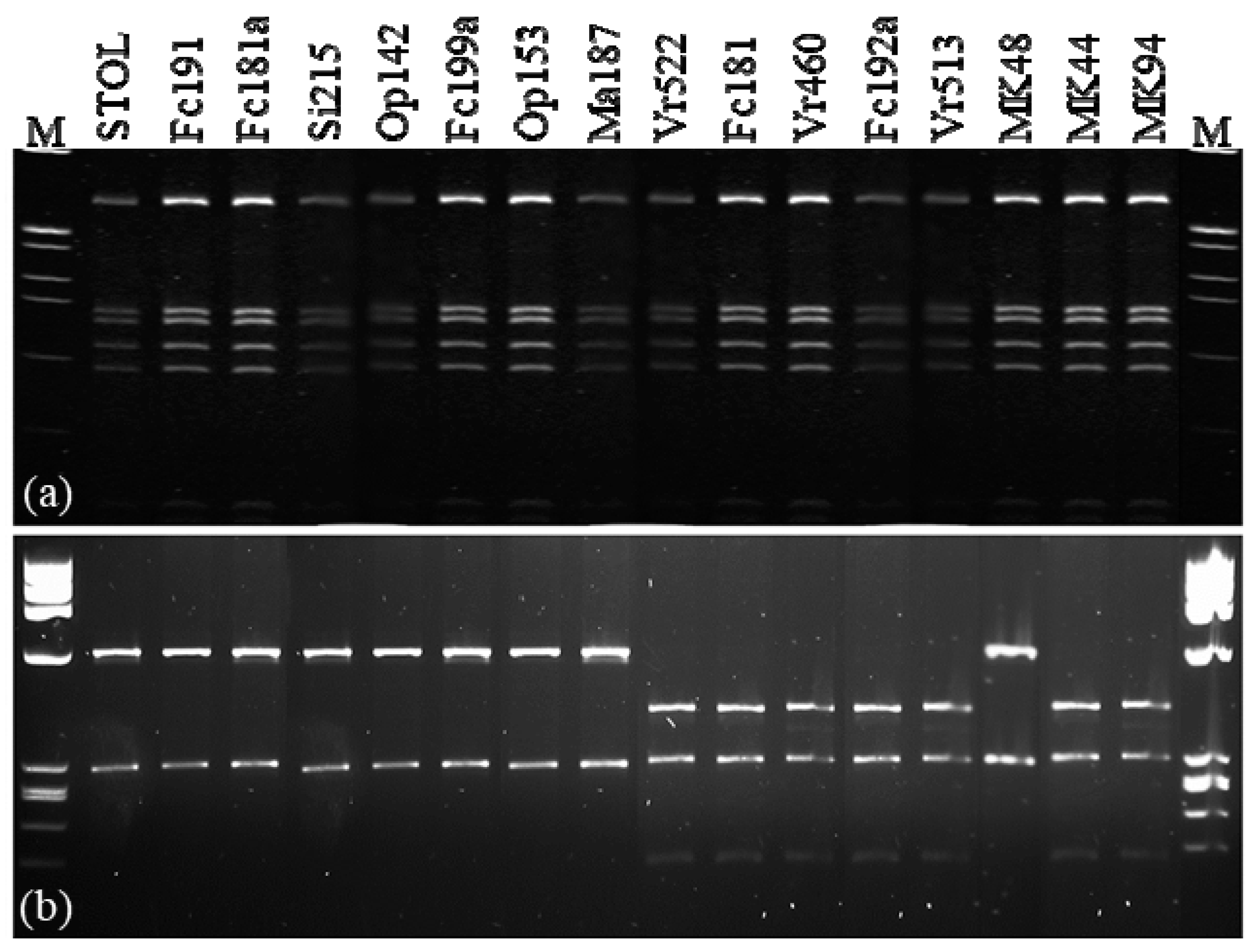{kind=link}
{kind=link}
{kind=link}
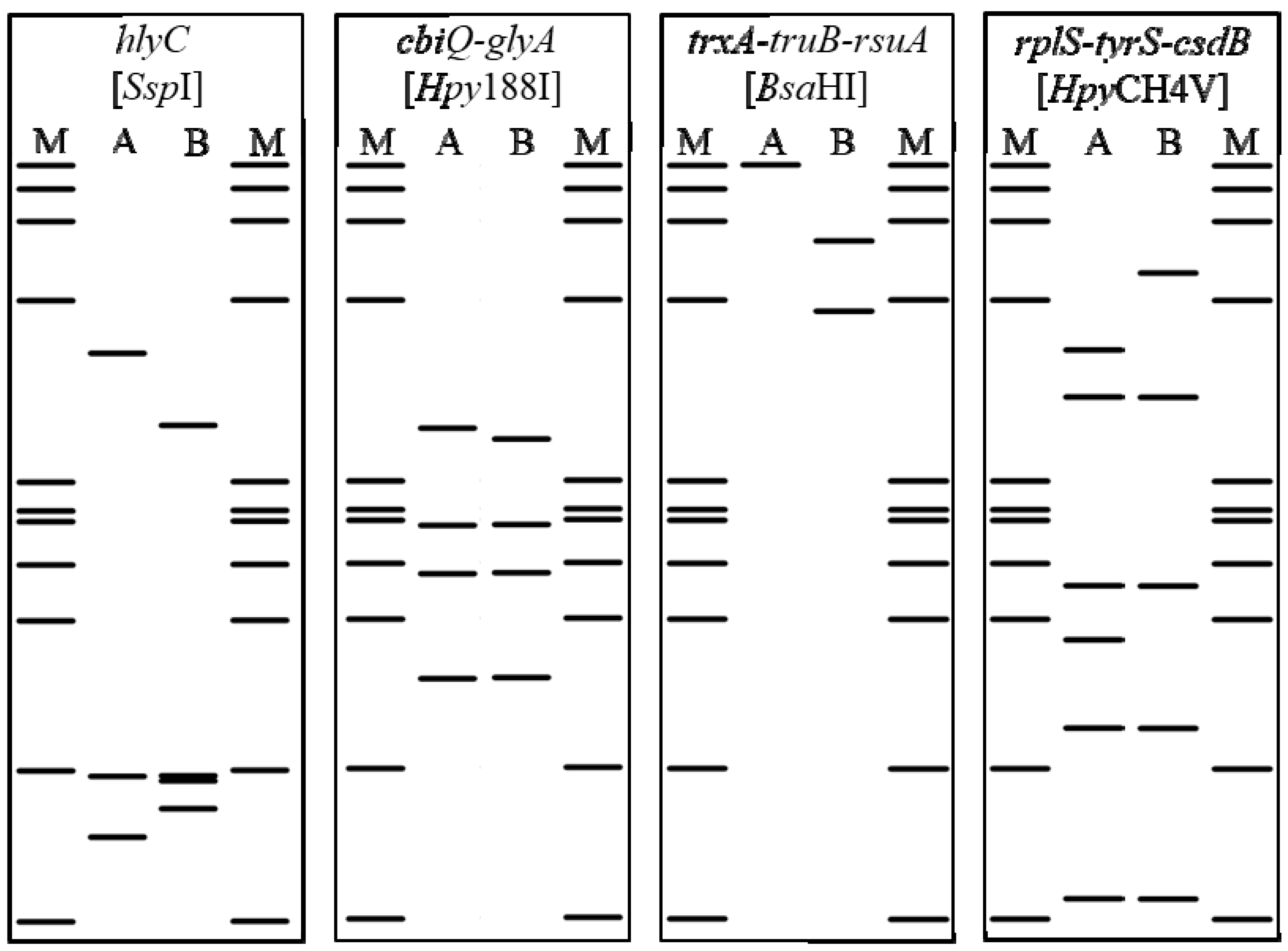{kind=link}
| Country | Region | No. of Samples | tuf-Type a | tuf-Type b | No. of CaPsol Strains in Each Lineage | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | |||||
| Italy (North) | Lombardy | 46 | 27 | 19 | 11 | 4 | 1 | 9 | 3 | 2 | 10 | 1 | 1 | 2 | 1 | 1 | |||
| Veneto | 39 | 36 | 3 | 13 | 2 | 11 | 1 | 2 | 10 | ||||||||||
| Italy (Center) | Marche | 16 | 2 | 14 | 2 | 14 | |||||||||||||
| Tuscany | 16 | 0 | 16 | 6 | 10 | ||||||||||||||
| Italy (South) | Apulia | 6 | 0 | 6 | 6 | ||||||||||||||
| Sicily | 3 | 0 | 3 | 3 | |||||||||||||||
| Macedonia (North) | 16 | 2 | 14 | 2 | 6 | 3 | 1 | 1 | 2 | 1 | |||||||||
| Overall | 142 | 67 | 75 | 24 | 6 | 1 | 20 | 11 | 18 | 15 | 1 | 1 | 14 | 12 | 15 | 1 | 2 | 1 | |
| Origin | CaPsol Strain | Sequence Variant/RFLP Profile | CaPsol | ||||
|---|---|---|---|---|---|---|---|
| fusA-tufB | hlyC | cbiQ-glyA | trxA-truB-rsuA | rplS-tyrS-csdB | Lineage | ||
| Lombardy | Fc180 | A (tuf-type a) | A | A | A | A | 1 |
| (North Italy) | Fc181 | A | A | A | B | A | 2 |
| Fc182 | A | A | A | A | A | 1 | |
| Fc183 | A | A | A | B | A | 2 | |
| Fc185 | A | A | A | A | A | 1 | |
| Fc191 | B (tuf-type b) | B | A | A | B | 3 | |
| Fc181a | B | B | B | B | A | 5 | |
| Fc192a | A | A | B | B | B | 8 | |
| Fc199a | B | B | B | A | B | 9 | |
| Op142 | B | B | A | B | B | 7 | |
| Op153 | B | B | B | B | B | 11 | |
| Op280 | A | A | B | B | A | 4 | |
| Op282 | A | A | B | B | A | 4 | |
| Op304 | A | A | B | B | A | 4 | |
| Veneto | Vr460 | A | A | B | B | A | 4 |
| (North Italy) | Vr462 | B | B | A | B | A | 6 |
| Vr501 | B | B | A | B | B | 7 | |
| Vr509 | B | B | A | B | B | 7 | |
| Vr513 | A | A | B | A | A | 10 | |
| Vr517 | A | A | B | A | A | 10 | |
| Vr522 | A | A | A | A | A | 1 | |
| Marche | Ma187 | B | B | B | A | A | 12 |
| (Center Italy) | Ma189 | B | B | B | A | A | 12 |
| Ma190 | A | A | B | A | A | 10 | |
| Ma191 | A | A | B | A | A | 10 | |
| Tuscany | San11/15 | B | B | B | B | B | 11 |
| (Center Italy) | San15/15 | B | B | B | B | B | 11 |
| San17/15 | B | B | B | B | B | 11 | |
| San5/16 | B | B | B | B | B | 11 | |
| Apulia | Pu123 | B | B | A | B | A | 6 |
| (South Italy) | Pu124 | B | B | A | B | A | 6 |
| Pu125 | B | B | A | B | A | 6 | |
| Sicily | Si212 | B | B | A | B | A | 6 |
| (South Italy) | Si214 | B | B | A | B | A | 6 |
| Si215 | B | B | A | B | A | 6 | |
| North | MK44 | A | A | A | A | B | 13 |
| Macedonia | MK48 | B | B | A | A | A | 14 |
| MK61 | B | B | A | B | A | 6 | |
| MK62 | B | B | A | B | A | 6 | |
| MK94 | A | A | A | B | B | 15 | |
| Gene | Primer | Sequence (5’–3’) | PCR | Nested PCR |
|---|---|---|---|---|
| Product Size (nt) | ||||
| fusA-tufB | fusAF1 | CTTTCTGARATGTTTGGMTATGCTAC | d | 1399 |
| fusAF2 | GCGTTCCAATACYCAAGGAAGAG | n | ||
| tufBR1 | ACAAAGCTCCAACGTTATCGCCTGC | d/n | ||
| cbiQ-glyA | cbiQF1 | AGAGGTTATGTATTGGGAGCG | d/n | 1024 |
| glyAR1 | CAAAGAACTTGCAAGAGTTTGGGC | d | ||
| glyAR2 | TGTTGATAATCTTTAAAGGCAGG | n | ||
| rplS-tyrS-csdB | rplSF1 | CCTGTGCACTCCCCTAATAACGA | d | 1529 |
| csdBR1 | ACCTTCTTGGAGTGTTTCGCCTAGAC | d | ||
| rplSF2 | CGTCGTGCTAAGTCGCATTACG | n | ||
| csdBR2 | GTTTCAAAGAGGTAGCCGCATTATCG | n | ||
| trxA-truB-rsuA | trxAF1 | TGCCAATTGGTGTGGTCCATGTC | d | 1363 |
| truBR3 | GCCTCTATGATCAAATCAAGGACAG | d | ||
| trxAF2 | GAATTATCACAATCAGAACAGGGTG | n | ||
| truBR4 | TCTTTGGCGGTCGAAAGGTAGCC | n | ||
| hlyC | hlysF1 | ATKATTVTGAAATGKBCTAC | d | 914 |
| ackAR2 | GAAATTTTAAAGAAGARCTAC | d | ||
| hlysF2 | ATKATTVTGAAATGKBCTACYAAAMMAAC | n | ||
| ackAR3 | AGAAGARCTACCWGAATTWACCGAC | n | ||
| potD | potF1 | ACGATTAATCCAACTGTTAATGC | d | 953 |
| potR1 | TACTTGGATAAGCAATGATGTC | d | ||
| potF2 | AGGGTGAATATTTAGACCCTCAAAC | n | ||
| potR2 | TGGATAAGCAATGATGTCATTCC | n | ||
| pnp | pnpF1 | TGCTAGAAATGTGGATGCTTCTG | d | 1448 |
| pnpR1 | TGACATTTCTTGGCGTGGAGTG | d | ||
| pnpF2 | GATACAGTAGTTTTATCGGCTAC | n | ||
| pnpR2 | GTAATACCATCTTTGCTGCCAGC | n | ||
| gyrA-gyrB | gyrAF2 | TGGGCTTCTTTGATGTCTGCTG | d | 1705 |
| gyrBR2 | TGACCGATGCTGACGTTGATGGTG | d | ||
| gyrAF3 | TCTAATTGCAGTATCGATGTC | n | ||
| gyrBR3 | TGCTGACGTTGATGGTGCTCAC | n | ||
| aspS-mesJ | aspSF1 | GTAGTTGAGATCAAGGGGTTAGTTG | d | 1333 |
| mesJR1 | TGAATCAACGCCGCCGCTAACAG | d | ||
| aspSF2 | TCGCAGCCAAGATAGTCTTGAAG | n | ||
| mesJR2 | CTTTTCGACTGTTCCGGGGGAATC | n |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passera, A.; Zhao, Y.; Murolo, S.; Pierro, R.; Arsov, E.; Mori, N.; Moussa, A.; Silletti, M.R.; Casati, P.; Panattoni, A.; et al. Multilocus Genotyping Reveals New Molecular Markers for Differentiating Distinct Genetic Lineages among “Candidatus Phytoplasma Solani” Strains Associated with Grapevine Bois Noir. Pathogens 2020, 9, 970. https://doi.org/10.3390/pathogens9110970
Passera A, Zhao Y, Murolo S, Pierro R, Arsov E, Mori N, Moussa A, Silletti MR, Casati P, Panattoni A, et al. Multilocus Genotyping Reveals New Molecular Markers for Differentiating Distinct Genetic Lineages among “Candidatus Phytoplasma Solani” Strains Associated with Grapevine Bois Noir. Pathogens. 2020; 9(11):970. https://doi.org/10.3390/pathogens9110970
Chicago/Turabian StylePassera, Alessandro, Yan Zhao, Sergio Murolo, Roberto Pierro, Emilija Arsov, Nicola Mori, Abdelhameed Moussa, Maria R. Silletti, Paola Casati, Alessandra Panattoni, and et al. 2020. "Multilocus Genotyping Reveals New Molecular Markers for Differentiating Distinct Genetic Lineages among “Candidatus Phytoplasma Solani” Strains Associated with Grapevine Bois Noir" Pathogens 9, no. 11: 970. https://doi.org/10.3390/pathogens9110970
APA StylePassera, A., Zhao, Y., Murolo, S., Pierro, R., Arsov, E., Mori, N., Moussa, A., Silletti, M. R., Casati, P., Panattoni, A., Wei, W., Mitrev, S., Materazzi, A., Luvisi, A., Romanazzi, G., Bianco, P. A., Davis, R. E., & Quaglino, F. (2020). Multilocus Genotyping Reveals New Molecular Markers for Differentiating Distinct Genetic Lineages among “Candidatus Phytoplasma Solani” Strains Associated with Grapevine Bois Noir. Pathogens, 9(11), 970. https://doi.org/10.3390/pathogens9110970