Efficient Confirmation of Plant Viral Proteins and Identification of Specific Viral Strains by nanoLC-ESI-Q-TOF Using Single-Leaf-Tissue Samples

 , ,
, ,  , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
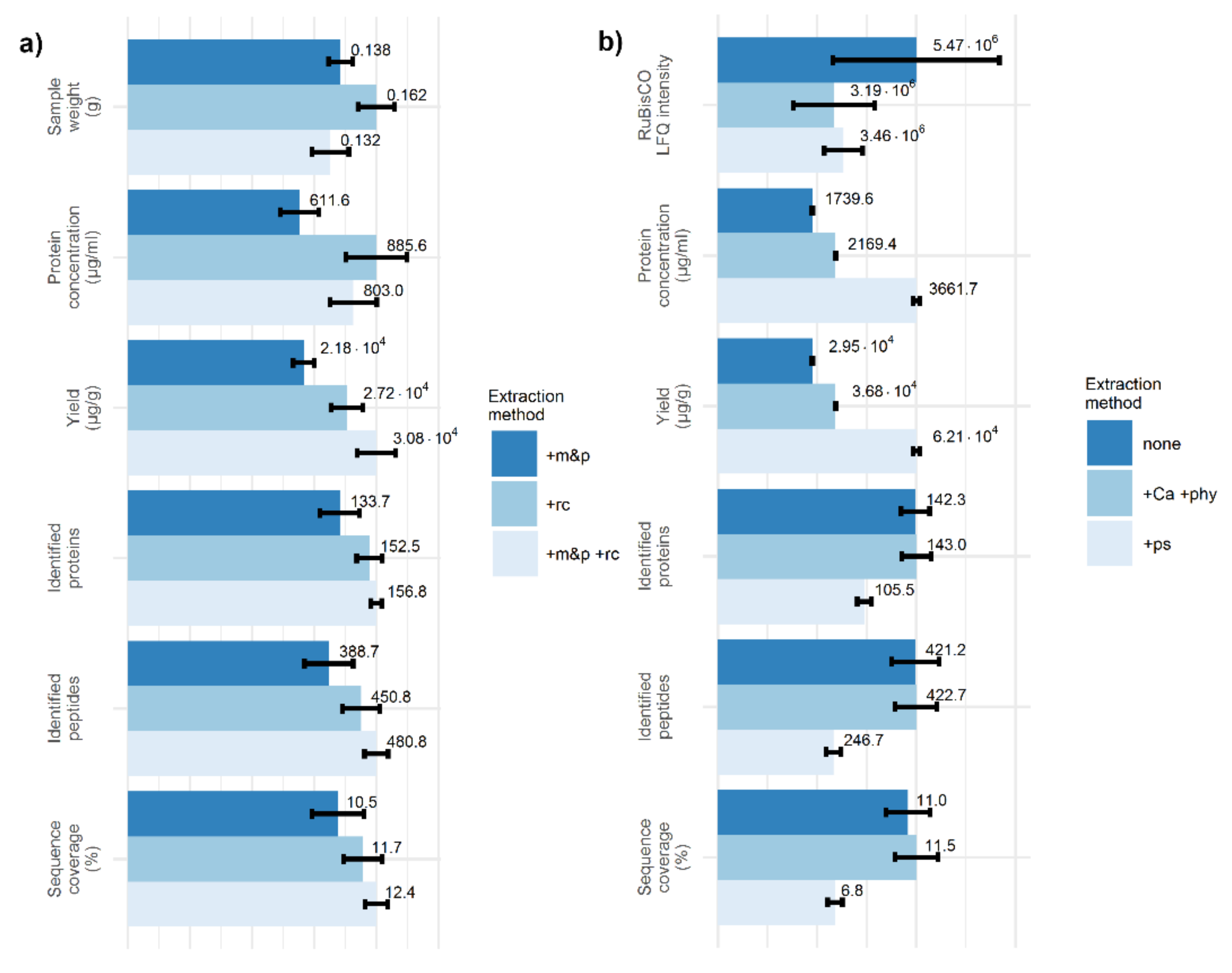
2.1. Two Crushing Steps Improve Identification of Plant Proteins
2.2. Plant Virus Pathogen Capsid Proteins Could Be Efficiently Confirmed in Samples of Plants with Strong Infection Using the Double Crushing Extraction Protocol Followed by nanoLC-ESI-Q-TOF
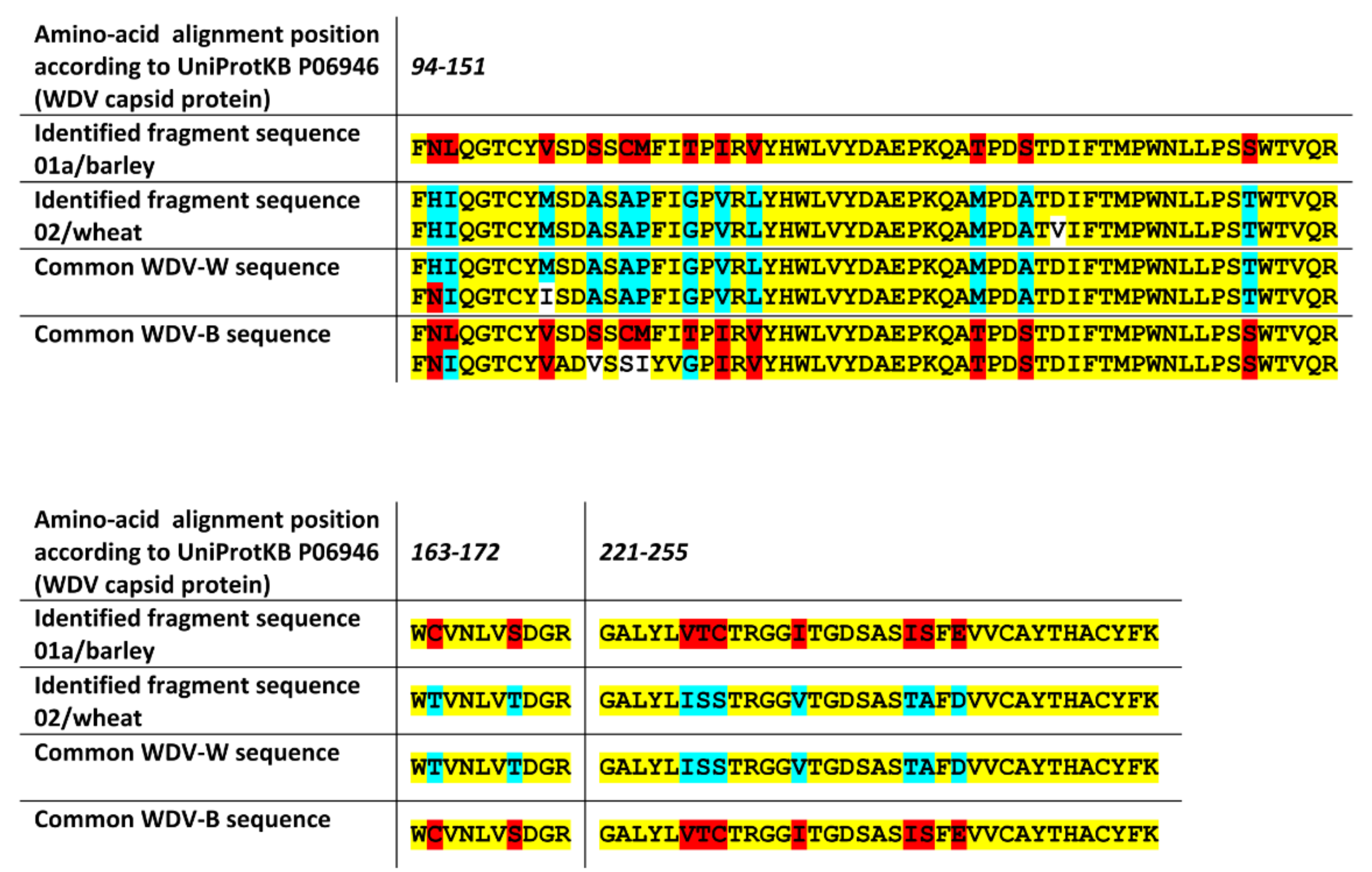
2.3. The nanoLC-ESI-Q-TOF Based Detection Method with Optimized Extraction Protocol Allows Discrimination of Virus Strains or Distinct Disease Species Based on Detected Fragments
3. Discussion
4. Materials and Methods
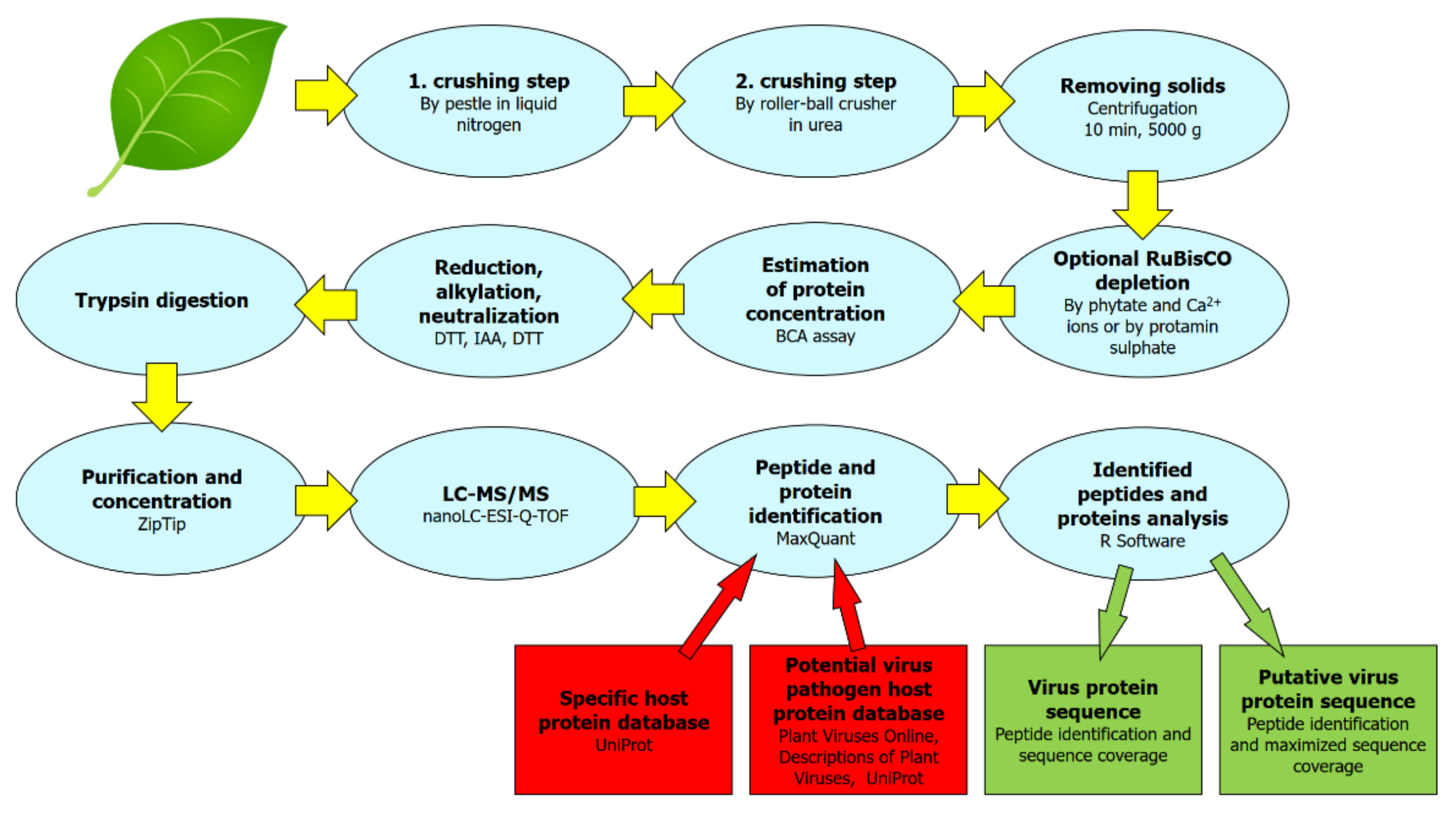
4.1. Protein Sample Extraction and Preparation Protocol
4.2. LC-MS/MS
4.3. Protein Identification and Protein Databases Searched
4.4. Virus Protein Sequence Coverage, Virus Protein Putative Sequence
4.5. Identification of Strain-Specific Sequences
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vanderschuren, H.; Stupak, M.; Futterer, J.; Gruissem, W.; Zhang, P. Engineering resistance to geminiviruses—Review and perspectives. Plant. Biotechnol. J. 2007, 5, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Oerke, E.C. Crop losses to pests. J. Agric. Sci. 2006, 144, 31–43. [Google Scholar] [CrossRef]
- Savary, S.; Ficke, A.; Aubertot, J.N.; Hollier, C. Crop losses due to diseases and their implications for global food production losses and food security. Food Secur. 2012, 4, 519–537. [Google Scholar] [CrossRef]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. G 2016, 17, 95–115. [Google Scholar] [CrossRef] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Kuckova, S.; Cejnar, P.; Santrucek, J.; Hynek, R. Characterization of proteins in cultural heritage using MALDI–TOF and LC–MS/MS mass spectrometric techniques. Phys. Sci. Rev. 2018, 4. [Google Scholar] [CrossRef]
- Chen, Y.; Vu, J.; Thompson, M.G.; Sharpless, W.A.; Chan, L.J.G.; Gin, J.W.; Keasling, J.D.; Adams, P.D.; Petzold, C.J. A rapid methods development workflow for high-throughput quantitative proteomic applications. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Wenger, C.D.; Coon, J.J. A Proteomics Search Algorithm Specifically Designed for High-Resolution Tandem Mass Spectra. J. Proteome Res. 2013, 12, 1377–1386. [Google Scholar] [CrossRef]
- Tu, C.J.; Sheng, Q.H.; Li, J.; Ma, D.J.; Shen, X.M.; Wang, X.; Shyr, Y.; Yi, Z.P.; Qu, J. Optimization of Search Engines and Postprocessing Approaches to Maximize Peptide and Protein Identification for High-Resolution Mass Data. J. Proteome Res. 2015, 14, 4662–4673. [Google Scholar] [CrossRef] [Green Version]
- Beck, S.; Michalski, A.; Raether, O.; Lubeck, M.; Kaspar, S.; Goedecke, N.; Baessmann, C.; Hornburg, D.; Meier, F.; Paron, I.; et al. The Impact II, a Very High-Resolution Quadrupole Time-of-Flight Instrument (QTOF) for Deep Shotgun Proteomics. Mol. Cell Proteomics 2015, 14, 2014–2029. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.J.; Bakhtiar, R.; Siuzdak, G. Mass spectrometry in viral proteomics. Acc. Chem. Res. 2000, 33, 179–187. [Google Scholar] [PubMed]
- Trauger, S.A.; Junker, T.; Siuzdak, G. Investigating viral proteins and intact viruses with mass spectrometry. Top. Curr. Chem. 2003, 225, 265–282. [Google Scholar] [CrossRef]
- Blouin, A.G.; Greenwood, D.R.; Chavan, R.R.; Pearson, M.N.; Clover, G.R.G.; MacDiarmid, R.M.; Cohen, D. A generic method to identify plant viruses by high-resolution tandem mass spectrometry of their coat proteins. J. Virol. Methods 2010, 163, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.; Eckert, D.; Andon, N.L.; Yates, J.R.; Haynes, P.A. Investigative proteomics: Identification of an unknown plant virus from infected plants using mass spectrometry. J. Am. Soc. Mass Spectr. 2003, 14, 736–741. [Google Scholar] [CrossRef] [Green Version]
- Di Carli, M.; Benvenuto, E.; Donini, M. Recent Insights into Plant-Virus Interactions through Proteomic Analysis. J. Proteome Res. 2012, 11, 4765–4780. [Google Scholar] [CrossRef]
- Fang, X.P.; Chen, J.P.; Dai, L.Y.; Ma, H.S.; Zhang, H.M.; Yang, J.; Wang, F.; Yan, C.Q. Proteomic dissection of plant responses to various pathogens. Proteomics 2015, 15, 1525–1543. [Google Scholar] [CrossRef]
- Jorrin-Novo, J.V.; Pascual, J.; Sanchez-Lucas, R.; Romero-Rodriguez, M.C.; Rodriguez-Ortega, M.J.; Lenz, C.; Valledor, L. Fourteen years of plant proteomics reflected in Proteomics: Moving from model species and 2DE-based approaches to orphan species and gel-free platforms. Proteomics 2015, 15, 1089–1112. [Google Scholar] [CrossRef]
- Crowell, A.M.J.; Wall, M.J.; Doucette, A.A. Maximizing recovery of water-soluble proteins through acetone precipitation. Anal. Chim. Acta 2013, 796, 48–54. [Google Scholar] [CrossRef]
- Feist, P.; Hummon, A.B. Proteomic Challenges: Sample Preparation Techniques for Microgram-Quantity Protein Analysis from Biological Samples. Int J. Mol. Sci. 2015, 16, 3537–3563. [Google Scholar] [CrossRef] [Green Version]
- Bodzon-Kulakowska, A.; Bierczynska-Krzysik, A.; Dylag, T.; Drabik, A.; Suder, P.; Noga, M.; Jarzebinska, J.; Silberring, J. Methods for samples preparation in proteomic research. J. Chromatogr. B 2007, 849, 1–31. [Google Scholar] [CrossRef]
- Burgess, R.R. Protein Precipitation Techniques. Methods Enzymol. 2009, 463, 331–342. [Google Scholar] [CrossRef]
- Gundry, R.L.; White, M.Y.; Murray, C.I.; Kane, L.A.; Fu, Q.; Stanley, B.A.; Van Eyk, J.E. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr. Protoc. Mol. Biol. 2009, 90, 10–25. [Google Scholar] [CrossRef] [Green Version]
- Suttapitugsakul, S.; Xiao, H.P.; Smeekens, J.; Wu, R.H. Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS- based proteomics. Mol. Biosyst. 2017, 13, 2574–2582. [Google Scholar] [CrossRef]
- Muller, T.; Winter, D. Systematic Evaluation of Protein Reduction and Alkylation Reveals Massive Unspecific Side Effects by Iodine-containing Reagents. Mol. Cell Proteomics 2017, 16, 1173–1187. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.S.; Zhou, J.Y.; Yang, W.M.; Zhang, H. Inhibition of protein carbamylation in urea solution using ammonium-containing buffers. Anal. Biochem. 2014, 446, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Wang, Y.M.; Agrawal, G.K.; Rakwal, R.; Jo, I.H.; Bang, K.H.; Kim, S.T. Time to dig deep into the plant proteome: A hunt for low-abundance proteins. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Lee, H.M.; Wang, Y.M.; Wu, J.; Kim, S.G.; Kang, K.Y.; Park, K.H.; Kim, Y.C.; Choi, I.S.; Agrawal, G.K.; et al. Depletion of abundant plant RuBisCO protein using the protamine sulfate precipitation method. Proteomics 2013, 13, 2176–2179. [Google Scholar] [CrossRef]
- Krishnan, H.B.; Natarajan, S.S. A rapid method for depletion of Rubisco from soybean (Glycine max) leaf for proteomic analysis of lower abundance proteins. Phytochemistry 2009, 70, 1958–1964. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell Proteomics 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Burger, T. Gentle Introduction to the Statistical Foundations of False Discovery Rate in Quantitative Proteomics. J. Proteome Res. 2018, 17, 12–22. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics 2010, 73, 2092–2123. [Google Scholar] [CrossRef] [Green Version]
- Plant Viruses Online. Plant Viruses Online: Descriptions and Lists from the VIDE Database. Version: 20th August 1996. Available online: http://bio-mirror.im.ac.cn/mirrors/pvo/vide/refs.htm (accessed on 3 January 2020).
- Adams, M.J.; Antoniw, J.F. DPVweb: A comprehensive database of plant and fungal virus genes and genomes. Nucleic Acids Res. 2006, 34, D382–D385. [Google Scholar] [CrossRef] [Green Version]
- Descriptions of Plant Viruses. Available online: http://www.dpvweb.net/dpv/dpvnameidx.php (accessed on 3 January 2020).
- Nesvizhskii, A.I. Protein identification by tandem mass spectrometry and sequence database searching. Methods Mol. Biol. 2007, 367, 87–119. [Google Scholar] [CrossRef]
- Tabb, D.L.; Friedman, D.B.; Ham, A.J.L. Verification of automated peptide identifications from proteomic tandem mass spectra. Nat. Protoc. 2006, 1, 2213–2222. [Google Scholar] [CrossRef] [Green Version]
- Kourelis, J.; Kaschani, F.; Grosse-Holz, F.M.; Homma, F.; Kaiser, M.; van der Hoorn, R.A.L. A homology-guided, genome-based proteome for improved proteomics in the alloploid Nicotiana benthamiana. Bmc Genomics 2019, 20. [Google Scholar] [CrossRef]
- Kundu, J.K.; Jarosova, J.; Gadiou, S.; Cervena, G. Discrimination of Three BYDV Species by One-step RT-PCR-RFLP and Sequence Based Methods in Cereal Plants from the Czech Republic. Cereal Res. Commun. 2009, 37, 541–550. [Google Scholar] [CrossRef]
- Mann, M.; Ong, S.E.; Gronborg, M.; Steen, H.; Jensen, O.N.; Pandey, A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 2002, 20, 261–268. [Google Scholar] [CrossRef]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A.; et al. Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509. [Google Scholar] [CrossRef]
- Tiwary, S.; Levy, R.; Gutenbrunner, P.; Soto, F.S.; Palaniappan, K.K.; Deming, L.; Berndl, M.; Brant, A.; Cimermancic, P.; Cox, J. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 2019, 16, 519. [Google Scholar] [CrossRef]
- CABI (CAB International). Invasive Species Compendium. Available online: https://www.cabi.org/isc (accessed on 1 July 2020).
- APS (The American Phytopathological Society). Common Names of Plant Diseases. Available online: https://www.apsnet.org/edcenter/resources/commonnames/Pages/default.aspx (accessed on 1 July 2020).
- ICTV (International Committee on Taxonomy of Viruses). Virus Taxonomy: 2019 Release. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 1 July 2020).
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.R. Target-Decoy Search Strategy for Mass Spectrometry-Based Proteomics. Methods Mol. Biol. 2010, 604, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Alpi, E.; Bely, B.; Bingley, M.; Britto, R.; Bursteinas, B.; Busiello, G.; et al. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005. [Google Scholar]
- NCBI (National Center for Biotechnology Information). Bethesda (MD): National Library of Medicine (US). Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 3 January 2020).
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Lisa, V.; Boccardo, G. Fabaviruses: Broad Bean Wilt and Allied Viruses. In The Plant Viruses, Volume 5: Polyhedral Virion and Bipartite RNA Genomes; Harrison, B.D., Murant, A.F., Eds.; Plenum Press: New York, NY, USA, 1996; pp. 229–250. [Google Scholar]
- Edwardson, J.R.; Christie, R.G. CRC Handbook of Viruses Infecting Legumes; CRC Press: Boca Raton, FL, USA, 1991; p. 504. [Google Scholar]
- Vaughan, R.; Tragesser, B.; Ni, P.; Ma, X.; Dragnea, B.; Kao, C.C. The Tripartite Virions of the Brome Mosaic Virus Have Distinct Physical Properties That Affect the Timing of the Infection Process. J. Virol. 2014, 88, 6483–6491. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.S.; Mannas, S.W.; Bishop, B.A.; Rao, X.L.; Lecoultre, M.; Kwon, S.; Nelson, R.S. An Improved Brome mosaic virus Silencing Vector: Greater Insert Stability and More Extensive VIGS. Plant. Physiol. 2018, 176, 496–510. [Google Scholar] [CrossRef] [Green Version]
- Hodge, B.A.; Salgado, J.D.; Paul, P.A.; Stewart, L.R. Characterization of an Ohio Isolate of Brome Mosaic Virus and Its Impact on the Development and Yield of Soft Red Winter Wheat. Plant. Dis. 2019, 103, 1101–1111. [Google Scholar] [CrossRef]
- Rastgou, M.; Khatabi, B.; Kvarnheden, A.; Izadpanah, K. Relationships of Barley yellow dwarf virus-PAV and Cereal yellow dwarf virus-RPV from Iran with viruses of the family Luteoviridae. Eur. J. Plant Pathol. 2005, 113, 321–326. [Google Scholar] [CrossRef]
- D’Arcy, C.J.; Domier, L.L. Luteoviridae. In Virus Taxonomy-Eighth Report of the ICTV; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Springer-Verlag: New York, NY, USA, 2005; pp. 891–900. [Google Scholar]
- D’Arcy, C.J. Symptomology and host range of barley yellow dwarf. In Barley Yellow Dwarf: 40 Years of Progress; D’Arcy, C.J., Burnett, P.A., Eds.; American Phytopathological Society: St. Paul, MN, USA, 1995; pp. 9–28. [Google Scholar]
- Haas, M.; Bureau, M.; Geldreich, A.; Yot, P.; Keller, M. Cauliflower mosaic virus: Still in the news. Mol. Plant Pathol. 2002, 3, 419–429. [Google Scholar] [CrossRef]
- Bak, A.; Gargani, D.; Macia, J.L.; Malouvet, E.; Vernerey, M.S.; Blanc, S.; Drucker, M. Virus Factories of Cauliflower Mosaic Virus Are Virion Reservoirs That Engage Actively in Vector Transmission. J. Virol. 2013, 87, 12207–12215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitfield, A.E.; Falk, B.W.; Rotenberg, D. Insect vector-mediated transmission of plant viruses. Virology 2015, 479, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.A.; Glasa, M.; Cambra, M.; Candresse, T. Plum pox virus and sharka: A model potyvirus and a major disease. Mol. Plant. Pathol. 2014, 15, 226–241. [Google Scholar] [CrossRef] [PubMed]
- Gan, D.F.; Zhang, J.A.; Jiang, H.B.; Jiang, T.; Zhu, S.W.; Cheng, B.J. Bacterially expressed dsRNA protects maize against SCMV infection. Plant Cell Rep. 2010, 29, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Pennerman, K.K.; Wang, H.X.; Yin, G.H. Characterization of a Sorghum mosaic virus (SrMV) isolate in China. Saudi J. Biol. Sci. 2016, 23, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Verma, N.; Kumar, K.; Kulshrestha, S.; Raikhy, G.; Hallan, V.; Ram, R.; Zaidi, A.A.; Garg, I.D. Molecular studies on Tomato aspermy virus isolates infecting chrysanthemums. Arch. Phytopathol. Plant Prot. 2009, 42, 99–111. [Google Scholar] [CrossRef]
- Zaitlin, M. The Discovery of the Causal Agent of the Tobacco Mosaic Disease. In Discoveries in Plant Biology; Kung, S.D., Yang, S.F., Eds.; World Publishing Co.: Hong Kong, China, 1998; pp. 105–110. [Google Scholar]
- Kundu, J.K.; Gadiou, S.; Schlesingerova, G.; Dziakova, M.; Cermak, V. Emergence of Quarantine Tobacco ringspot virus in Impatiens walleriana in the Czech Republic. Plant Prot. Sci. 2015, 51, 115–122. [Google Scholar] [CrossRef] [Green Version]
- EPPO (European and Mediterranean Plant Protection Organization). EPPO A2 List of pests recommended for regulation as quarantine pests—Version 2019-09. Available online: https://www.eppo.int/ACTIVITIES/plant_quarantine/A2_list (accessed on 1 July 2020).
- Walsh, J.A.; Jenner, C.E. Turnip mosaic virus and the quest for durable resistance. Mol. Plant Pathol. 2002, 3, 289–300. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Tomitaka, Y.; Ho, S.Y.W.; Duchene, S.; Vetten, H.J.; Lesemann, D.; Walsh, J.A.; Gibbs, A.J.; Ohshima, K. Turnip Mosaic Potyvirus Probably First Spread to Eurasian Brassica Crops from Wild Orchids about 1000 Years Ago. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Melcher, U. Turnip vein-clearing virus, from pathogen to host expression profile. Mol. Plant Pathol. 2003, 4, 133–140. [Google Scholar] [CrossRef]
- Lindblad, M.; Waern, P. Correlation of wheat dwarf incidence to winter wheat cultivation practices. Agric. Ecosyst. Environ. 2002, 92, 115–122. [Google Scholar] [CrossRef]
- Sirlová, L.; Vacke, J.; Chaloupová, M. Reaction of selected winter wheat varieties to autumnal infection with Wheat dwarf virus. Plant. Prot. Sci. 2005, 41, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lindsten, K.; Lindsten, B.; Abdelmoeti, M.; Junti, N. Purification and some properties of wheat dwarf virus. In Proceedings of the 3rd Conference on Virus Diseases of Gramineae in Europe, Rothamsted, UK, 28–30 May 1980; pp. 27–34. [Google Scholar]
- Lindsten, K.; Vacke, J. A possible barley adapted strain of wheat dwarf virus (WDV). Acta Phytopathol. Entomol. Hung. 1991, 26, 175–180. [Google Scholar]
- Muhire, B.; Martin, D.P.; Brown, J.K.; Navas-Castillo, J.; Moriones, E.; Zerbini, F.M.; Rivera-Bustamante, R.; Malathi, V.G.; Briddon, R.W.; Varsani, A. A genome-wide pairwise-identity-based proposal for the classification of viruses in the genus Mastrevirus (family Geminiviridae). Arch. Virol. 2013, 158, 1411–1424. [Google Scholar] [CrossRef] [Green Version]
- Svoboda, J.; Leisova-Svobodova, L. First Report of Broad bean wilt virus-2 in Pepper in the Czech Republic. Plant. Dis. 2013, 97, 1261. [Google Scholar] [CrossRef]
- Glasa, M.; Palkovics, L.; Kominek, P.; Kabonne, G.; Pittnerova, S.; Kudela, O.; Candresse, T.; Subr, Z. Geographically and temporally distant natural recombinant isolates of Plum pox virus (PPV) are genetically very similar and form a unique PPV subgroup. J. Gen. Virol. 2004, 85, 2671–2681. [Google Scholar] [CrossRef]
- Viktorova, J.; Klcova, B.; Rehorova, K.; Vlcko, T.; Stankova, L.; Jelenova, N.; Cejnar, P.; Kundu, J.K.; Ohnoutkova, L.; Macek, T. Recombinant expression of osmotin in barley improves stress resistance and food safety during adverse growing conditions. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.F.; Adams, A.N. Characteristics of the microplate method of enzyme-linked immunosorbent assay for the detection of plant viruses. J. Gen. Virol. 1977, 34, 475–483. [Google Scholar] [CrossRef]
- Predajna, L.; Subr, Z.; Candresse, T.; Glasa, M. Evaluation of the genetic diversity of Plum pox virus in a single plum tree. Virus Res. 2012, 167, 112–117. [Google Scholar] [CrossRef]
- Ferrer, R.M.; Ferriol, I.; Moreno, P.; Guerri, J.; Rubio, L. Genetic variation and evolutionary analysis of broad bean wilt virus 2. Arch. Virol. 2011, 156, 1445–1450. [Google Scholar] [CrossRef]
- Gadiou, S.; Ripl, J.; Janourova, B.; Jarosova, J.; Kundu, J.K. Real-time PCR assay for the discrimination and quantification of wheat and barley strains of Wheat dwarf virus. Virus Genes 2012, 44, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Subr, Z.; Pittnerova, S.; Glasa, M. A simplified RT-PCR-based detection of recombinant Plum pox virus isolates. Acta Virol. 2004, 48, 173–176. [Google Scholar] [PubMed]
- Virus Collection CRI. Virus Collection of the Crop Research Institute. Available online: http://www.vurv.cz/collections/vurv.exe/search?org=VI (accessed on 3 January 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | 01a/barley *1 | 01b/barley | 01c/barley | 02/wheat *1 | 03a/Chinese cabbage | 03b/Chinese cabbage | 04a/tobacco | 04b/tobacco | 04c/tobacco | 04d/tobacco | 04e/tobacco | 05/apricot | 06a/plum | 06b/plum | 07/bean | 08/goosefoot | 09/sorghum | 10/maize | |
| PREPARATION | Sample weight (g) | 0.14 | 0.34 | 0.46 | 0.34 | 0.52 | 0.47 | 0.47 | 0.46 | 0.45 | 0.55 | 0.63 | 0.60 | 0.60 | 0.97 | 0.48 | 0.63 | 0.32 | 0.13 |
| Host taxonomic classification | Hordeum vulgare | Hordeum vulgare | Hordeum vulgare | Triticum aestivum | Brassica rapa subsp. pekinensis | Brassica rapa subsp. pekinensis | Nicotiana benthamiana | Nicotiana benthamiana | Nicotiana benthamiana | Nicotiana clevelandia x N. glutinosa | Nicotiana tabacum | Prunus armeniaca | Prunus domestica | Prunus Pdomestica | Phaseolus vulgaris | Chenopodium amaranticolor | Sorghum bicolor | Zea mays | |
| UniProt reference proteome used | UP000011116 | UP000019116 | UP000011750 | UP000084051 *2 | *3 | UP000000226 | *4 | UP000000768 | UP000007305 | ||||||||||
| Reference genome proteins | 189,799 | 130,673 | 40,809 | 73,605 + 53,411 + 74,802 *2 | 155,017 *3 | 30,501 | 1063 *4 | 41,380 | 99,254 | ||||||||||
| Known host viruses in online databases | 66 | 63 | 29 | 369 | 17 | 227 | 378 | 22 | 69 | ||||||||||
| Tested viruses for sample (UniProt protein sequences available) | 47 | 45 | 24 | 232 | 13 | 151 | 232 | 18 | 52 | ||||||||||
| RESULTS | Identified peptides in sample | 3233 | 2871 | 3963 | 3888 | 1844 | 1094 | 3176 | 2772 | 3412 | 3426 | 1270 | 2344 | 1648 | 1047 | 4364 | 346 | 1942 | 1956 |
| Identified host proteins * | 886 | 810 | 1082 | 1076 | 537 | 339 | 756 | 668 | 865 | 865 | 287 | 593 | 435 | 362 | 1163 | 71 | 570 | 549 | |
| Identified virus | Wheat dwarf virus (WDV) | Barley yellow dwarf virus (BYDV) | Brome mosaic virus (BMV) | Wheat dwarf virus (WDV) | Cauliflower mosaic virus (CaMV) | Turnip vein clearing virus (TVCV) | Plum pox virus (PPV) | Turnip mosaic virus (TuMV) | Tobacco ringspot virus (TRSV) | Tomato aspermy virus (TAV) | Tobacco mosaic virus (TMV) | Plum pox virus (PPV) | Plum pox virus (PPV) | Plum pox virus (PPV) | Bean common mosaic virus (BCMV) | Broad Bean Wilt Virus 2 (BBWV-2) | Sorghum mosaic virus (SrMV) | Sugarcane mosaic virus (SCMV) | |
| Identified virus family | Geminiviridae (DNA) | Luteoviridae (RNA) | Bromoviridae (RNA) | Geminiviridae (DNA) | Caulimoviridae (DNA) | Virgaviridae (RNA) | Potyviridae (RNA) | Potyviridae (RNA) | Secoviridae (RNA) | Bromoviridae (RNA) | Virgaviridae (RNA) | Potyviridae (RNA) | Potyviridae (RNA) | Potyviridae (RNA) | Potyviridae (RNA) | Secoviridae (RNA) | Potyviridae (RNA) | Potyviridae (RNA) | |
| Identified capsid protein peptides ** | 10 | 2 | 11 | 10 | 10 | 6 | 11 | 14 | 8 | 12 | 12 | 6 | 6 | 5 | 4 | 17 | 9 | 4 | |
| Capsid protein sequence coverage (AA) ** | 147 of 260 | 36 of 199 | 147 of 189 | 147 of 260 | 110 of 489 | 82 of 157 | 119 of 330 | 154 of 286 | 104 of 513 | 134 of 218 | 92 of 159 | 86 of 330 | 65 of 330 | 57 of 330 | 71 of 287 | 247 of 599 | 95 of 320 | 59 of 313 | |
| (56.5%) | (18.1%) | (77.8%) | (56.5%) | (22.5%) | (52.2%) | (36.1%) | (53.8%) | (20.3%) | (61.5%) | (57.9%) | (26.0%) | (19.7%) | (17.2%) | (24.7%) | (41.2%) | (29.7%) | (18.8%) | ||
| Capsid protein sequence coverage on ≥ 7 amino acid sequences (AA) ** | 147 of 189 | 36 of 148 | 147 of 152 | 147 of 189 | 110 of 333 | 82 of 142 | 119 of 278 | 154 of 210 | 104 of 437 | 134 of 187 | 92 of 148 | 86 of 278 | 65 of 278 | 57 of 278 | 71 of 232 | 247 of 506 | 95 of 211 | 59 of 247 | |
| (77.8%) | (24.3%) | (96.7%) | (77.8%) | (33.0%) | (57.7%) | (42.8%) | (73.3%) | (23.8%) | (71.7%) | (62.2%) | (30.1%) | (23.4%) | (20.5%) | (30.6%) | (48.8%) | (45.0%) | (23.9%) | ||
| Other viral proteins identified ** | − | 2 fragments of movement protein | − | − | more than 10 fragments of other viral proteins - movement protein, reverse transcriptase, aphid transmission protein, etc. | − | other more than 10 fragments of genome polyprotein | other more than 10 fragments of genome polyprotein | − | − | more than 10 fragments of replication protein | other more than 10 fragments of genome polyprotein | other 6 fragments of genome polyprotein | − | other more than 10 fragments of genome polyprotein | one other fragment of genome polyprotein from RNA 1 component | other more than 10 fragments of genome polyprotein | other more than 10 fragments of genome polyprotein | |
| Sequence covered (aa) ** | 147 | 87 | 147 | 147 | 794 | 82 | 936 | 695 | 104 | 134 | 405 | 291 | 126 | 57 | 380 | 275 | 486 | 343 | |
| Putative sequence covered (aa) *** | 147 | 87 | 147 | 147 | 807 | 82 | 1073 | 734 | 104 | 142 | 405 | 291 | 126 | 57 | 407 | 327 | 522 | 398 | |
| Virus presence also confirmed by **** | DAS-ELISA qPCR | DAS-ELISA RT-PCR RFLP | DAS-ELISA | DAS-ELISA qPCR | DAS-ELISA | DAS-ELISA | DAS-ELISA qPCR | DAS-ELISA qPCR | DAS-ELISA | DAS-ELISA | DAS-ELISA | DAS-ELISA qPCR | DAS-ELISA qPCR | DAS-ELISA qPCR | DAS-ELISA | electron microscopy RT-PCR | DAS-ELISA | DAS-ELISA | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cejnar, P.; Kučková, Š.; Šantrůček, J.; Glasa, M.; Komínek, P.; Mihálik, D.; Slavíková, L.; Leišová-Svobodová, L.; Smirnova, T.; Hynek, R.; et al. Efficient Confirmation of Plant Viral Proteins and Identification of Specific Viral Strains by nanoLC-ESI-Q-TOF Using Single-Leaf-Tissue Samples. Pathogens 2020, 9, 966. https://doi.org/10.3390/pathogens9110966
Cejnar P, Kučková Š, Šantrůček J, Glasa M, Komínek P, Mihálik D, Slavíková L, Leišová-Svobodová L, Smirnova T, Hynek R, et al. Efficient Confirmation of Plant Viral Proteins and Identification of Specific Viral Strains by nanoLC-ESI-Q-TOF Using Single-Leaf-Tissue Samples. Pathogens. 2020; 9(11):966. https://doi.org/10.3390/pathogens9110966
Chicago/Turabian StyleCejnar, Pavel, Štěpánka Kučková, Jiří Šantrůček, Miroslav Glasa, Petr Komínek, Daniel Mihálik, Lucie Slavíková, Leona Leišová-Svobodová, Tatiana Smirnova, Radovan Hynek, and et al. 2020. "Efficient Confirmation of Plant Viral Proteins and Identification of Specific Viral Strains by nanoLC-ESI-Q-TOF Using Single-Leaf-Tissue Samples" Pathogens 9, no. 11: 966. https://doi.org/10.3390/pathogens9110966
APA StyleCejnar, P., Kučková, Š., Šantrůček, J., Glasa, M., Komínek, P., Mihálik, D., Slavíková, L., Leišová-Svobodová, L., Smirnova, T., Hynek, R., Kundu, J. K., & Ryšánek, P. (2020). Efficient Confirmation of Plant Viral Proteins and Identification of Specific Viral Strains by nanoLC-ESI-Q-TOF Using Single-Leaf-Tissue Samples. Pathogens, 9(11), 966. https://doi.org/10.3390/pathogens9110966