Taxonomy and Phylogenetic Research on Ralstonia solanacearum Species Complex: A Complex Pathogen with Extraordinary Economic Consequences


Abstract
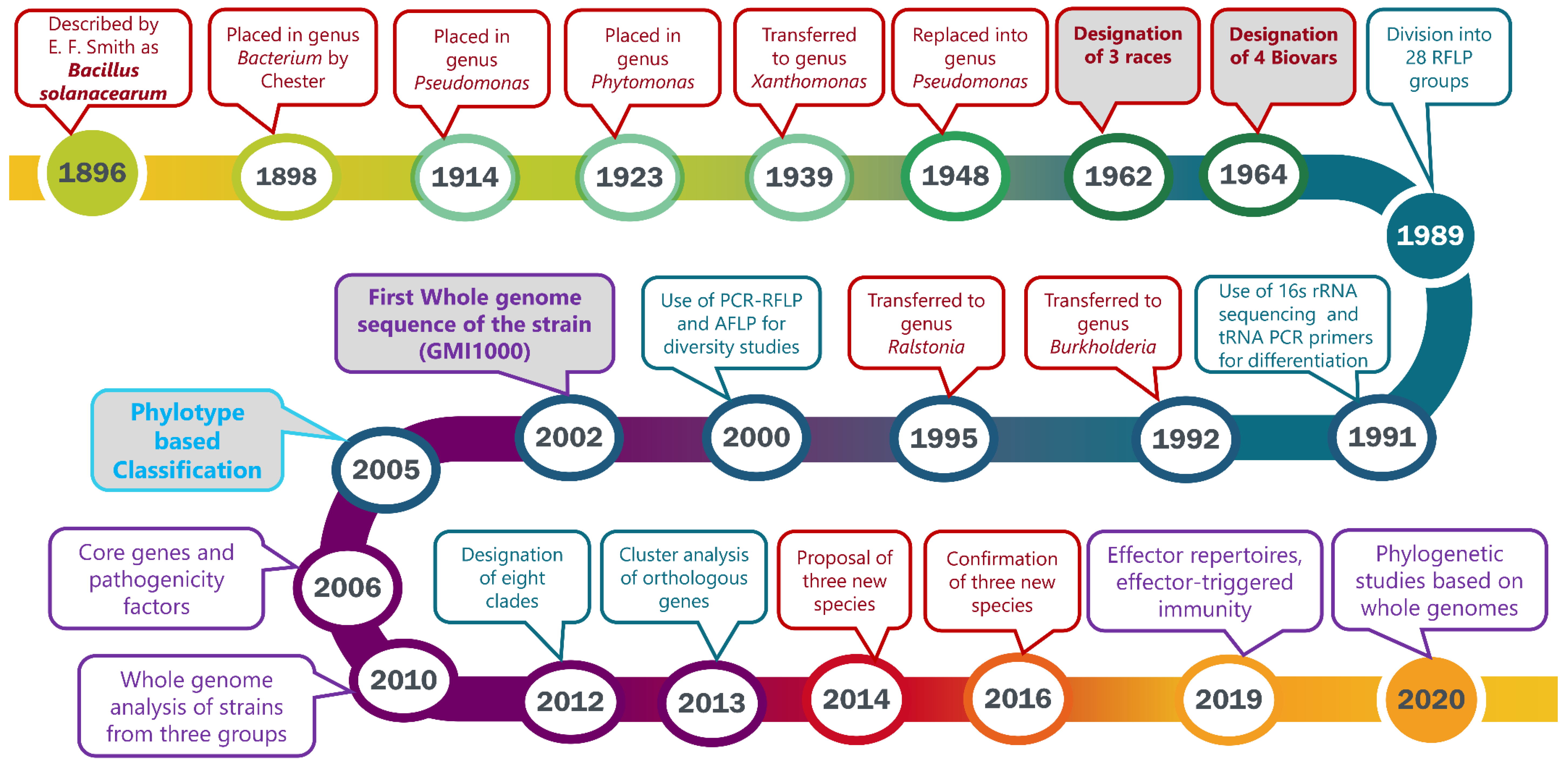
1. Introduction
2. 1896–1963: The Discovery and Early Classification
3. 1964: Sub-Classification of Pseudomonas solanacearum into Races and Biovars
4. 1989: Classification into Two Divisions Based on RFLP Patterns
5. General Taxonomic Revision of the Genus Pseudomonas
6. 1992: Determination of Pseudomonas solanacearum Subgroups Using PCR Amplification and t-RNA Consensus Primers
7. 1992: Transfer of Pseudomonas Homology Group II into the New Genus Burkholderia
8. 1995: Transfer of Burkholderia solanacearum into the New Genus Ralstonia
9. 1994–1996: Diversity Studies of the Pseudomonas/Burkholderia/Ralstonia solanacearum Species Complex
10. 2000: Identification of the African Sub-Division
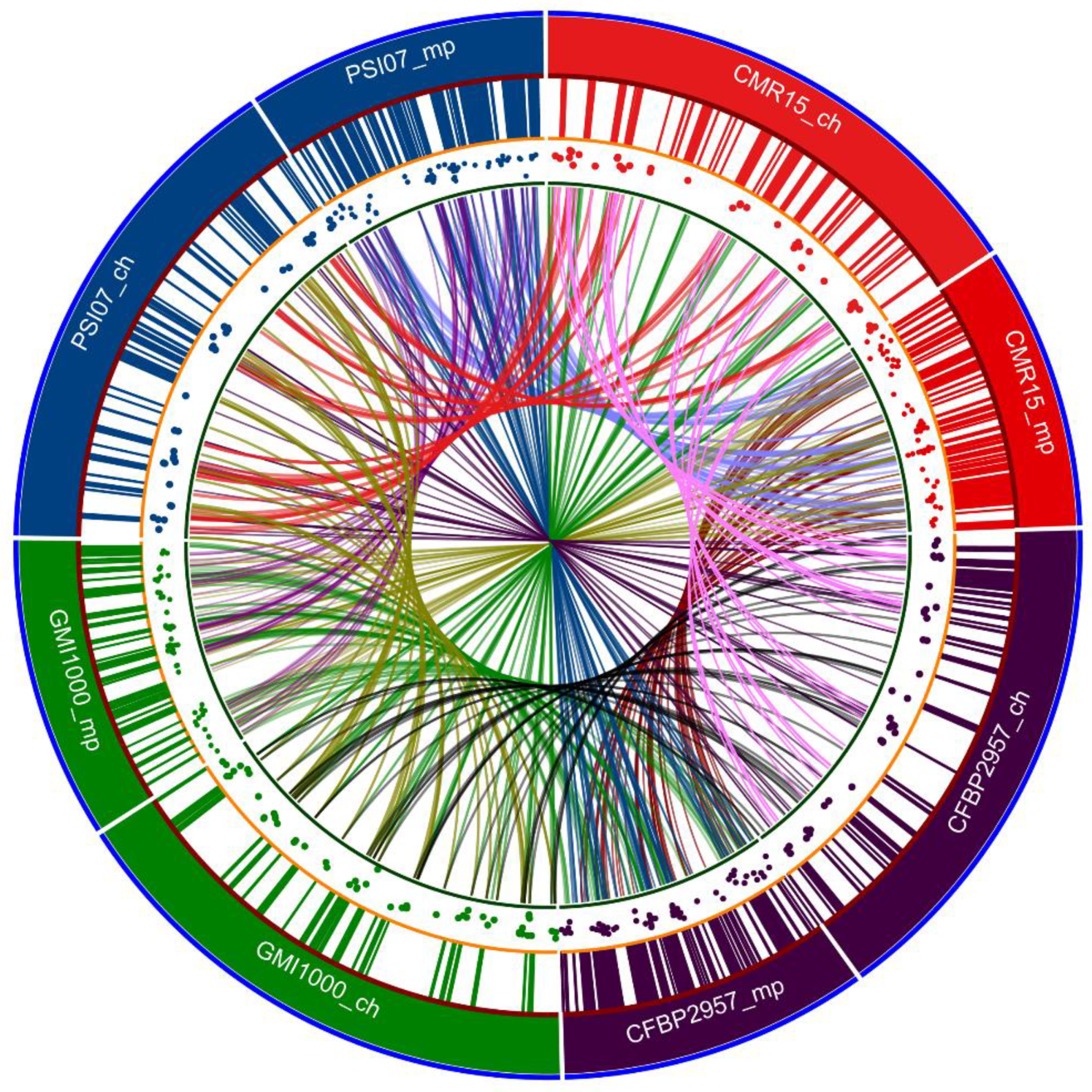
11. 2002: First Whole Genome Sequence of the Reference Strain GMI1000: General Structure of the Chromosome and Megaplasmid
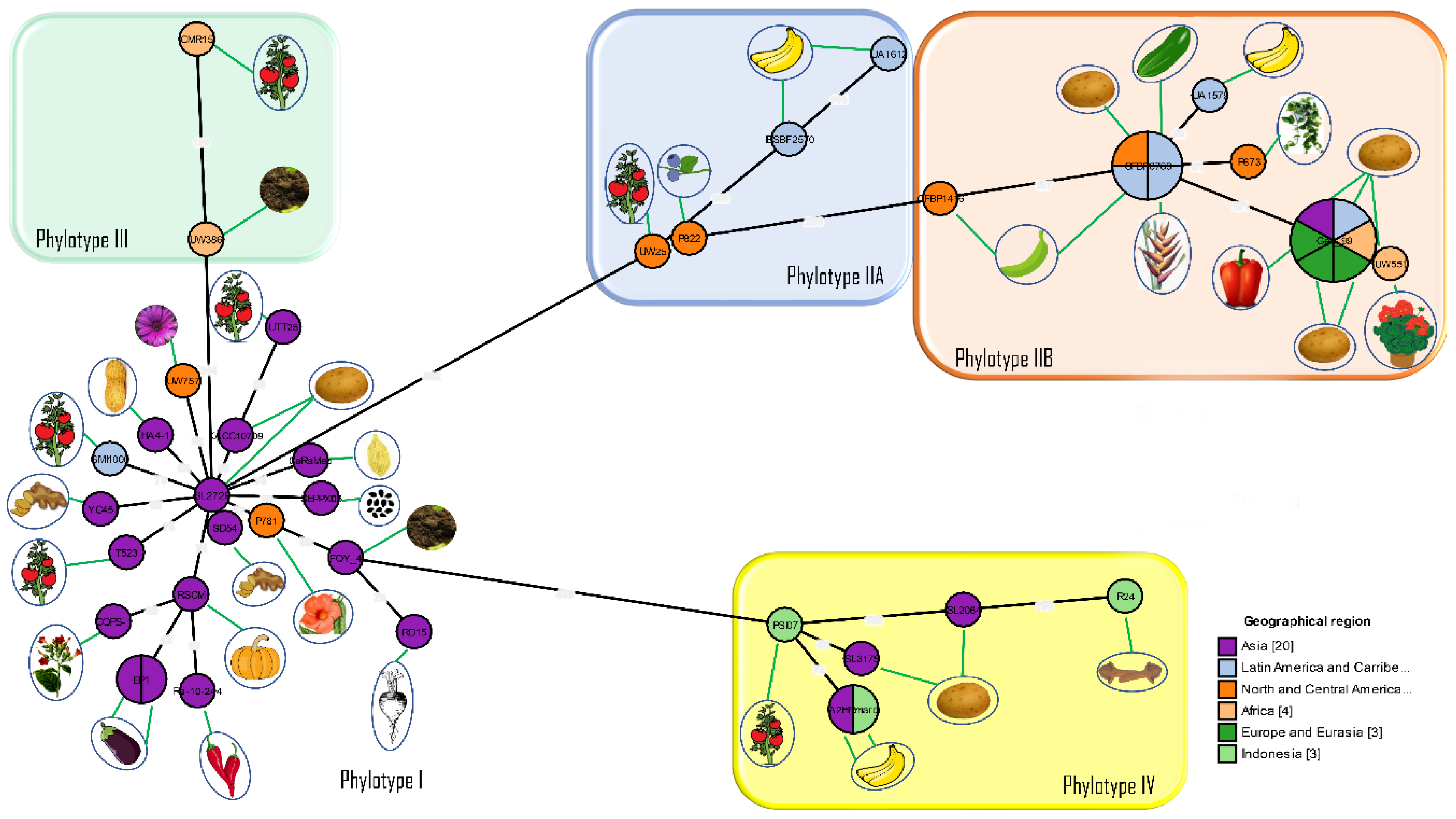
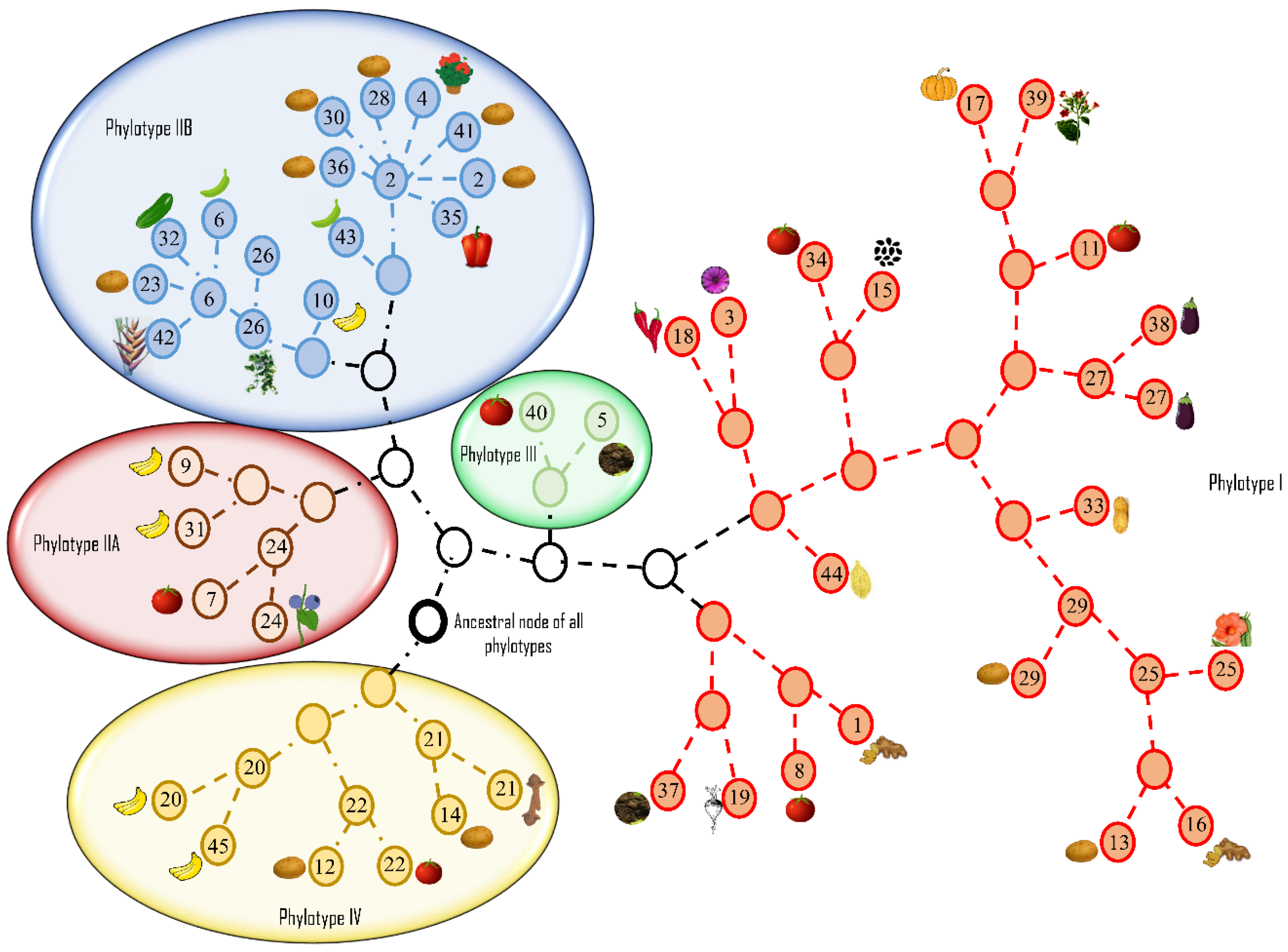
12. 2005: Introduction of a Phylotype-Based Classification of the Ralstonia solanacearum Species Complex
13. 2006–2007: Core Genes and, Pathogenicity Determinants
14. 2010: Whole-Genome Analyses Further Highlight the Genomic Diversity across the Different Phylotypes
15. 2012: Evolutionary History and Contrasting Recombination Patterns among Phylotypes
16. Pathogenicity Functions Elucidated through Genomic Studies
17. 2013: Gene Gain and Loss Contributing to Adaption and Bacterial Fitness
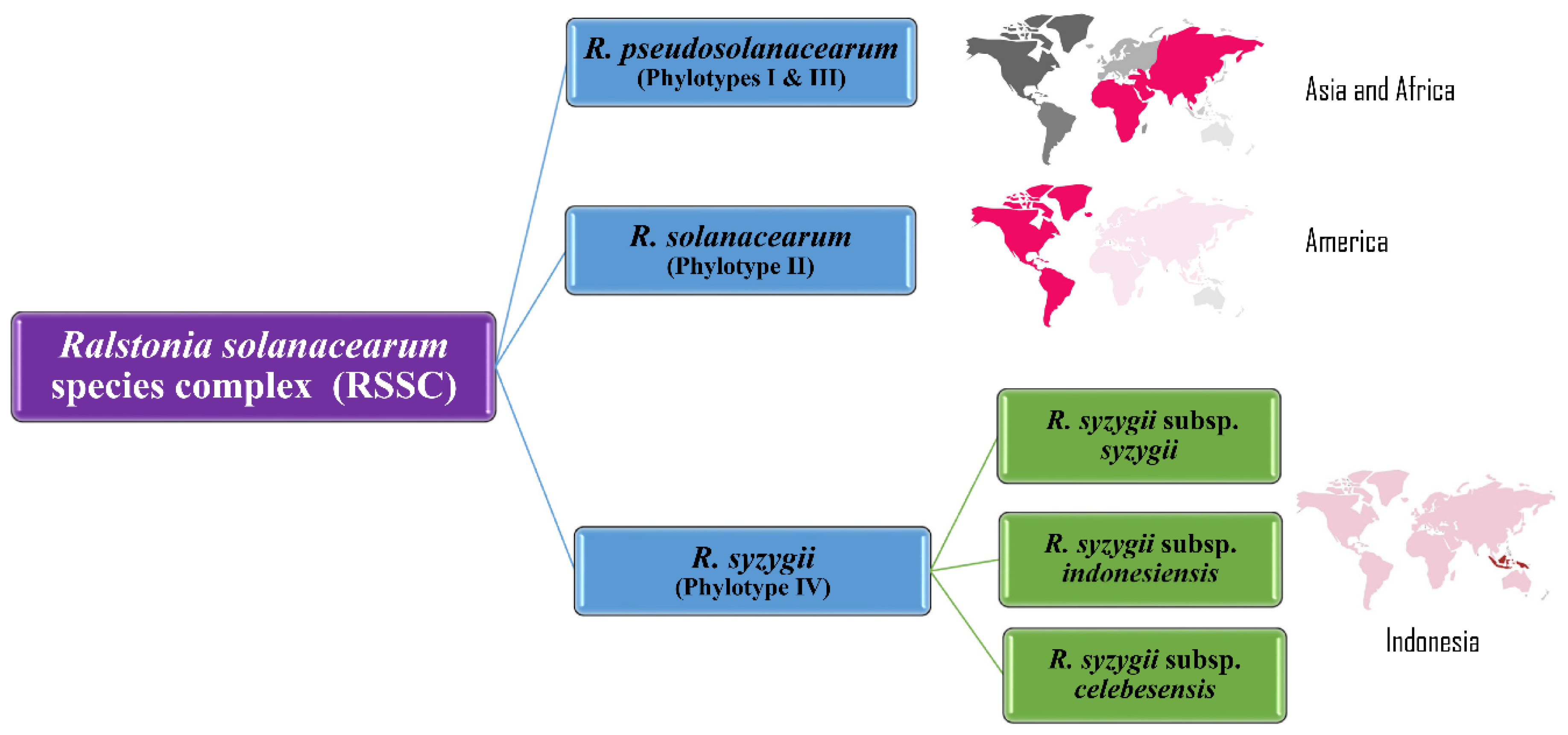
18. 2014–2020: Division into Three Genomic Species, Phylogenomics and Effector Repertoires
19. Conclusions and Future Research
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kannan, V.; Bastas, K.; Antony, R. Plant pathogenic bacteria: An overview. In Sustainable Approaches to Controlling Plant Pathogenic Bacteria; CRC Press: Boca Raton, FL, USA, 2015; pp. 1–16. [Google Scholar]
- Hayward, A.C. Biology and Epidemiology of Bacterial Wilt Caused by Pseudomonas solanacearum. Annu. Rev. Phytopathol. 1991, 29, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Prior, P.; Ailloud, F.; Dalsing, B.L.; Remenant, B.; Sanchez, B.; Allen, C. Genomic and proteomic evidence supporting the division of the plant pathogen Ralstonia solanacearum into three species. BMC Genom. 2016, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Genin, S. Molecular traits controlling host range and adaptation to plants in Ralstonia solanacearum. New Phytol. 2010, 187, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Van Overbeek, L.S.; Bergervoet, J.H.W.; Jacobs, F.H.H.; Van Elsas, J.D. The low-temperature-induced viable-but-nonculturable State affects the virulence of Ralstonia solanacearum biovar 2. Phytopathology 2004, 94, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, J.; Genin, S.; Magori, S.; Citovsky, V.; Sriariyanum, M.; Ronald, P.; Dow, M.; Verdier, V.; Beer, S.V.; Machado, M.A.; et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 614–629. [Google Scholar] [CrossRef]
- Elphinstone, J.G. The current bacterial wilt situation: A global overview. In Bacterial wilt Disease and the Ralstonia solanacearum Species Complex; APS Press: Saint Paul, MN, USA, 2005; pp. 9–28. [Google Scholar]
- Wicker, E.; Lefeuvre, P.; De Cambiaire, J.-C.; Lemaire, C.; Poussier, S.; Prior, P. Contrasting recombination patterns and demographic histories of the plant pathogen Ralstonia solanacearum inferred from MLSA. ISME J. 2011, 6, 961–974. [Google Scholar] [CrossRef]
- Floyd, J. New Pest Response Guidelines: Ralstonia solanacearum race 3 biovar 2. USDAAPHIS-PPQ, Emergency and Domestic Programs, Riverdale, MD. 2007. Available online: https://www.ippc.int/static/media/uploads/resources/new_pest_response_guidelines_ralstonia_solanacearum_race_3_biovar_2.pdf (accessed on 20 September 2020).
- National Potato Council. 2020 Annual Potato Yearbook; National Potato Council: Washington, DC, USA, 2020; p. 50. [Google Scholar]
- AgMRC. Tomatoes; Agricultural Marketing Research Center, Iowa State University: Ames, IA, USA, 2018; Available online: https://www.agmrc.org/commodities-products/vegetables/tomatoes (accessed on 21 September 2020).
- Champoiseau, P. Ralstonia solanacearum Race 3 biovar 2. In From the Field to the Lab: Towards Accurate Identification of a Select Agent Pathogen; National Research Initiative (NRI) Program of the USDA Cooperative State Research, Education, and Extension Services (CSREES): Miami, FL, USA, 2009. [Google Scholar]
- Smith, E.F. A Bacterial Disease of the Tomato, Eggplant, and Irish Potato (Bacillus solanacearum n. sp.); G.P.O.: Washington, DC, USA, 1896. [Google Scholar]
- Burrill, T.J. Preliminary notes upon the rotting of potatoes. In Proceedings of the 11th Annual Meeting Society for Promotion of Agricultural Sciences, Indianapolis, IN, USA, 18–19 August 1890. [Google Scholar]
- Burrill, T.J. Additional note on the rot of potatoes. In Proceedings of the Washington Meeting of the Society for the Promotion of Agricultural Science, Washington, DC, USA, 18–19 August 1891; p. 29. [Google Scholar]
- Halsted, B.D. An Investigation of Tomato Blight, a Blight of Potatoes, Bacterial Melon Blight; New Jersey State Agricultural Experiment Station 12th Annual Report; The John L. Murphy Publishing Company: Trenton, NJ, USA, 1892; pp. 267–276. [Google Scholar]
- Kelman, A. The Bacterial Wilt Caused by Pseudomonas Solanacearum: A Literature Review and Bibliography; North Carolina Agricultural Experiment Station: Raleigh, NC, USA, 1953. [Google Scholar]
- Smith, E.F. The southern tomato blight. In Proceedings of the American American Association for the Advancement of Science, Springfield, MA, USA, 28 August 1895; p. 191. [Google Scholar]
- Chester, F.D. A preliminary arrangement of the species of the genus Bacterium. JAMA 1898, XXX, 1480. [Google Scholar] [CrossRef]
- Smith, E.F. The Granville Tobacco Wilt; Bulletin 141, Part II; US Government Printing Office: Washington, DC, USA, 1909.
- Rorer, J.B. A bacterial disease of bananas and plantains. Phytopathology 1911, 1, 45–49. [Google Scholar]
- Smith, E.F. Bacteria in Relation to Plant Diseases; Carnegie Institution: Washington, DC, USA, 1905; Volume 3. [Google Scholar]
- Bergey, D.H. Manual of Determinative Bacteriology; William and Wilkins Co.: Baltimore, MD, USA, 1923. [Google Scholar]
- Dowson, W. On the generic names Pseudomonas, Xanthomonas and Bacterium for certain bacterial plant pathogens. Trans. Br. Mycol. Soc. 1943, 26, 4–14. [Google Scholar] [CrossRef]
- Dowson, W.J. Manual of Bacterial Plant Diseases; Adam and Charles Black: London, UK, 1949. [Google Scholar]
- Breed, R.S.; Murray, E.G.D.; Hitchens, A.P. Bergey’s Manual of Determinative Bacteriology, 6th ed.; Williams and Wilkins Co.: Baltimore, MD, USA, 1948. [Google Scholar]
- Savulescu, T. Contribution a la classification des bacteriacees phytopathogenes. Rev. Appl. Mycol. 1948, 27, 512–513. (In French) [Google Scholar]
- Buddenhagen, I.; Sequeira, L.; Kelman, A. Designation of races in Pseudomonas solanacearum. Phytopathology 1962, 52, 726. [Google Scholar]
- Buddenhagen, I.; Kelman, A. Biological and physiological aspects of bacterial wilt caused by Pseudomonas solanacearum. Annu. Rev. Phytopathol. 1964, 2, 203–230. [Google Scholar] [CrossRef]
- Dye, D.W.; Bradbury, J.F.; Goto, M.; Hayward, A.C.; Leelliott, R.A.; Schroth, M.N. International standards for naming pathovars of phytopathogenic bacteria and a list of pathovar names and pathotype strains. Rev. Plant Pathol. 1980, 59, 153–168. [Google Scholar]
- Hayward, A.C. Characteristics of Pseudomonas solanacearum. J. Appl. Bacteriol. 1964, 27, 265–277. [Google Scholar] [CrossRef]
- He, L.Y. Characteristics of Strains of Pseudomonas solanacearum from China. Plant Dis. 1983, 67, 1357–1361. [Google Scholar] [CrossRef]
- Aragaki, M.; Quinon, V.L. Bacterial wilt of ornamental gingers (Hedychium spp.) caused by Pseudomonas solanacearum. Plant Dis. Rep. 1965, 49, 378–379. [Google Scholar]
- Pegg, K.; Moffett, M. Host range of the ginger strain of Pseudomonas solanacearum in Queensland. Aust. J. Exp. Agric. 1971, 11, 696–698. [Google Scholar] [CrossRef]
- Pan, Z.C.; Xu, J.S.; Prior, P.; Zhang, H.; Chen, K.Y.; Tian, Q.; Zhang, L.Q.; Liu, L.; He, L.Y.; Feng, J. Development of a specific molecular tool for the detection of epidemiologically active mulberry causing-disease strains of Ralstonia solanacearum phylotype I (historically race 5-biovar 5) in China. Eur. J. Plant Pathol. 2013, 137, 377–391. [Google Scholar] [CrossRef]
- Cook, D. Genetic Diversity of Pseudomonas solanacearum: Detection of restriction fragment length polymorphisms with DNA probes that specify virulence and the hypersensitive response. Mol. Plant-Microbe Interact. 1989, 2, 113. [Google Scholar] [CrossRef]
- Palleroni, N.J.; Doudoroff, M. Phenotypic characterization and deoxyribonucleic acid homologies of Pseudomonas solanacearum. J. Bacteriol. 1971, 107, 690–696. [Google Scholar] [CrossRef]
- Doi, R.H.; Igarashi, R.T. Conservation of ribosomal and messenger ribonucleic acid cistrons in Bacillus species. J. Bacteriol. 1965, 90, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Dubnau, D.; Smith, I.; Morell, P.; Marmur, J. Gene conservation in Bacillus species. I. Conserved genetic and nucleic acid base sequence homologies. Proc. Natl. Acad. Sci. USA 1965, 54, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Palleroni, N.J.; Kunisawa, R.; Contopoulou, R.; Doudoroff, M. Nucleic acid homologies in the genus Pseudomonas. Int. J. Syst. Bacteriol. 1973, 23, 333–339. [Google Scholar] [CrossRef]
- Li, X.; Dorsch, M.; Del Dot, T.; Sly, L.; Stackebrandt, E.; Hayward, A. Phylogenetic studies of the rRNA group II pseudomonads based on 16S rRNA gene sequences. J. Appl. Bacteriol. 1993, 74, 324–329. [Google Scholar] [CrossRef]
- Byng, G.S.; Johnson, J.L.; Whitaker, R.J.; Gherna, R.L.; Jensen, R.A. The evolutionary pattern of aromatic amino acid biosynthesis and the emerging phylogeny of pseudomonad bacteria. J. Mol. Evol. 1983, 19, 272–282. [Google Scholar] [CrossRef]
- De Vos, P.; De Ley, J. Intra- and intergeneric similarities of Pseudomonas and Xanthomonas ribosomal ribonucleic acid cistrons. Int. J. Syst. Bacteriol. 1983, 33, 487–509. [Google Scholar] [CrossRef][Green Version]
- De Vos, P.; Goor, M.; Gillis, M.; De Ley, J. Ribosomal ribonucleic acid cistron similarities of phytopathogenic Pseudomonas species. Int. J. Syst. Bacteriol. 1985, 35, 169–184. [Google Scholar] [CrossRef]
- De Vos, P. Intrageneric and intergeneric similarities of ribosomal RNA cistrons of the genus Pseudomonas and the implications for taxonomy. Antonie van Leeuwenhoek 1980, 46, 96. [Google Scholar] [CrossRef]
- Seal, S.E.; Jackson, L.A.; Daniels, M.J. Use of tRNA consensus primers to indicate subgroups of Pseudomonas solanacearum by polymerase chain reaction amplification. Appl. Environ. Microbiol. 1992, 58, 3759–3761. [Google Scholar] [CrossRef]
- Welsh, J.; McClelland, M. Genomic fingerprints produced by PCR with consensus tRNA gene primers. Nucleic Acids Res. 1991, 19, 861–866. [Google Scholar] [CrossRef]
- Eden-Green, S.J.; Adhi, E. Sumatra Disease of Cloves and Pseudomonas solanacearum; ACIAR Bacterial Wilt Newsletter: Canberra, Australia, 1987; pp. 2–3. [Google Scholar]
- Eden-Green, S.; Sastraatmadja, H. Blood Disease in Indonesia; FAO Plant Protection Bulletin 38; Food and Agricultural Organization of the United Nations: Rome, Italy, 1990; pp. 49–50.
- Roberts, S.; Eden-Green, S.; Jones, P.; Ambler, D. Pseudomonas syzygii, sp. nov., the cause of Sumatra disease of cloves. Syst. Appl. Microbiol. 1990, 13, 34–43. [Google Scholar] [CrossRef]
- Seal, S.E.; Jackson, L.A.; Young, J.P.W.; Daniels, M.J. Differentiation of Pseudomonas solanacearum, Pseudomonas syzygii, Pseudomonas pickettii and the Blood Disease Bacterium by partial 16S rRNA sequencing: Construction of oligonucleotide primers for sensitive detection by polymerase chain reaction. J. Gen. Microbiol. 1993, 139, 1587–1594. [Google Scholar] [CrossRef]
- Yabuuchi, E.; Kosako, Y.; Oyaizu, H.; Yano, I.; Hotta, H.; Hashimoto, Y.; Ezaki, T.; Arakawa, M. Proposal of Burkholderia gen. nov. and transfer of seven species of the genus Pseudomonas homology group II to the new genus, with the type species Burkholderia cepacia (Palleroni and Holmes 1981) comb. nov. Microbiol. Immunol. 1992, 36, 1251–1275. [Google Scholar] [CrossRef]
- Fegan, M.; Taghavi, M.; Sly, L.I.; Hayward, A.C. Phylogeny, diversity, and molecular diagnostics of Ralstonia solanacearum. In Bacterial Wilt Disease: Molecular and Ecological Aspects; Prior, P., Allen, C., Elphinstone, J., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 19–33. [Google Scholar]
- Taghavi, M.; Hayward, C.; Sly, L.I.; Fegan, M. Analysis of the phylogenetic relationships of strains of Burkholderia solanacearum, Pseudomonas syzygii, and the Blood Disease Bacterium of banana based on 16S rRNA gene sequences. Int. J. Syst. Bacteriol. 1996, 46, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Gillis, M.; Van Van, T.; Bardin, R.; Goor, M.; Hebbar, P.; Willems, A.; Segers, P.; Kersters, K.; Heulin, T.; Fernandez, M.P. Polyphasic taxonomy in the genus Burkholderia leading to an emended description of the genus and proposition of Burkholderia vietnamiensis sp. nov. for N2-Fixing isolates from rice in Vietnam. Int. J. Syst. Bacteriol. 1995, 45, 274–289. [Google Scholar] [CrossRef]
- Yabuuchi, E.; Kosako, Y.; Yano, I.; Hotta, H.; Nishiuchi, Y. Transfer of two Burkholderia and an Alcaligenes species to Ralstonia Gen. Nov. Microbiol. Immunol. 1995, 39, 897–904. [Google Scholar] [CrossRef]
- Ralston, E.; Palleroni, N.J.; Doudoroff, M. Pseudomonas pickettii, a new species of clinical origin related to Pseudomonas solanacearum. Int. J. Syst. Bacteriol. 1973, 23, 15–19. [Google Scholar] [CrossRef][Green Version]
- Gillings, M.; Fahy, P. Genomic fingerprinting: Towards a unified view of the Pseudomonas solanacearum species complex. In Bacterial Wilt: The Disease and Its Causative Agent, Pseudomonas solanacearum; CAB International: Wallingford, UK, 1994; pp. 95–112. [Google Scholar]
- Poussier, S.; Prior, P.; Luisetti, J.; Hayward, C.; Fegan, M. Partial Sequencing of the hrpB and Endoglucanase genes confirms and expands the known diversity within the Ralstonia solanacearum species complex. Syst. Appl. Microbiol. 2000, 23, 479–486. [Google Scholar] [CrossRef]
- Poussier, S.; Trigalet-Demery, D.; Vandewalle, P.; Goffinet, B.; Luisetti, J.; Trigalet, A. Genetic diversity of Ralstonia solanacearum as assessed by PCR-RFLP of the hrp gene region, AFLP and 16S rRNA sequence analysis, and identification of an African subdivision. Microbiology 2000, 146, 1679–1692. [Google Scholar] [CrossRef][Green Version]
- Salanoubat, M.; Genin, S.; Artiguenave, F.; Gouzy, J.; Mangenot, S.; Arlat, M.; Billault, A.; Brottier, P.; Camus, J.C.; Cattolico, L.; et al. Genome sequence of the plant pathogen Ralstonia solanacearum. Nature 2002, 415, 497–502. [Google Scholar] [CrossRef]
- Eckhardt, T. A rapid method for the identification of plasmid desoxyribonucleic acid in bacteria. Plasmid 1978, 1, 584–588. [Google Scholar] [CrossRef]
- Rosenberg, C.; Casse-Delbart, F.; Dusha, I.; David, M.; Boucher, C. Megaplasmids in the plant-associated bacteria Rhizobium meliloti and Pseudomonas solanacearum. J. Bacteriol. 1982, 150, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Coenye, T.; Vandamme, P. Simple sequence repeats and compositional bias in the bipartite Ralstonia solanacearum GMI1000 genome. BMC Genom. 2003, 4, 10. [Google Scholar] [CrossRef]
- Guidot, A.; Prior, P.; Schoenfeld, J.; Carrère, S.; Genin, S.; Boucher, C. Genomic Structure and Phylogeny of the Plant Pathogen Ralstonia solanacearum Inferred from Gene Distribution Analysis. J. Bacteriol. 2006, 189, 377–387. [Google Scholar] [CrossRef]
- Genin, S.; Boucher, C. Lessons learned from the genome analysis of Ralstonia solanacearum. Annu. Rev. Phytopathol. 2004, 42, 107–134. [Google Scholar] [CrossRef] [PubMed]
- Genin, S.; Boucher, C. Ralstonia solanacearum: Secrets of a major pathogen unveiled by analysis of its genome. Mol. Plant Pathol. 2002, 3, 111–118. [Google Scholar] [CrossRef]
- Alfano, J.R.; Collmer, A. Type III secretion system effector proteins: Double agents in bacterial disease and plant defense. Annu. Rev. Phytopathol. 2004, 42, 385–414. [Google Scholar] [CrossRef]
- Cohan, F.M. What are bacterial species? Annu. Rev. Microbiol. 2002, 56, 457–487. [Google Scholar] [CrossRef]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef]
- Sarkar, S.F.; Guttman, D.S. Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic elant pathogen. Appl. Environ. Microbiol. 2004, 70, 1999–2012. [Google Scholar] [CrossRef]
- Prior, P.; Fegan, M. Recent developments in the phylogeny and classification of Ralstonia solanacearum. Acta Hortic. 2005, 127–136. [Google Scholar] [CrossRef]
- Fegan, M.; Prior, P. How complex is the Ralstonia solanacearum species complex. In Bacterial Wilt Disease and the Ralstonia solanacearum Species Complex; APS Press: St. Paul, MN, USA, 2005; p. 510. [Google Scholar]
- Fegan, M. Bacterial wilt Diseases of Banana: Evolution and Ecology in Bacterial Wilt Disease and the Ralstonia solanacearum Species Complex; APS Press: St. Paul, MN, USA, 2005; pp. 379–386. [Google Scholar]
- Zhou, Z.; Alikhan, N.-F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.A.; Greenberg, J.T. Evolutionary dynamics of Ralstonia solanacearum. Appl. Environ. Microbiol. 2006, 73, 1225–1238. [Google Scholar] [CrossRef] [PubMed]
- Orgambide, G.; Montrozier, H.; Servin, P.; Roussel, J.; Trigalet-Demery, D.; Trigalet, A. High heterogeneity of the exopolysaccharides of Pseudomonas solanacearum strain GMI 1000 and the complete structure of the major polysaccharide. J. Biol. Chem. 1991, 266, 8312–8321. [Google Scholar]
- Schell, M.; Denny, T.; Clough, S.; Huang, J. Further characterization of genes encoding extracellular polysaccharide of Pseudomonas solanacearum and their regulation. In Advances in Molecular Genetics of Plant-Microbe Interactions, Volume 2, Proceedings of the 6th International Symposium on Molecular Plant-Microbe Interactions, Seattle, WA, USA, July 1992; Nester, E.W., Verma, D.P.S., Eds.; Springer: Dordrecht, The Netherlands, 1993; pp. 231–239. [Google Scholar]
- Schell, M.A. Control of virulence and pathogenicity genes of Ralstonia solanacearum by an elaborate sensory network. Annu. Rev. Phytopathol. 2000, 38, 263–292. [Google Scholar] [CrossRef]
- Huang, J.; A Schell, M. Molecular characterization of the eps gene cluster of Pseudomonas solanacearum and its transcriptional regulation at a single promoter. Mol. Microbiol. 1995, 16, 977–989. [Google Scholar] [CrossRef]
- González, E.T.; Allen, C. Characterization of a Ralstonia solanacearum operon required for polygalacturonate degradation and uptake of galacturonic acid. Mol. Plant Microbe Interact. 2003, 16, 536–544. [Google Scholar] [CrossRef]
- Kang, Y. Dramatically reduced virulence of mutants of Pseudomonas solanacearum defective in export of extracellular proteins across the outer membrane. Mol. Plant Microbe Interact. 1994, 7, 370. [Google Scholar] [CrossRef]
- Allen, C. Cloning of genes affecting polygalacturonase production in Pseudomonas solanacearum. Mol. Plant Microbe Interact. 1991, 4, 147–154. [Google Scholar] [CrossRef]
- Huang, Q.; Allen, C. An exo-poly-alpha-d-galacturonosidase, PehB, is required for wild-type virulence of Ralstonia solanacearum. J. Bacteriol. 1997, 179, 7369–7378. [Google Scholar] [CrossRef]
- Meng, F.; Babujee, L.; Jacobs, J.M.; Allen, C. Comparative transcriptome analysis reveals cool virulence factors of Ralstonia solanacearum race 3 biovar 2. PLoS ONE 2015, 10, e0139090. [Google Scholar] [CrossRef] [PubMed]
- Remenant, B.; Coupat-Goutaland, B.; Guidot, A.; Cellier, G.; Wicker, E.; Allen, C.; Fegan, M.; Pruvost, O.; Elbaz, M.; Calteau, A.; et al. Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genom. 2010, 11, 379. [Google Scholar] [CrossRef] [PubMed]
- Remenant, B.; De Cambiaire, J.-C.; Cellier, G.; Jacobs, J.M.; Mangenot, S.; Barbe, V.; Lajus, A.; Vallenet, D.; Medigue, C.; Fegan, M.; et al. Ralstonia syzygii, the Blood Disease Bacterium and some asian R. solanacearum strains form a single genomic species despite divergent lifestyles. PLoS ONE 2011, 6, e24356. [Google Scholar] [CrossRef] [PubMed]
- Boucher, C.A.; Barberis, P.A.; Demery, D.A. Transposon mutagenesis of Pseudomonas solanacearum: Isolation of Tn5-induced avirulent mutants. Microbiology 1985, 131, 2449–2457. [Google Scholar] [CrossRef]
- Prior, P.; Steva, H. Characteristics of strains of Pesudomonas solanacearum from the French West Indies. Plant Dis. 1990, 74, 13–17. [Google Scholar] [CrossRef]
- Van Elsas, J.D.; Kastelein, P.; De Vries, P.M.; Van Overbeek, L.S. Effects of ecological factors on the survival and physiology of Ralstonia solanacearum bv. 2 in irrigation water. Can. J. Microbiol. 2001, 47, 842–854. [Google Scholar] [CrossRef]
- Lefeuvre, P.; Cellier, G.; Remenant, B.; Chiroleu, F.; Prior, P. Constraints on genome dynamics revealed from gene distribution among the Ralstonia solanacearum species. PLoS ONE 2013, 8, e63155. [Google Scholar] [CrossRef]
- Seal, S.; Taghavi, M.; Fegan, N.; Hayward, A.C.; Fegan, M. Determination of Ralstonia (Pseudomonas) solanacearum rDNA subgroups by PCR tests. Plant Pathol. J. 1999, 48, 115–120. [Google Scholar] [CrossRef]
- Smith, J.; Offord, L.; Holderness, M.; Saddler, G. Genetic diversity of Burkholderia solanacearum (synonym Pseudomonas solanacearum) race 3 in Kenya. Appl. Environ. Microbiol. 1996, 61, 4263–4268. [Google Scholar] [CrossRef]
- Toukam, G.M.S.; Cellier, G.; Wicker, E.; Guilbaud, C.; Kahane, R.; Allen, C.; Prior, P. Broad diversity of Ralstonia solanacearum strains in Cameroon. Plant Dis. 2009, 93, 1123–1130. [Google Scholar] [CrossRef]
- Coupat, B.; Chaumeille-Dole, F.; Fall, S.; Prior, P.; Simonet, P.; Nesme, X.; Bertolla, F.; Coupat-Goutaland, B. Natural transformation in the Ralstonia solanacearum species complex: Number and size of DNA that can be transferred. FEMS Microbiol. Ecol. 2008, 66, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Loper, J.E.; Henkels, M.D.; Shaffer, B.T.; Valeriote, F.A.; Gross, H. Isolation and identification of rhizoxin analogs from Pseudomonas fluorescens Pf-5 by using a genomic mining strategy. Appl. Environ. Microbiol. 2008, 74, 3085–3093. [Google Scholar] [CrossRef]
- Partida-Martinez, L.P.; Hertweck, C. A Gene cluster encoding rhizoxin biosynthesis in “Burkholderia rhizoxina”, the bacterial endosymbiont of the fungus Rhizopus microsporus. ChemBioChem 2007, 8, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zheng, H.-J.; Liu, L.; Pan, Z.; Prior, P.; Tang, B.; Zhang, H.; Tian, Q.; Zhang, L.-Q.; Feng, J.; et al. Complete genome sequence of the plant pathogen Ralstonia solanacearum strain Po82. J. Bacteriol. 2011, 193, 4261–4262. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Studholme, D.J.; Kemen, E.; MacLean, D.; Schornack, S.; Aritua, V.; Thwaites, R.; Grant, M.; Smith, J.; Jones, J.D. Genome-wide sequencing data reveals virulence factors implicated in banana xanthomonas wilt. FEMS Microbiol. Lett. 2010, 310, 182–192. [Google Scholar] [CrossRef]
- El Yacoubi, B.; Brunings, A.M.; Yuan, Q.; Shankar, S.; Gabriel, D.W. In planta horizontal transfer of a major pathogenicity effector gene. Appl. Environ. Microbiol. 2007, 73, 1612–1621. [Google Scholar] [CrossRef]
- Mira, A.; Ochman, H.; Moran, N.A. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001, 17, 589–596. [Google Scholar] [CrossRef]
- Moran, N.A. Microbial minimalism. Cell 2002, 108, 583–586. [Google Scholar] [CrossRef]
- Levin, B.R. Periodic selection, infectious gene exchange and the genetic structure of E. coli populations. Genetics 1981, 99, 1–23. [Google Scholar]
- Didelot, X.; Falush, D. Inference of bacterial microevolution using multilocus sequence data. Genetics 2006, 175, 1251–1266. [Google Scholar] [CrossRef]
- Darling, A.; Mau, B.; Perna, N. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Brumbley, S.M.; Carney, B.F.; Denny, T.P. Phenotype conversion in Pseudomonas solanacearum due to spontaneous inactivation of PhcA, a putative LysR transcriptional regulator. J. Bacteriol. 1993, 175, 5477–5487. [Google Scholar] [CrossRef]
- Brumbley, S.M.; Denny, T.P. Cloning of wild-type Pseudomonas solanacearum phcA, a gene that when mutated alters expression of multiple traits that contribute to virulence. J. Bacteriol. 1990, 172, 5677–5685. [Google Scholar] [CrossRef]
- Denny, T.P. Inactivation of multiple virulence genes reduces the ability of Pseudomonas solanacearum to cause wilt symptoms. Mol. Plant Microbe Interact. 1990, 3, 293. [Google Scholar] [CrossRef]
- Genin, S.; Denny, T. Pathogenomics of the Ralstonia solanacearum species complex. Annu. Rev. Phytopathol. 2012, 50, 67–89. [Google Scholar] [CrossRef]
- Jacobs, J.M.; Allen, C. Virulence mechanisms of plant-pathogenic Ralstonia spp. In Virulence Mechanisms of Plant-Pathogenic Bacteria; APS Press: Saint Paul, MN, USA, 2016; pp. 365–380. [Google Scholar]
- Peeters, N.; Carrère, S.; Anisimova, M.; Plener, L.; Cazalé, A.-C.; Genin, S. Repertoire, unified nomenclature and evolution of the Type III effector gene set in the Ralstonia solanacearum species complex. BMC Genom. 2013, 14, 859. [Google Scholar] [CrossRef]
- Deslandes, L.; Genin, S. Opening the Ralstonia solanacearum type III effector tool box: Insights into host cell subversion mechanisms. Curr. Opin. Plant Biol. 2014, 20, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Hajri, A.; Brin, C.; Hunault, G.; Lardeux, F.; Lemaire, C.; Manceau, C.; Boureau, T.; Poussier, S. A «Repertoire for Repertoire» Hypothesis: Repertoires of type three effectors are candidate determinants of host specificity in Xanthomonas. PLoS ONE 2009, 4, e6632. [Google Scholar] [CrossRef]
- Collmer, A.; Badel, J.L.; Charkowski, A.O.; Deng, W.-L.; Fouts, D.E.; Ramos, A.R.; Rehm, A.H.; Anderson, D.M.; Schneewind, O.; Van Dijk, K.; et al. Pseudomonas syringae Hrp type III secretion system and effector proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 8770–8777. [Google Scholar] [CrossRef]
- Cunnac, S.; Lindeberg, M.; Collmer, A. Pseudomonas syringae type III secretion system effectors: Repertoires in search of functions. Curr. Opin. Microbiol. 2009, 12, 53–60. [Google Scholar] [CrossRef]
- Fouts, D.E.; Badel, J.L.; Ramos, A.R.; Rapp, R.A.; Collmer, A. A Pseudomonas syringae pv. tomato DC3000 Hrp (type III secretion) deletion mutant expressing the Hrp system of bean pathogen P. syringae pv. syringae 61 retains normal host specificity for tomato. Mol. Plant Microbe Interact. 2003, 16, 43–52. [Google Scholar] [PubMed]
- Lindeberg, M.; Cunnac, S.; Collmer, A. The evolution of Pseudomonas syringae host specificity and type III effector repertoires. Mol. Plant Pathol. 2009, 10, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Pensec, F.; Lebeau, A.; Daunay, M.-C.; Chiroleu, F.; Guidot, A.; Wicker, E. Towards the identification of type III effectors associated with Ralstonia solanacearum virulence on tomato and eggplant. Phytopathology 2015, 105, 1529–1544. [Google Scholar] [CrossRef]
- Safni, I.; Cleenwerck, I.; De Vos, P.; Fegan, M.; Sly, L.; Kappler, U. Polyphasic taxonomic revision of the Ralstonia solanacearum species complex: Proposal to emend the descriptions of Ralstonia solanacearum and Ralstonia syzygii and reclassify current R. syzygii strains as Ralstonia syzygii subsp. syzygii subsp. nov., R. solanacearum phylotype IV strains as Ralstonia syzygii subsp. indonesiensis subsp. nov., banana blood disease bacterium strains as Ralstonia syzygii subsp. celebesensis subsp. nov. and R. solanacearum phylotype I and III strains as Ralstonia pseudosolanacearum sp. nov. Int. J. Syst. Evol. Microbiol. 2014, 64, 3087–3103. [Google Scholar] [CrossRef]
- Dalsing, B.L.; Truchon, A.N.; Gonzalez-Orta, E.T.; Milling, A.S.; Allen, C. Ralstonia solanacearum uses inorganic nitrogen metabolism for virulence, ATP production, and detoxification in the oxygen-limited host xylem environment. mBio 2015, 6, e02471-14. [Google Scholar] [CrossRef] [PubMed]
- Ailloud, F.; Lowe-Power, T.; Robène, I.; Cruveiller, S.; Allen, C.; Prior, P. In planta comparative transcriptomics of host-adapted strains of Ralstonia solanacearum. PeerJ 2016, 4, e1549. [Google Scholar] [CrossRef]
- Ailloud, F.; Lowe, T.; Cellier, G.; Roche, D.; Allen, C.; Prior, P. Comparative genomic analysis of Ralstonia solanacearum reveals candidate genes for host specificity. BMC Genom. 2015, 16, 270. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, S. Phylogenomic analysis of the genus Ralstonia based on 686 single-copy genes. Antonie van Leeuwenhoek 2015, 109, 71–82. [Google Scholar] [CrossRef]
- Castillo, J.A.; Agathos, S.N. A genome-wide scan for genes under balancing selection in the plant pathogen Ralstonia solanacearum. BMC Evol. Biol. 2019, 19, 1–16. [Google Scholar] [CrossRef]
- Cho, H.; Song, E.-S.; Heu, S.; Baek, J.; Lee, Y.K.; Lee, S.; Lee, S.-W.; Park, D.S.; Lee, T.-H.; Kim, J.-G.; et al. Prediction of host-specific genes by pan-genome analyses of the Korean Ralstonia solanacearum species complex. Front. Microbiol. 2019, 10, 506. [Google Scholar] [CrossRef]
- Perrier, A.; Barlet, X.; Rengel, D.; Prior, P.; Poussier, S.; Genin, S.; Guidot, A. Spontaneous mutations in a regulatory gene induce phenotypic heterogeneity and adaptation of Ralstonia solanacearum to changing environments. Environ. Microbiol. 2019, 21, 3140–3152. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, C.R.R.; Carrère, S.; Lonjon, F.; Vailleau, F.; Macho, A.P.; Genin, S.; Peeters, N. Pangenomic type III effector database of the plant pathogenic Ralstonia spp. PeerJ 2019, 7, e7346. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Yu, W.; Zhuang, H.; Wei, Y.; Derevnina, L.; Yu, G.; Luo, J.; Macho, A.P. Intra-strain elicitation and suppression of plant immunity by Ralstonia solanacearum type-III effectors in Nicotiana benthamiana. Plant Commun. 2020, 1, 100025. [Google Scholar] [CrossRef]
- Zheng, X.; Li, X.; Wang, B.; Cheng, D.; Li, Y.; Li, W.; Huang, M.; Tan, X.; Zhao, G.; Song, B.; et al. A systematic screen of conserved Ralstonia solanacearum effectors reveals the role of RipAB, a nuclear-localized effector that suppresses immune responses in potato. Mol. Plant Pathol. 2019, 20, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Mukaihara, T. Comprehensive identification of PTI suppressors in type III effector repertoire reveals that Ralstonia solanacearum activates jasmonate signaling at two different steps. Int. J. Mol. Sci. 2019, 20, 5992. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Jiang, H.; Liao, B.; Ren, X. Progress on groundnut genetic enhancement for bacterial wilt resistance. Chin. Agric. Sci. Bull. 2007, 23, 369–372. [Google Scholar]
- Deslandes, L.; Pileur, F.; Liaubet, L.; Camut, S.; Can, C.; Williams, K.; Holub, E.; Beynon, J.L.; Arlat, M.; Marco, Y. Genetic characterization of RRS1, a recessive locus in Arabidopsis thaliana that confers resistance to the bacterial soilborne pathogen Ralstonia solanacearum. Mol. Plant Microbe Interact. 1998, 11, 659–667. [Google Scholar] [CrossRef]
- Elsayed, T.R.; Jacquiod, S.; Nour, E.H.; Sørensen, S.J.; Smalla, K. Biocontrol of bacterial wilt disease through complex interaction between tomato plant, antagonists, the indigenous rhizosphere microbiota, and Ralstonia solanacearum. Front. Microbiol. 2020, 10, 2835. [Google Scholar] [CrossRef]
- Fujiwara, A.; Fujisawa, M.; Hamasaki, R.; Kawasaki, T.; Fujie, M.; Yamada, T. Biocontrol of Ralstonia solanacearum by treatment with lytic bacteriophages. Appl. Environ. Microbiol. 2011, 77, 4155–4162. [Google Scholar] [CrossRef]
- Huet, G. Breeding for resistances to Ralstonia solanacearum. Front. Plant Sci. 2014, 5, 715. [Google Scholar] [CrossRef]
- Lemessa, F.; Zeller, W. Screening rhizobacteria for biological control of Ralstonia solanacearum in Ethiopia. Biol. Control 2007, 42, 336–344. [Google Scholar] [CrossRef]
- Mansfield, J.W. From bacterial avirulence genes to effector functions via the hrp delivery system: An overview of 25 years of progress in our understanding of plant innate immunity. Mol. Plant Pathol. 2009, 10, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Ranamukhaarachchi, S. Soil-borne antagonists for biological control of bacterial wilt disease caused by Ralstonia solanacearum in tomato and pepper. J. Plant Pathol. 2010, 92, 395–405. [Google Scholar]
- Wang, J.-F.; Ho, F.-I.; Truong, H.T.H.; Huang, S.-M.; Balatero, C.H.; Dittapongpitch, V.; Hidayati, N. Identification of major QTLs associated with stable resistance of tomato cultivar ‘Hawaii 7996’ to Ralstonia solanacearum. Euphytica 2012, 190, 241–252. [Google Scholar] [CrossRef]
- Wei, Z.; Huang, J.; Yang, T.; Jousset, A.; Xu, Y.; Shen, Q.; Friman, V.-P. Seasonal variation in the biocontrol efficiency of bacterial wilt is driven by temperature-mediated changes in bacterial competitive interactions. J. Appl. Ecol. 2017, 54, 1440–1448. [Google Scholar] [CrossRef]
- Wei, Z.; Huang, J.-F.; Hu, J.; Gu, Y.-A.; Yang, C.-L.; Mei, X.-L.; Shen, Q.-R.; Xu, Y.; Friman, V.-P. Altering transplantation time to avoid periods of high temperature can efficiently reduce bacterial wilt disease incidence with tomato. PLoS ONE 2015, 10, e0139313. [Google Scholar] [CrossRef]
- Wei, Z.; Yang, X.; Yin, S.; Shen, Q.; Ran, W.; Xu, Y. Efficacy of Bacillus-fortified organic fertilizer in controlling bacterial wilt of tomato in the field. Appl. Soil Ecol. 2011, 48, 152–159. [Google Scholar] [CrossRef]
- Yang, L.; Ding, W.; Xu, Y.; Wu, D.; Li, S.; Chen, J.; Guo, B. New insights into the antibacterial activity of hydroxycoumarins against Ralstonia solanacearum. Molecules 2016, 21, 468. [Google Scholar] [CrossRef]
- Yuliar; Nion, Y.A.; Toyota, K. Recent trends in control methods for bacterial wilt diseases caused by Ralstonia solanacearum. Microbes Environ. 2015, 30, 1–11. [Google Scholar] [CrossRef]
- Zheng, X.; Zhu, Y.; Wang, J.; Wang, Z.; Liu, B. Combined use of a microbial restoration substrate and avirulent Ralstonia solanacearum for the control of tomato bacterial wilt. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biotype a | Disaccharides | Hexose Alcohols | References | ||||
|---|---|---|---|---|---|---|---|
| Lactose | Maltose | Cellobiose | Dulcitol | Mannitol | Sorbitol | ||
| 1 | − | − | − | − | − | − | [31] |
| 2 | + | + | + | − | − | − | [31] |
| 3 | + | + | + | + | + | + | [31] |
| 4 | − | − | − | + | + | + | [31] |
| 5 | + | + | + | − | + | − | [32] b |
| Method Used to Create the Dendrogram | Position of Strain ACH0732 in the Dendrogram |
|---|---|
| 16S-23S rRNA intergenic spacer region sequences | Outside of all three clusters |
| Polygalacturonase gene sequences | Outside of all three clusters |
| Endogluconase gene sequences | Subdivision 2b |
| 16S rDNA-based PCR | Division 1 |
| ITS multiplex PCR | Subdivision 2b |
| Phylotype | General Geographical Origin | Phenotypic Characteristics |
|---|---|---|
| I | Asia | Biovars 3, 4 and 5 |
| II | America | Biovars 1, 2 and 2T a |
| III | Africa and surrounding islands | Biovars 1 and 2T |
| IV | Indonesia | Biovars 1, 2 and 2T; P. syzygii and BDB b |
| Operons/Gene Products | Name | Function | References |
|---|---|---|---|
| Eps operon | Extracellular polysaccharide | Production of EPS I | [78,79] |
| Cel, PME, PG | Cellulase, pectin methylesterase, cellobiosidase and polygalacturonases | Plant cell wall degradation | [79,81,82] |
| PehA | Pectin methylesterase | Exposes pectin to degradation by removing its methoxy groups | [79,83] |
| PehB | Endo-polygalacturonic acid esterase | Pectin degradation | [79,84] |
| PehC | Exo-polygalacturonic acid esterase | Pectin degradation | [79,81] |
| PehX | Endo-polygalacturonic acid esterase (like PehB) | Aids conversion of complex polygalacturonic acid and pectin into simple monomeric, dimeric, or oligomeric units | [79] |
| LecM | Mannose–fucose binding lectin | Contributes to cool temperature virulence of R3bv2 strains | [85] |
| AidA, AidC | Quorum-sensing protein and a hypothetical protein | Contributes to cool temperature virulence of R3bv2 strains | [85] |
| Strain No. | Race, Biovar, Phylotype a | Host | Origin | Reference |
|---|---|---|---|---|
| GMI1000 | R 1, bv 3, phylotype I | Tomato | French Guyana | [88] |
| K60 | R 1, bv 1, phylotype IIA | Tomato | North Carolina, USA | [17] |
| CFBP2957 | phylotype IIA | Tomato | French West Indies | [86,89] |
| IPO 1609 | R 3, bv 2, phylotype IIB | Potato | The Netherlands | [90] |
| Molk2 b | R 2, bv 1, phylotype IIB | Banana | Philippines | [65,91] |
| ACH0732 | R1, bv 2 | Tomato | Australia | [53] |
| R240 | R1, bv N2 | Potato | Nepal | [92,93] |
| R780 | R1, bv N2 | Potato | Indonesia | [53] |
| CMR15 | phylotype III | Tomato | Cameroon | [86,94] |
| PSI07 | phylotype IV | Tomato | Indonesia | [74,86] |
| R229 | BDB c phylotype IV | Banana | Indonesia | [87] |
| R24 | R. syzygii phylotype IV | Clove | Indonesia | [87] |
| Phylotype | Complete | Chromosome | Scaffold | Contig | Total |
|---|---|---|---|---|---|
| I | 67 | 6 | 37 | 18 | 128 |
| III | 1 | 1 | 1 | 3 | |
| IIA | 1 | 3 | 7 | 6 | 17 |
| IIB | 11 | 4 | 20 | 18 | 53 |
| IV | 13 | 1 | 2 | 16 | |
| Total | 93 | 15 | 64 | 45 | 217 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paudel, S.; Dobhal, S.; Alvarez, A.M.; Arif, M. Taxonomy and Phylogenetic Research on Ralstonia solanacearum Species Complex: A Complex Pathogen with Extraordinary Economic Consequences. Pathogens 2020, 9, 886. https://doi.org/10.3390/pathogens9110886
Paudel S, Dobhal S, Alvarez AM, Arif M. Taxonomy and Phylogenetic Research on Ralstonia solanacearum Species Complex: A Complex Pathogen with Extraordinary Economic Consequences. Pathogens. 2020; 9(11):886. https://doi.org/10.3390/pathogens9110886
Chicago/Turabian StylePaudel, Sujan, Shefali Dobhal, Anne M. Alvarez, and Mohammad Arif. 2020. "Taxonomy and Phylogenetic Research on Ralstonia solanacearum Species Complex: A Complex Pathogen with Extraordinary Economic Consequences" Pathogens 9, no. 11: 886. https://doi.org/10.3390/pathogens9110886
APA StylePaudel, S., Dobhal, S., Alvarez, A. M., & Arif, M. (2020). Taxonomy and Phylogenetic Research on Ralstonia solanacearum Species Complex: A Complex Pathogen with Extraordinary Economic Consequences. Pathogens, 9(11), 886. https://doi.org/10.3390/pathogens9110886