Staphylococcus aureus Lipoprotein Induces Skin Inflammation, Accompanied with IFN-γ-Producing T Cell Accumulation through Dermal Dendritic Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. S. aureus Lipoprotein Activates Dendritic Cells through TLR2
2.2. S. aureus Lipoprotein Promotes Dendritic Cell Migration
2.3. SA-LP Induce Skin Inflammation Accompanied with T Cell Accumulation
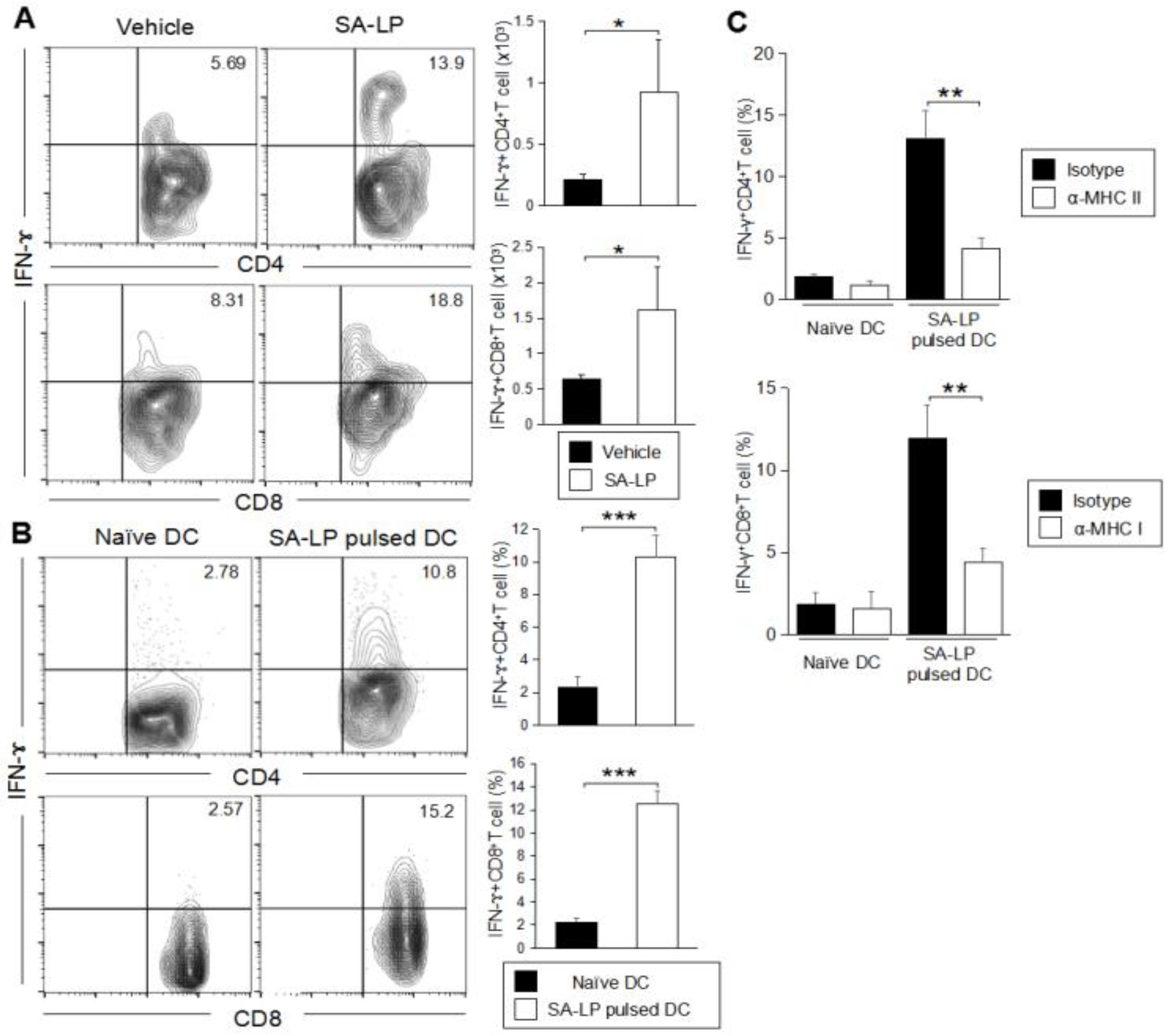
2.4. SA-LP Induces IFN-ɤ-Producing CD4+ and CD8+T Cell Generation
2.5. DDC Plays an Important Role for the Induction of SA-LP-Originated Skin Inflammation
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cogen, A.L.; Nizet, V.; Gallo, R.L. Skin microbiota: A source of disease or defence? Br. J. Dermatol. 2008, 158, 442–455. [Google Scholar] [PubMed]
- Naik, S.; Bouladoux, N.; Linehan, J.L.; Han, S.J.; Harrison, O.J.; Wilhelm, C.; Conlan, S.; Himmelfarb, S.; Byrd, A.L.; Deming, C.; et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 2015, 520, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.A.; Connolly, J.; Hourihane, J.O.; Fallon, P.G.; McLean, W.H.I.; Murray, D.; Jo, J.H.; Segre, J.A.; Kong, H.H.; Irvine, A.D. Skin microbiome before development of atopic dermatitis: Early colonization with commensal staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J. Allergy Clin. Immunol. 2017, 139, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9, eaah4680. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, T.; Skabytska, Y.; Kaesler, S.; Volz, T. Regulation of T Cell Immunity in Atopic Dermatitis by Microbes: The Yin and Yang of Cutaneous Inflammation. Front. Immunol. 2015, 6, 353. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Archer, N.K.; Dillen, C.A.; Wang, Y.; Ashbaugh, A.G.; Ortines, R.V.; Kao, T.; Lee, S.K.; Cai, S.S.; Miller, R.J.; et al. Staphylococcus aureus Epicutaneous Exposure Drives Skin Inflammation via IL-36-Mediated T Cell Responses. Cell Host Microbe 2017, 8, 653–666. [Google Scholar]
- Laborel-Préneron, E.; Bianchi, P.; Boralevi, F.; Lehours, P.; Fraysse, F.; Morice-Picard, F.; Sugai, M.; Sato’o, Y.; Badiou, C.; Lina, G.; et al. Effects of the Staphylococcus aureus and Staphylococcus epidermidis Secretomes Isolated from the Skin Microbiota of Atopic Children on CD4+ T Cell Activation. PLoS ONE 2015, 10, e0141067. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Boguniewicz, M.; Leung, D.Y. Atopic dermatitis: A disease of altered skin barrier and immune dysregulation. Immunol. Rev. 2011, 242, 233–246. [Google Scholar] [PubMed]
- De Benedetto, A.; Kubo, A.; Beck, L.A. Skin barrier disruption: A requirement for allergen sensitization? J. Investig. Dermatol. 2012, 132, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Atilano, M.L.; Pereira, P.M.; Yates, J.; Reed, P.; Veiga, H.; Pinho, M.G.; Filipe, S.R. Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2010, 107, 18991–18996. [Google Scholar] [CrossRef] [PubMed]
- Qamar, A.; Golemi-Kotra, D. Dual roles of FmtA in Staphylococcus aureus cell wall biosynthesis and autolysis. Antimicrob. Agents Chemother. 2012, 56, 3797–3805. [Google Scholar] [CrossRef] [PubMed]
- Sobhanifar, S.; Worrall, L.J.; Gruninger, R.J.; Wasney, G.A.; Blaukopf, M.; Baumann, L.; Lameignere, E.; Solomonson, M.; Brown, E.D.; Withers, S.G.; et al. Structure and mechanism of Staphylococcus aureus TarM, the wall teichoic acid α-glycosyltransferase. Proc. Natl. Acad. Sci. USA 2015, 112, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Bubeck Wardenburg, J.; Williams, W.A.; Missiakas, D. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc. Natl. Acad. Sci. USA 2006, 103, 13831–13836. [Google Scholar] [CrossRef] [PubMed]
- Shahmirzadi, S.V.; Nguyen, M.T.; Götz, F. Evaluation of Staphylococcus aureus Lipoproteins: Role in Nutritional Acquisition and Pathogenicity. Front. Microbiol. 2016, 7, 1404. [Google Scholar] [CrossRef] [PubMed]
- Dziarski, R.; Gupta, D. Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: A reevaluation. Infect. Immun. 2005, 73, 5212–5216. [Google Scholar] [CrossRef] [PubMed]
- Schwandner, R.; Dziarski, R.; Wesche, H.; Rothe, M.; Kirschning, C.J. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 1999, 274, 17406–17409. [Google Scholar] [CrossRef] [PubMed]
- Tawaratsumida, K.; Furuyashiki, M.; Katsumoto, M.; Fujimoto, Y.; Fukase, K.; Suda, Y.; Hashimoto, M. Characterization of N-terminal structure of TLR2-activating lipoprotein in Staphylococcus aureus. J. Biol. Chem. 2009, 284, 9147–9152. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Tawaratsumida, K.; Kariya, H.; Aoyama, K.; Tamura, T.; Suda, Y. Lipoprotein is a predominant Toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. Int. Immunol. 2006, 18, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Schmaler, M.; Jann, N.J.; Ferracin, F.; Landolt, L.Z.; Biswas, L.; Götz, F.; Landmann, R. Lipoproteins in Staphylococcus aureus mediate inflammation by TLR2 and iron-dependent growth in vivo. J. Immunol. 2009, 182, 7110–7118. [Google Scholar] [CrossRef] [PubMed]
- Zaba, L.C.; Krueger, J.G.; Lowes, M.A. Resident and “inflammatory” dendritic cells in human skin. J. Investig. Dermatol. 2009, 129, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Kapsenberg, M.L. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 2003, 3, 984–993. [Google Scholar] [PubMed]
- Stoll, H.; Dengjel, J.; Nerz, C.; Götz, F. Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect. Immun. 2005, 73, 2411–2423. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B. The function of TLR2 during staphylococcal diseases. Front. Cell. Infect. Microbiol. 2013, 2, 167. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Wu, Q.; Jiang, J.; Duan, J.; Wang, C.; Smith, M.D.; Lu, H.; Wang, Q.; Nagarkatti, P.; Fan, D. Characterization of sparstolonin B, a Chinese herb-derived compound, as a selective Toll-like receptor antagonist with potent anti-inflammatory properties. J. Biol. Chem. 2011, 286, 26470–26479. [Google Scholar] [CrossRef] [PubMed]
- Clatworthy, M.R.; Aronin, C.E.; Mathews, R.J.; Morgan, N.Y.; Smith, K.G.; Germain, R.N. Immune complexes stimulate CCR7-dependent dendritic cell migration to lymph nodes. Nat. Med. 2014, 20, 1458–1463. [Google Scholar] [PubMed]
- Acton, S.E.; Astarita, J.L.; Malhotra, D.; Lukacs-Kornek, V.; Franz, B.; Hess, P.R.; Jakus, Z.; Kuligowski, M.; Fletcher, A.L.; Elpek, K.G.; et al. Podoplanin-rich stromal networks induce dendritic cell motility via activation of the C-type lectin receptor CLEC-2. Immunity 2012, 24, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Bröker, B.M.; Mrochen, D.; Péton, V. The T Cell Response to Staphylococcus aureus. Pathogens 2016, 5, 31. [Google Scholar] [CrossRef]
- Shen, W.; Li, W.; Hixon, J.A.; Bouladoux, N.; Belkaid, Y.; Dzutzev, A.; Durum, S.K. Adaptive immunity to murine skin commensals. Proc. Natl. Acad. Sci. USA 2014, 111, E2977–E2986. [Google Scholar] [PubMed]
- Leung, D.Y.; Gately, M.; Trumble, A.; Ferguson-Darnell, B.; Schlievert, P.M.; Picker, L.J. Bacterial superantigens induce T cell expression of the skin-selective homing receptor, the cutaneous lymphocyte-associated antigen, via stimulation of interleukin 12 production. J. Exp. Med. 1995, 181, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.Z.; Evrard, M.; Ng, L.G. Lights, camera, and action: Vertebrate skin sets the stage for immune cell interaction with arthropod-vectored pathogens. Front. Immunol. 2013, 4, 286. [Google Scholar] [CrossRef] [PubMed]
- Münz, C.; Steinman, R.M.; Fujii, S. Dendritic cell maturation by innate lymphocytes: Coordinated stimulation of innate and adaptive immunity. J. Exp. Med. 2005, 202, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Clausen, B.E.; Stoitzner, P. Functional Specialization of Skin Dendritic Cell Subsets in Regulating T Cell Responses. Front. Immunol. 2015, 6, 534. [Google Scholar] [CrossRef] [PubMed]
- Henri, S.; Poulin, L.F.; Tamoutounour, S.; Ardouin, L.; Guilliams, M.; de Bovis, B.; Devilard, E.; Viret, C.; Azukizawa, H.; Kissenpfennig, A.; et al. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J. Exp. Med. 2010, 207, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Lien, E.; Ingalls, R.R.; Tuomanen, E.; Dziarski, R.; Golenbock, D. Cutting edge: Recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol. 1999, 163, 1–5. [Google Scholar] [PubMed]
- Li, C.; Li, H.; Jiang, Z.; Zhang, T.; Wang, Y.; Li, Z.; Wu, Y.; Ji, S.; Xiao, S.; Ryffel, B.; et al. Interleukin-33 increases antibacterial defense by activation of inducible nitric oxide synthase in skin. PLoS Pathog. 2014, 10, e1003918. [Google Scholar] [CrossRef] [PubMed]
- Weidenmaier, C.; Peschel, A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 2008, 6, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Lacey, K.A.; Geoghegan, J.A.; McLoughlin, R.M. The Role of Staphylococcus aureus Virulence Factors in Skin Infection and Their Potential as Vaccine Antigens. Pathogens 2016, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Oscherwitz, J.; Cease, K.B.; Chan, S.M.; Muñoz-Planillo, R.; Hasegawa, M.; Villaruz, A.E.; Cheung, G.Y.; McGavin, M.J.; Travers, J.B.; et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature 2013, 503, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, L.; Hijnen, D.J.; Sellman, B.R.; Mustelin, T.; Sleeman, M.A.; May, R.D.; Strickland, I. The complex biology and contribution of Staphylococcus aureus in atopic dermatitis, current and future therapies. Br. J. Dermatol. 2017, 177, 63–71. [Google Scholar] [PubMed]
- Nemoto, Y.; Kanai, T.; Kameyama, K.; Shinohara, T.; Sakamoto, N.; Totsuka, T.; Okamoto, R.; Tsuchiya, K.; Nakamura, T.; Sudo, T.; et al. Long-lived colitogenic CD4+ memory T cells residing outside the intestine participate in the perpetuation of chronic colitis. J. Immunol. 2009, 183, 5059–5068. [Google Scholar] [CrossRef] [PubMed]
- Haniffa, M.; Gunawan, M.; Jardine, L. Human skin dendritic cells in health and disease. J. Dermatol. Sci. 2015, 77, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Forrest, L.; Kepler, T.B.; Parker, I.; Cahalan, M.D. Selective and site-specific mobilization of dermal dendritic cells and Langerhans cells by Th1- and Th2-polarizing adjuvants. Proc. Natl. Acad. Sci. USA 2010, 107, 8334–8339. [Google Scholar] [CrossRef] [PubMed]
- Nizza, S.T.; Campbell, J.J. CD11b+ migratory dendritic cells mediate CD8 T cell cross-priming and cutaneous imprinting after topical immunization. PLoS ONE 2014, 9, e91054. [Google Scholar] [CrossRef]
- Bedoui, S.; Whitney, P.G.; Waithman, J.; Eidsmo, L.; Wakim, L.; Caminschi, I.; Allan, R.S.; Wojtasiak, M.; Shortman, K.; Carbone, F.R.; et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat. Immunol. 2009, 10, 488–495. [Google Scholar] [PubMed]
- Singh, T.P.; Zhang, H.H.; Borek, I.; Wolf, P.; Hedrick, M.N.; Singh, S.P.; Kelsall, B.L.; Clausen, B.E.; Farber, J.M. Monocyte-derived inflammatory Langerhans cells and dermal dendritic cells mediate psoriasis-like inflammation. Nat. Commun. 2016, 7, 13581. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Kawamura, T.; Higuchi, M.; Kobayashi, T.; Yoshita-Takahashi, M.; Yamazaki, M.; Abe, M.; Sakimura, K.; Kanda, Y.; Kawamura, H.; et al. RASAL3, a novel hematopoietic RasGAP protein, regulates the number and functions of NKT cells. Eur. J. Immunol. 2015, 45, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Kukutsch, N.; Ogilvie, A.L.; Rössner, S.; Koch, F.; Romani, N.; Schuler, G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 1999, 223, 77–92. [Google Scholar] [CrossRef]
- Suurmond, J.; van Heemst, J.; van Heiningen, J.; Dorjée, A.L.; Schilham, M.W.; van der Beek, F.B.; Huizinga, T.W.; Schuerwegh, A.J.; Toes, R.E. Communication between human mast cells and CD4+T cells through antigen-dependent interactions. Eur. J. Immunol. 2013, 43, 1758–1768. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, S.; F. Quadery, A. Staphylococcus aureus Lipoprotein Induces Skin Inflammation, Accompanied with IFN-γ-Producing T Cell Accumulation through Dermal Dendritic Cells. Pathogens 2018, 7, 64. https://doi.org/10.3390/pathogens7030064
Saito S, F. Quadery A. Staphylococcus aureus Lipoprotein Induces Skin Inflammation, Accompanied with IFN-γ-Producing T Cell Accumulation through Dermal Dendritic Cells. Pathogens. 2018; 7(3):64. https://doi.org/10.3390/pathogens7030064
Chicago/Turabian StyleSaito, Suguru, and Ali F. Quadery. 2018. "Staphylococcus aureus Lipoprotein Induces Skin Inflammation, Accompanied with IFN-γ-Producing T Cell Accumulation through Dermal Dendritic Cells" Pathogens 7, no. 3: 64. https://doi.org/10.3390/pathogens7030064
APA StyleSaito, S., & F. Quadery, A. (2018). Staphylococcus aureus Lipoprotein Induces Skin Inflammation, Accompanied with IFN-γ-Producing T Cell Accumulation through Dermal Dendritic Cells. Pathogens, 7(3), 64. https://doi.org/10.3390/pathogens7030064