EBV-Positive Lymphoproliferations of B- T- and NK-Cell Derivation in Non-Immunocompromised Hosts



Abstract
1. Introduction
2. EBV Associated B-Cell Lymphoproliferative Disorders
2.1. Infectious Mononucleosis
2.1.1. Clinical Features
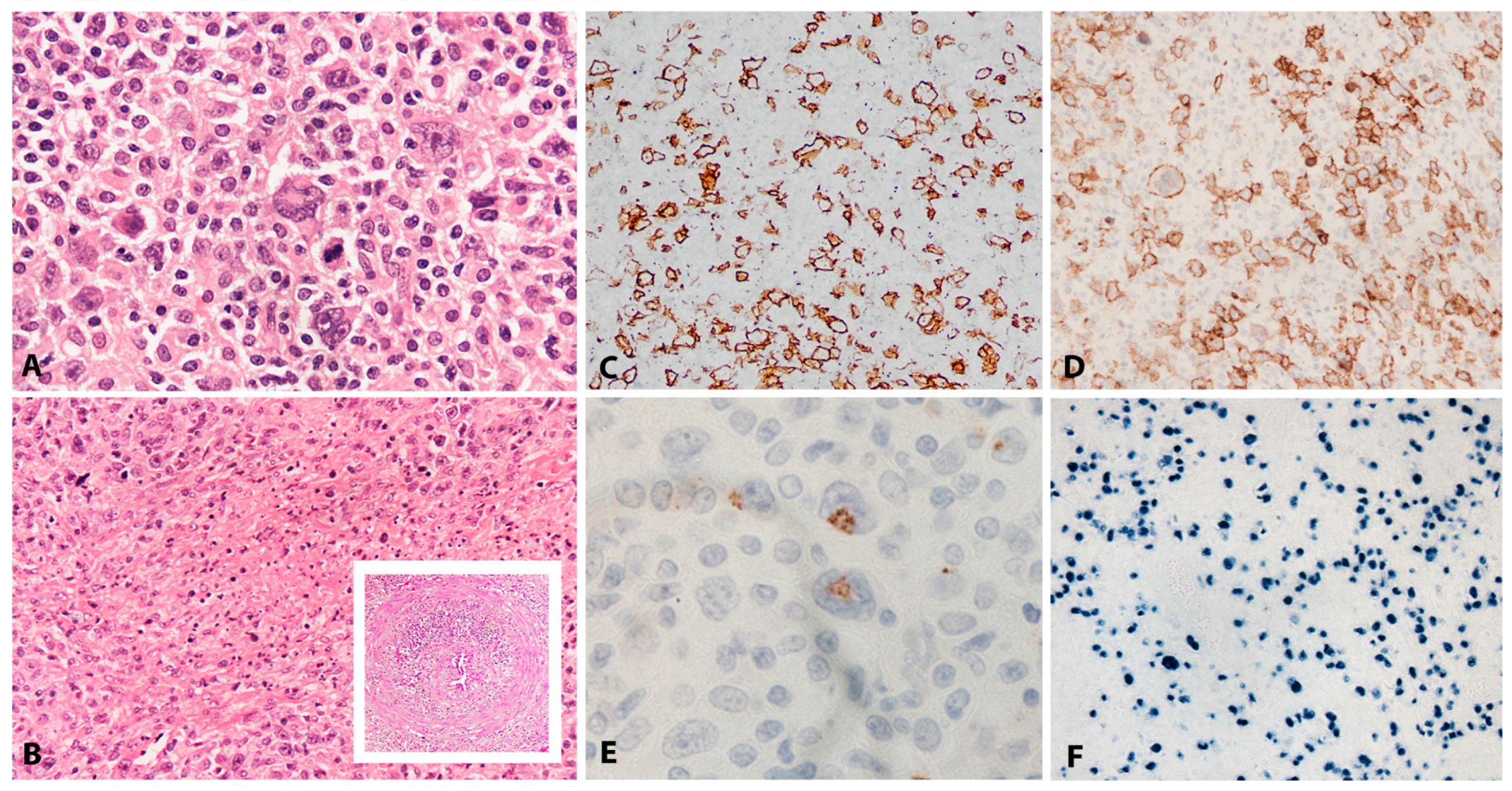
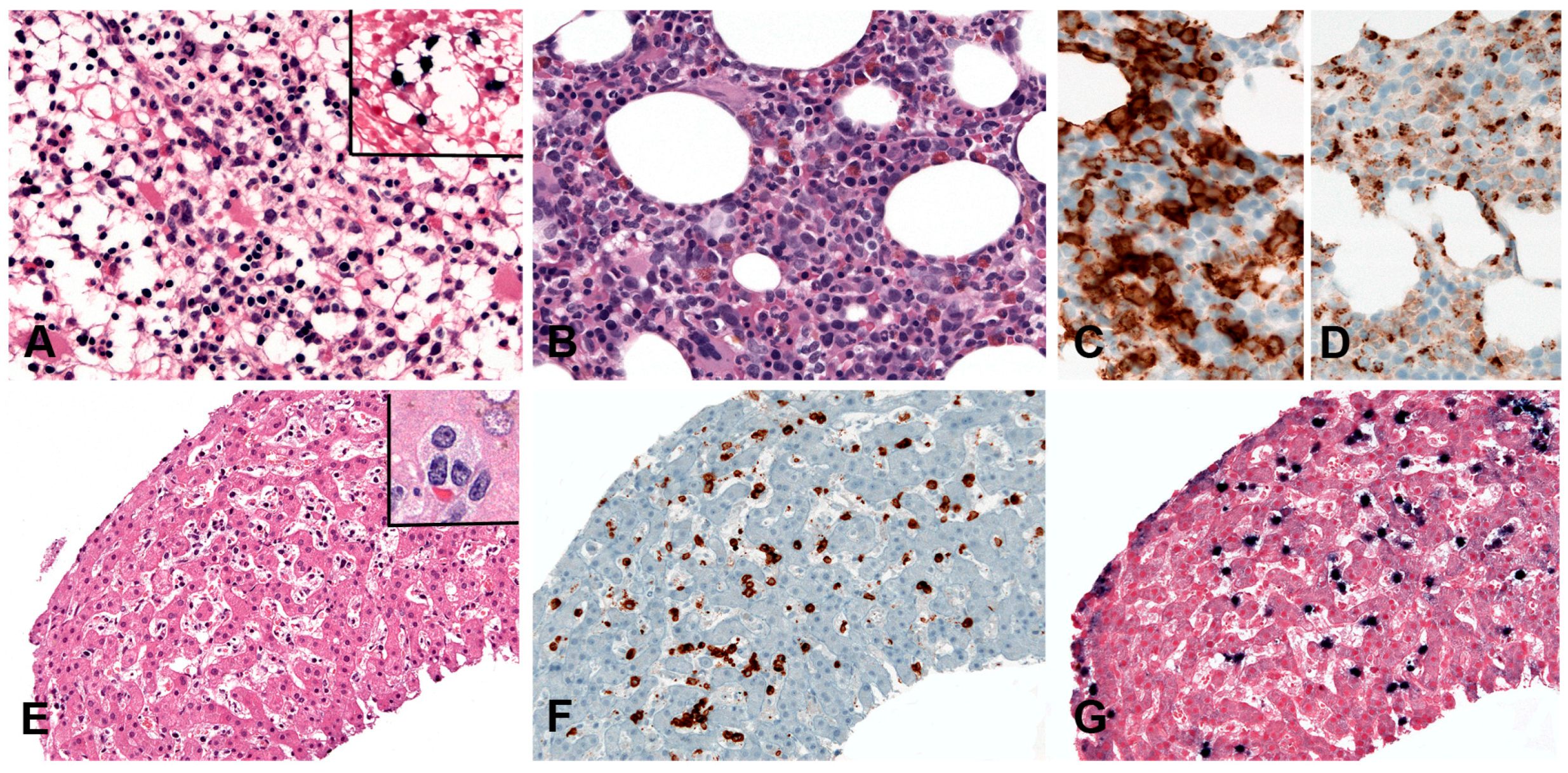
2.1.2. Morphology
2.1.3. Pathogenesis and Molecular Findings
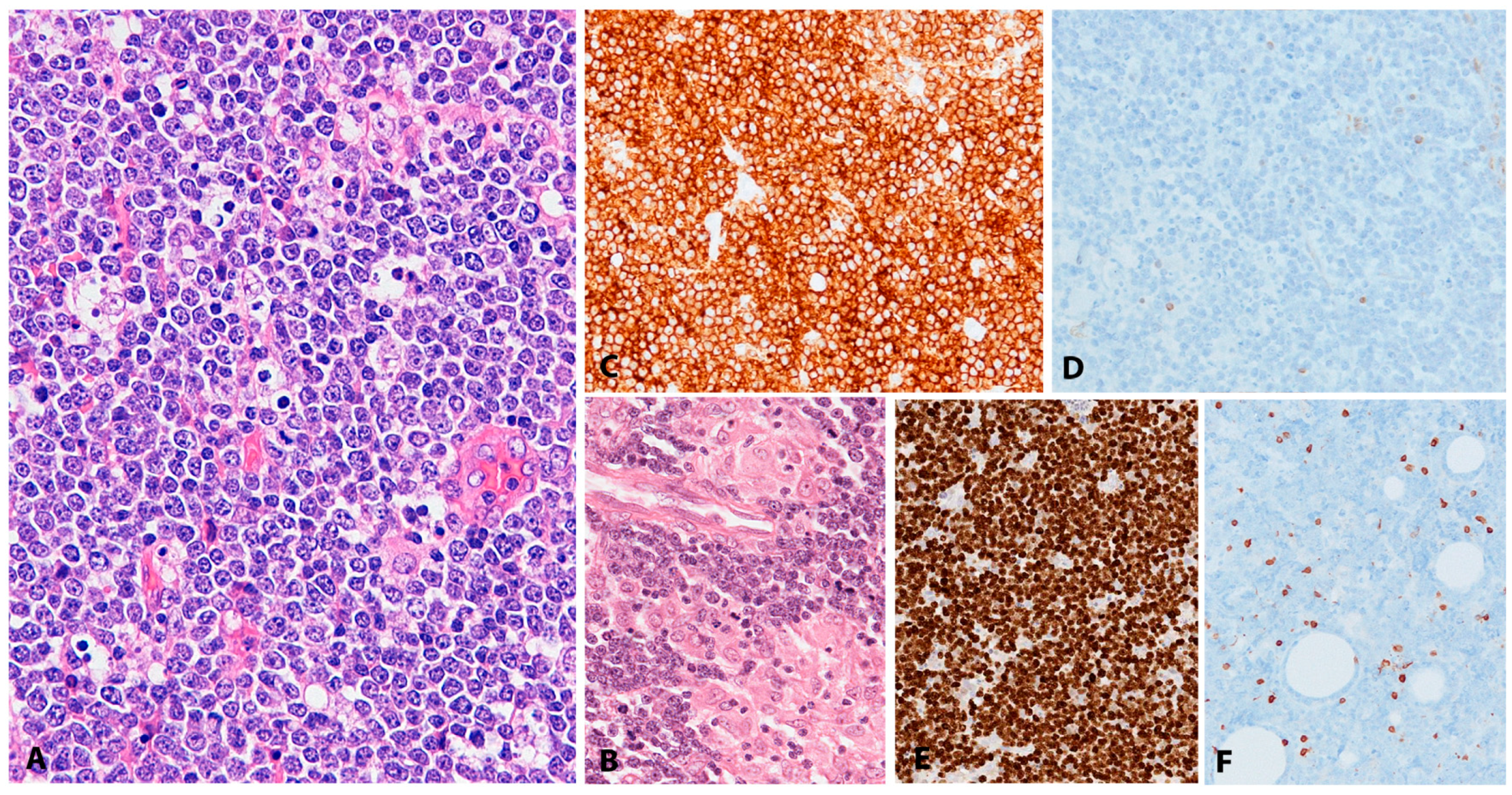
2.2. EBV Positive Diffuse Large B-Cell Lymphoma, Not Otherwise Specified
2.2.1. Clinical Features
2.2.2. Morphology
2.2.3. Pathogenesis and Molecular Findings
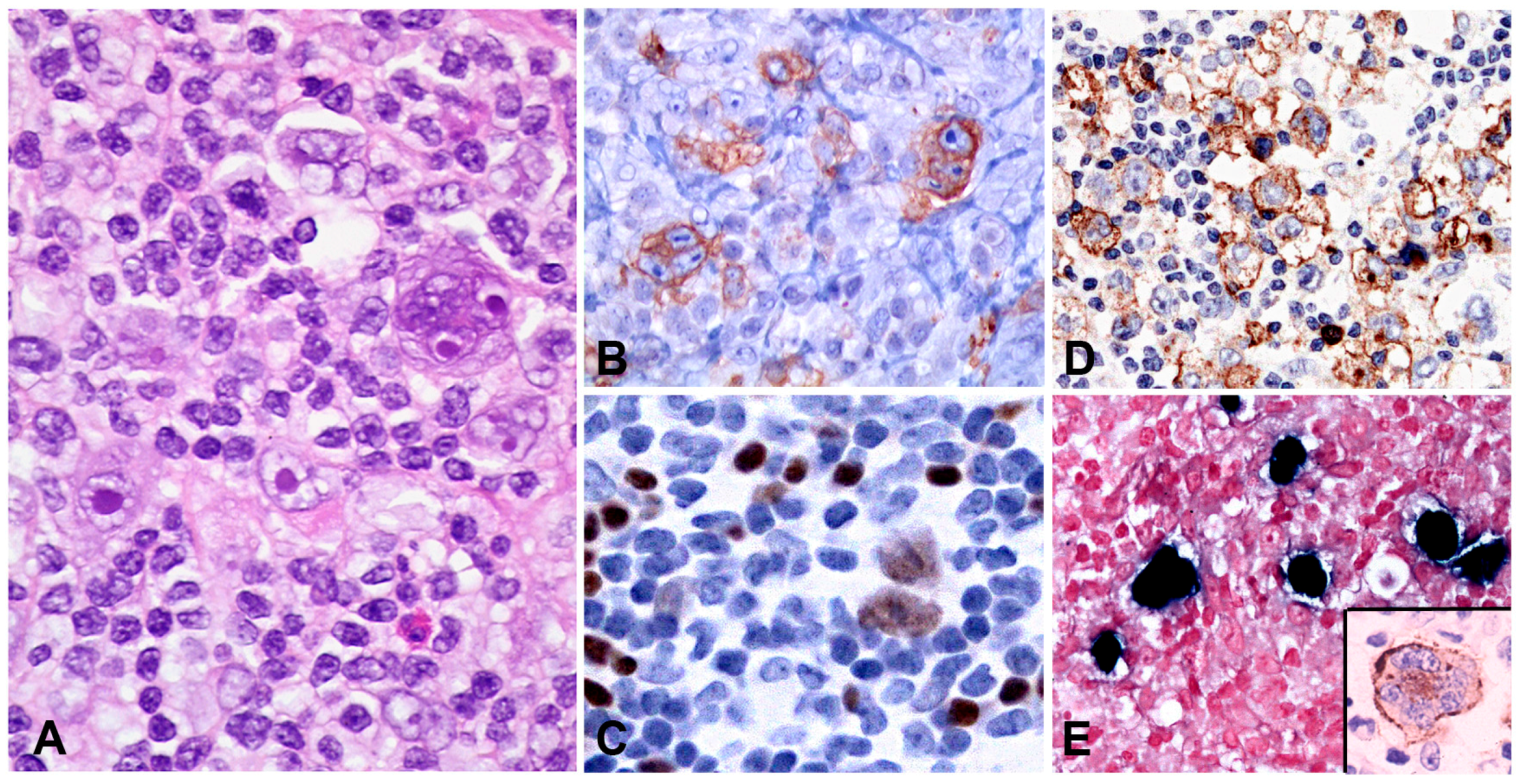
2.3. EBV Positive Mucocutaneous Ulcer
2.3.1. Clinical Features
2.3.2. Morphology
2.3.3. Pathogenesis and Molecular Findings
2.4. Diffuse Large B-Cell Lymphoma Associated with Chronic Inflammation
2.4.1. Clinical Presentation
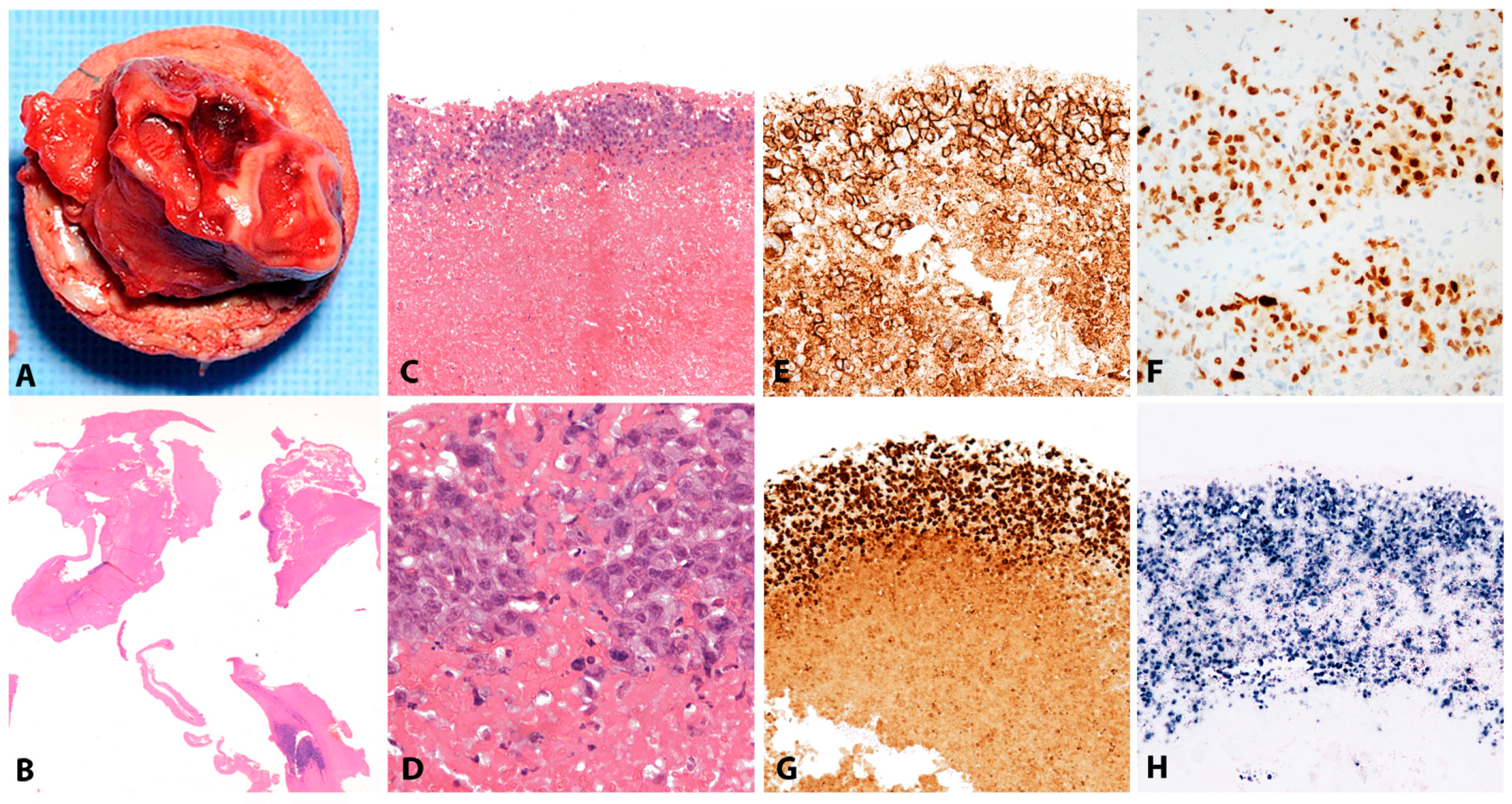
2.4.2. Morphology
2.4.3. Pathogenesis and Molecular Findings
2.5. Fibrin Associated Diffuse Large B-Cell Lymphoma
2.5.1. Clinical Presentation
2.5.2. Morphology
2.5.3. Pathogenesis and Molecular Findings
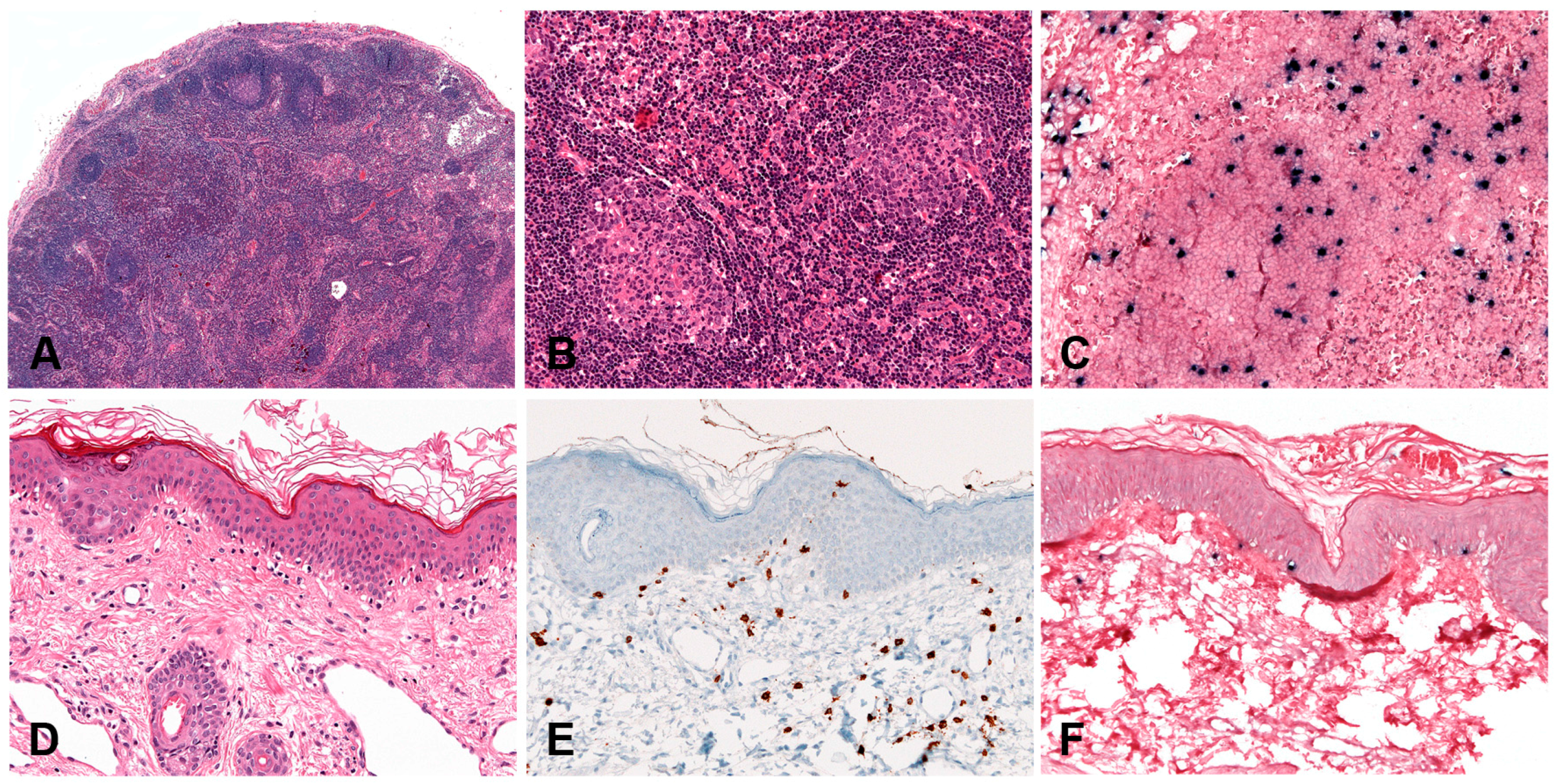
2.6. Lymphomatoid Granulomatosis
2.6.1. Clinical Presentation
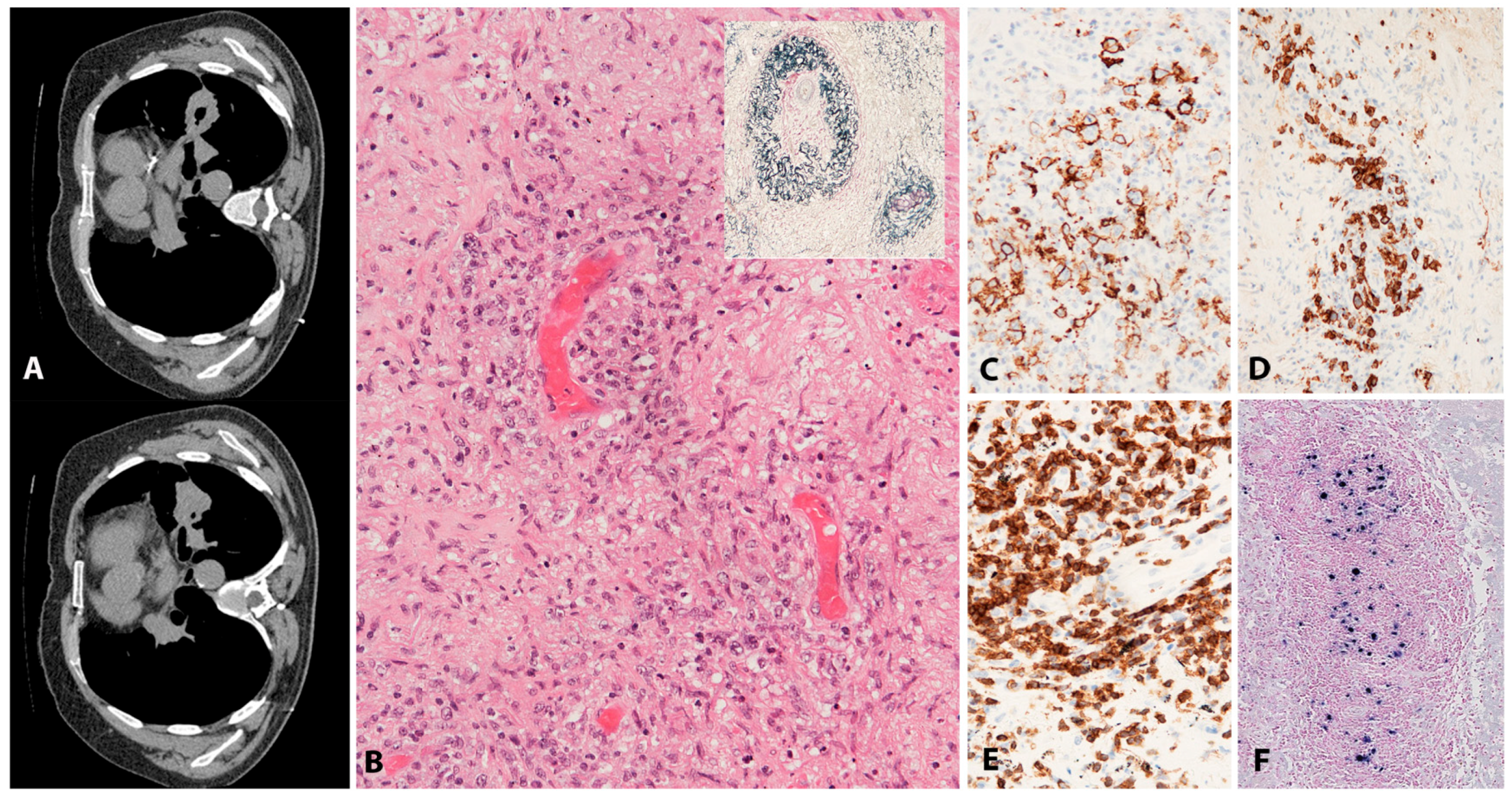
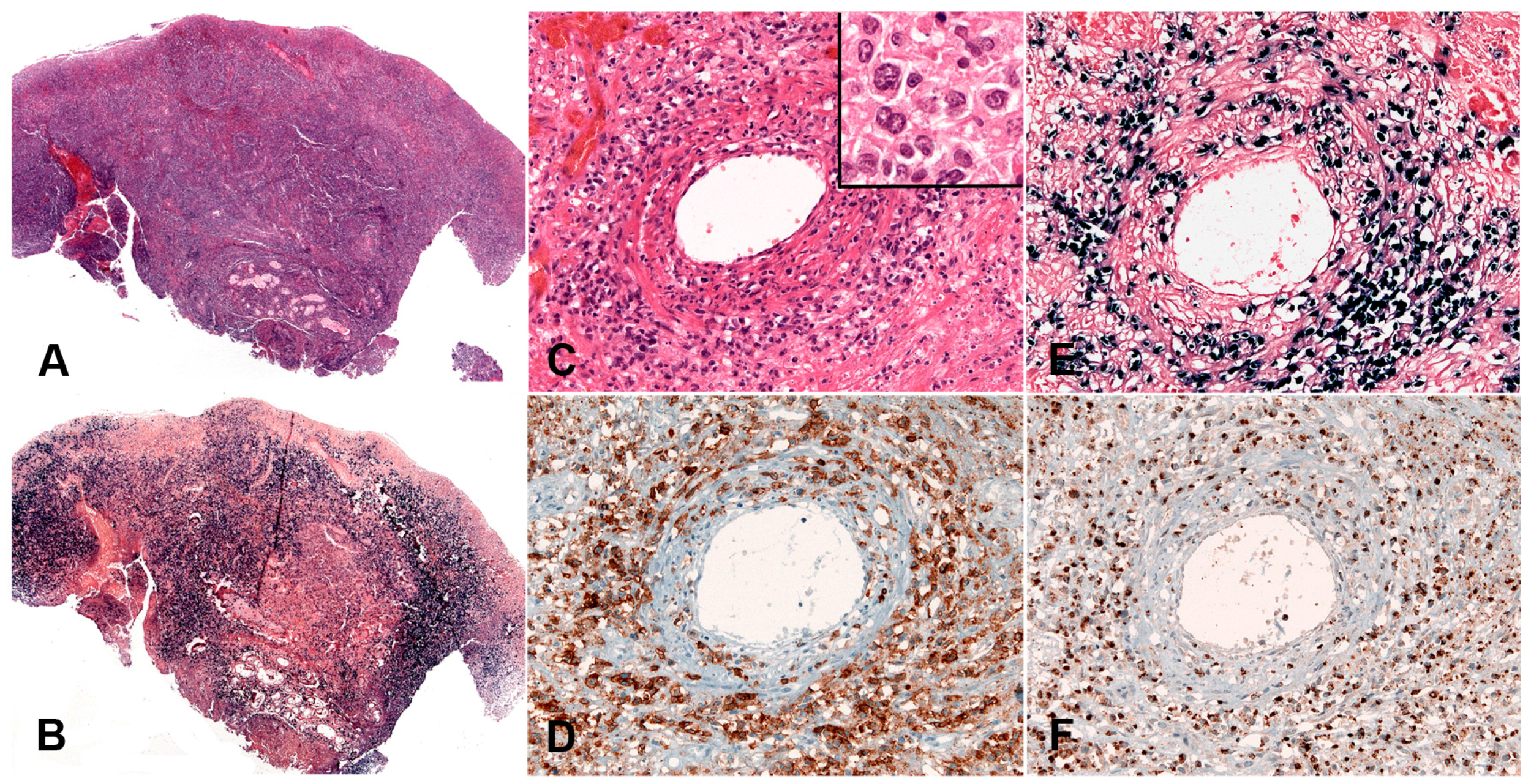
2.6.2. Morphology
2.6.3. Pathogenesis and Molecular Findings
3. Other B-Cell Lymphomas That Can Be EBV-Associated
3.1. Plasmablastic Lymphoma
3.1.1. Clinical Features
3.1.2. Morphology
3.1.3. Pathogenesis and Molecular Findings
3.2. Burkitt Lymphoma
3.2.1. Clinical Features
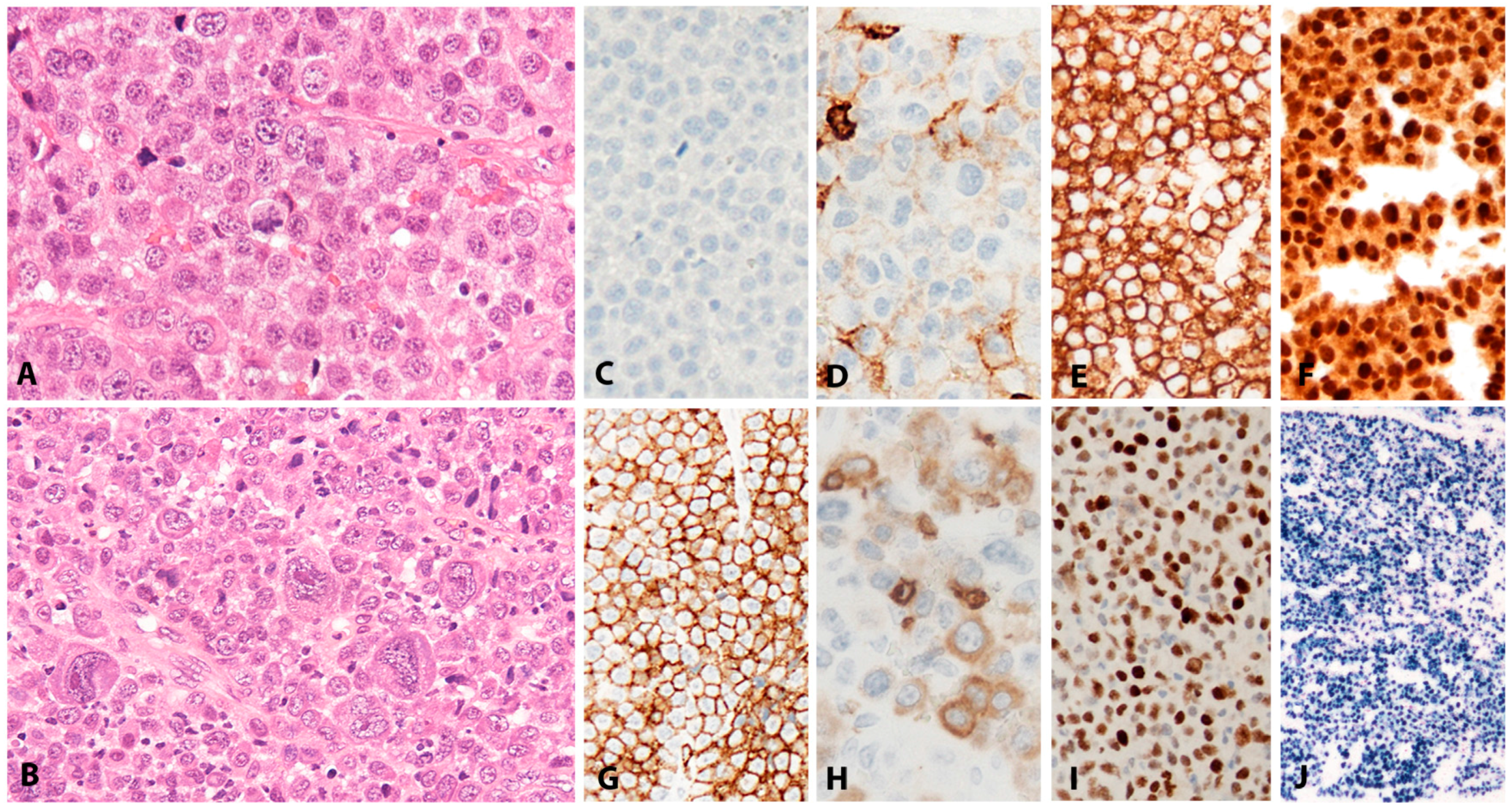
3.2.2. Morphology
3.2.3. Pathogenesis and Molecular Findings
3.3. Classic Hodgkin Lymphoma
3.3.1. Clinical Features
3.3.2. Morphology
3.3.3. Pathogenesis and Molecular Findings
4. Other B-Cell Lymphomas Rarely EBV-Associated
4.1. Chronic Lymphocytic Leukaemia
4.2. Plasma Cell Myeloma
5. EBV-Associated T and NK-Cell Lymphoproliferative Diseases
5.1. Chronic Active EBV Infection of T and NK Cell Type, Systemic Form
5.1.1. Clinical Features
5.1.2. Morphology
5.1.3. Pathogenesis and Molecular Findings
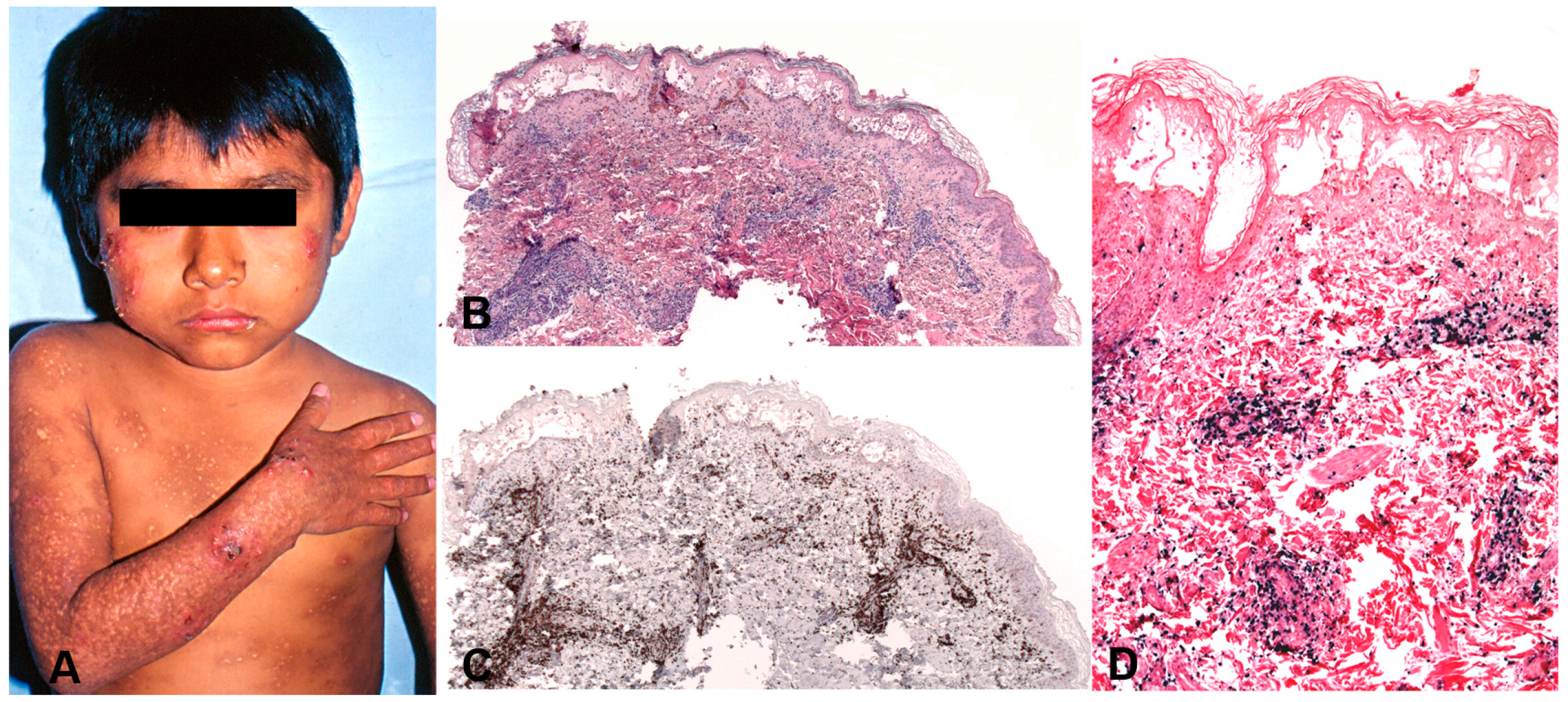
5.2. Hydroa Vacciniforme-Like Lymphoproliferative Disease
5.2.1. Clinical Features
5.2.2. Morphology
5.2.3. Pathogenesis and Molecular Findings
5.3. Severe Mosquito Bite Allergy
5.3.1. Clinical Features
5.3.2. Morphology
5.3.3. Pathogenesis and Molecular Findings
5.4. Systemic EBV-Positive T-Cell Lymphoma of Childhood
5.4.1. Clinical Features
5.4.2. Morphology
5.4.3. Pathogenesis and Molecular Findings
5.5. Aggressive NK-Cell Leukaemia
5.5.1. Clinical Features
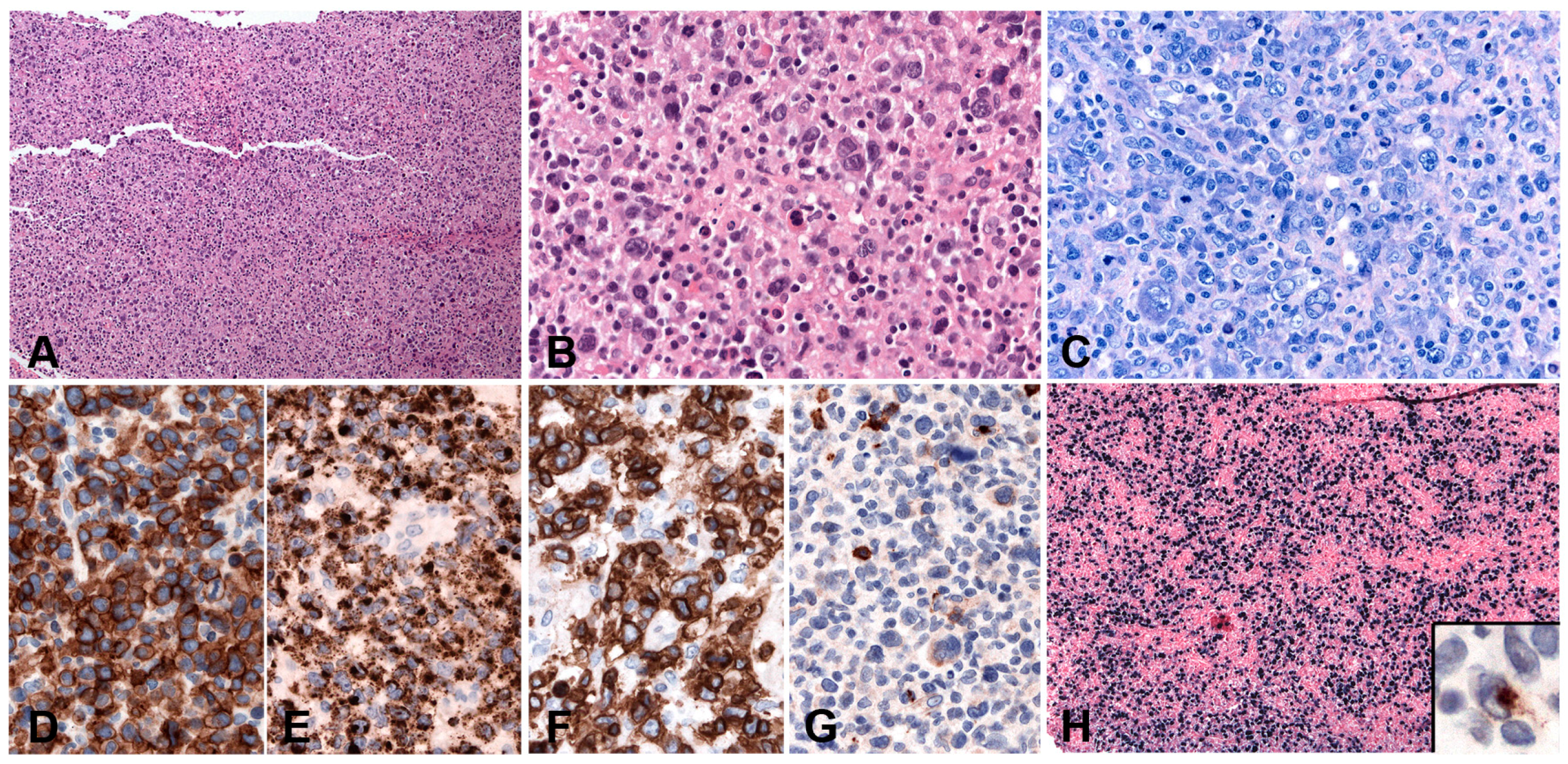
5.5.2. Morphology
5.5.3. Pathogenesis and Molecular Findings
5.6. Extranodal NK/T-Cell Lymphoma, Nasal Type
5.6.1. Clinical Features
5.6.2. Morphology
5.6.3. Pathogenesis and Molecular Findings
5.7. Primary EBV-Positive Nodal Peripheral T- or NK-Cell Lymphoma
5.7.1. Clinical Features
5.7.2. Morphology
5.7.3. Pathogenesis and Molecular Findings
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A. EBV persistence-introducing the virus. Curr. Top. Microbiol. 2015, 390, 151–209. [Google Scholar] [CrossRef]
- Dunmire, S.K.; Hogquist, K.A.; Balfour, H.H. Infectious mononucleosis. Curr. Top. Microbiol. 2015, 390, 211–240. [Google Scholar] [CrossRef]
- Abbott, R.J.M.; Quinn, L.L.; Leese, A.M.; Scholes, H.M.; Pachnio, A.; Rickinson, A.B. Cd8(+) T cell responses to lytic EBV infection: Late antigen specificities as subdominant components of the total response. J. Immunol. 2013, 191, 5398–5409. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Taylor, G.S.; Sauce, D.; Rickinson, A.B. Cellular responses to viral infection in humans: Lessons from Epstein-Barr virus. Annu. Rev. Immunol. 2007, 25, 587–617. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Kimura, H.; Nakamura, S.; Ko, Y.H.; Jaffe, E.S. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: A status report and summary of an international meeting, 8–9 September 2008. Ann. Oncol. 2009, 20, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. Mech. 2014, 9, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E. How do viruses trick B cells into becoming lymphomas? Curr. Opin. Hematol. 2014, 21, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Dolcetti, R.; Dal Col, J.; Martorelli, D.; Carbone, A.; Klein, E. Interplay among viral antigens, cellular pathways and tumor microenvironment in the pathogenesis of EBV-driven lymphomas. Semin. Cancer Biol. 2013, 23, 441–456. [Google Scholar] [CrossRef] [PubMed]
- Linke-Serinsoz, E.; Fend, F.; Quintanilla-Martinez, L. Human immunodeficiency virus (HIV) and Epstein-Barr virus (EBV) related lymphomas, pathology view point. Semin. Diagn. Pathol. 2017, 34, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. Who Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC: Lyon, France, 2017. [Google Scholar]
- Altschuler, E.L. Detection of Epstein-Barr virus in invasive breast cancers. J. Natl. Cancer Inst. 1999, 91, 2126–2127. [Google Scholar] [CrossRef] [PubMed]
- Henle, G.; Henle, W.; Diehl, V. Relation of Burkitts tumor-associated herpes-type virus to infectious mononucleosis. Proc. Natl. Acad. Sci. USA 1968, 59, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, K.; Sullivan, J.L. Infectious mononucleosis. N. Engl. J. Med. 2010, 363, 1993–2000. [Google Scholar] [CrossRef] [PubMed]
- Louissaint, A.; Ferry, J.A.; Soupir, C.P.; Hasserjian, R.P.; Harris, N.L.; Zukerberg, L.R. Infectious mononucleosis mimicking lymphoma: Distinguishing morphological and immunophenotypic features. Mod. Pathol. 2012, 25, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Vockerodt, M.; Yap, L.F.; Shannon-Lowe, C.; Curley, H.; Wei, W.B.; Vrzalikova, K.; Murray, P.G. The Epstein-Barr virus and the pathogenesis of lymphoma. J. Pathol. 2015, 235, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A.; Hawkins, J.B.; Tracy, S.I.; Shapiro, M. The pathogenesis of Epstein-Barr virus persistent infection. Curr. Opin. Virol. 2013, 3, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Oyama, T.; Yamamoto, K.; Asano, N.; Oshiro, A.; Suzuki, R.; Kagami, Y.; Morishima, Y.; Takeuchi, K.; Izumo, T.; Mori, S.; et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: A study of 96 patients. Clin. Cancer Res. 2007, 13, 5124–5132. [Google Scholar] [CrossRef] [PubMed]
- Dojcinov, S.D.; Venkataraman, G.; Pittaluga, S.; Wlodarska, I.; Schrager, J.A.; Raffeld, M.; Hills, R.K.; Jaffe, E.S. Age-related EBV-associated lymphoproliferative disorders in the western population: A spectrum of reactive lymphoid hyperplasia and lymphoma. Blood 2011, 117, 4726–4735. [Google Scholar] [CrossRef] [PubMed]
- Oyama, T.; Ichimura, K.; Suzuki, R.; Suzumiya, J.; Ohshima, K.; Yatabe, Y.; Yokoi, T.; Kojima, M.; Kamiya, Y.; Taji, H.; et al. Senile EBV plus B-cell lymphoproliferative disorders—A clinicopathologic study of 22 patients. Am. J. Surg. Pathol. 2003, 27, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. Who Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC: Lyon, France, 2008. [Google Scholar]
- Nicolae, A.; Pittaluga, S.; Abdullah, S.; Steinberg, S.M.; Pham, T.A.; Davies-Hill, T.; Xi, L.Q.; Raffeld, M.; Jaffe, E.S. EBV-positive large B-cell lymphomas in young patients: A nodal lymphoma with evidence for a tolerogenic immune environment. Blood 2015, 126, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Uccini, S.; Al-Jadiry, M.F.; Scarpino, S.; Ferraro, D.; Alsaadawi, A.R.; Al-Darraji, A.F.; Moleti, M.L.; Testi, A.M.; Al-Hadad, S.A.; Ruco, L. Epstein-Barr virus-positive diffuse large B-cell lymphoma in children: A disease reminiscent of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly. Hum. Pathol. 2015, 46, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; De Matteo, E.; Narbaitz, M.; Carreno, F.A.; Preciado, M.V.; Chabay, P.A. Epstein-Barr virus presence in pediatric diffuse large B-cell lymphoma reveals a particular association and latency patterns: Analysis of viral role in tumor microenvironment. Int. J. Cancer 2013, 132, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Hoeller, S.; Tzankov, A.; Pileri, S.A.; Went, P.; Dirnhofer, S. Epstein-Barr virus positive diffuse large B-cell lymphoma in elderly patients is rare in western populations. Hum. Pathol. 2010, 41, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Hofscheier, A.; Ponciano, A.; Bonzheim, I.; Adam, P.; Lome-Maldonado, C.; Vela, T.; Cortes, E.; Ortiz-Hidalgo, C.; Fend, F.; Quintanilla-Martinez, L. Geographic variation in the prevalence of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: A comparative analysis of a mexican and a german population. Mod. Pathol. 2011, 24, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Papathomas, T.G.; Medeiros, L.J.; Young, K.H. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood 2013, 122, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.H.; Lu, T.X.; Tian, T.; Wang, L.; Fan, L.; Xu, J.; Zhang, R.; Gong, Q.X.; Zhang, Z.H.; Li, J.Y.; et al. Epstein-Barr virus (EBV) DNA in whole blood as a superior prognostic and monitoring factor than EBV-encoded small RNA in situ hybridization in diffuse large B-cell lymphoma. Clin. Microbiol. Infect. 2015, 21, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Ko, Y.H.; Kim, S.J.; Kim, W.S. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: A concise review and update. Curr. Opin. Oncol. 2015, 27, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Narbaitz, M.; Metrebian, F.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Epstein-Barr virus-positive diffuse large B-cell lymphoma association is not only restricted to elderly patients. Int. J. Cancer 2014, 135, 2816–2824. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Yamamoto, K.; Tamaru, J.I.; Oyama, T.; Ishida, F.; Ohshima, K.; Yoshino, T.; Nakamura, N.; Mori, S.; Yoshie, O.; et al. Age-related Epstein-Barr virus (EBV)-associated B-cell lymphoproliferative disorders: Comparison with EBV-positive classic Hodgkin lymphoma in elderly patients. Blood 2009, 113, 2629–2636. [Google Scholar] [CrossRef] [PubMed]
- Montes-Moreno, S.; Odqvist, L.; Diaz-Perez, J.A.; Lopez, A.B.; de Villambrosia, S.G.; Mazorra, F.; Castillo, M.E.; Lopez, M.; Pajares, R.; Garcia, J.F.; et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod. Pathol. 2012, 25, 968–982. [Google Scholar] [CrossRef] [PubMed]
- Olsson, J.; Wikby, A.; Johansson, B.; Lofgren, S.; Nilsson, B.O.; Ferguson, F.G. Age-related change in peripheral blood T-lymphocyte subpopulations and cytomegalovirus infection in the very old: The swedish longitudinal OCTO immune study. Mech. Ageing Dev. 2000, 121, 187–201. [Google Scholar] [CrossRef]
- Vescovini, R.; Telera, A.; Fagnoni, F.F.; Biasini, C.; Medici, M.C.; Valcavi, P.; di Pede, P.; Lucchini, G.; Zanlari, L.; Passeri, G.; et al. Different contribution of EBV and cmv infections in very long-term carriers to age-related alterations of CD8(+) T cells. Exp. Gerontol. 2004, 39, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, D.; Velez, G.; Orfao, A.; Herrera, M.V.; Solano, J.; Olaya, M.; Uribe, A.M.; Saavedra, C.; Duarte, M.; Rodriguez, M.; et al. Epstein-Barr virus-specific CD8(+) t lymphocytes from diffuse large B cell lymphoma patients are functionally impaired. Clin. Exp. Immunol. 2015, 182, 173–183. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Ramer, P.C.; Naresh, K.N.; Meixlsperger, S.; Pinaud, L.; Rooney, C.; Savoldo, B.; Coutinho, R.; Bodor, C.; Gribben, J.; et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Investig. 2012, 122, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- Gibier, J.B.; Bouchindhomme, B.; Dubois, R.; Hivert, B.; Grardel, N.; Copin, M.C. Coexistence of age-related EBV-associated follicular hyperplasia and large B-cell EBV plus lymphoma of the elderly. Two distinct features of the same T-cell dysfunction related to senescence? Pathol. Res. Pract. 2017, 213, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Kunitomi, A.; Hasegawa, Y.; Asano, N.; Kato, S.; Tokunaga, T.; Miyata, Y.; Iida, H.; Nagai, H. EBV-positive reactive hyperplasia progressed into EBV-positive diffuse large B-cell lymphoma of the elderly over a 6-year period. Intern. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- De la Hera Magallanes, A.I.; Montes-Moreno, S.; Hernandez, S.G.; Hernandez-Leon, C.N.; Lopez, M.; Pajares, R.; Pinilla, S.M.; Piris, M.A. Early phase of Epstein-Barr virus (EBV)-positive diffuse large B cell lymphoma of the elderly mimicking EBV-positive reactive follicular hyperplasia. Histopathology 2011, 59, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Park, S.; Ju, H.; Ha, S.Y.; Sohn, I.; Jo, J.; Do, I.G.; Min, S.; Kim, S.J.; Kim, W.S.; et al. Integrated copy number and gene expression profiling analysis of Epstein-Barr virus-positive diffuse large B-cell lymphoma. Genes Chromosome Cancer 2015, 54, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Karube, K.; Yamamoto, K.; Takizawa, J.; Tsuzuki, S.; Yatabe, Y.; Kanda, T.; Katayama, M.; Ozawa, Y.; Ishitsuka, K.; et al. Gene expression profiling of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly reveals alterations of characteristic oncogenetic pathways. Cancer Sci. 2014, 105, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Li, L.; Xu-Monette, Z.Y.; Visco, C.; Tzankov, A.; Manyam, G.C.; Dybkaer, K. Prevalence and clinical implications of Epstein-Barr virus infection in de novo diffuse large B-cell lymphoma in western countries. Clin. Cancer Res. 2014, 20, 2338–2349. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, N.; Gebauer, J.; Hardel, T.T.; Bernard, V.; Biersack, H.; Lehnert, H.; Rades, D.; Feller, A.C.; Thorns, C. Prevalence of targetable oncogenic mutations and genomic alterations in Epstein-Barr virus-associated diffuse large B-cell lymphoma of the elderly. Leuk. Lymphoma 2015, 56, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Dojcinov, S.D.; Venkataraman, G.; Raffeld, M.; Pittaluga, S.; Jaffe, E.S. EBV positive mucocutaneous ulcer-a study of 26 cases associated with various sources of immunosuppression. Am. J. Surg. Pathol. 2010, 34, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Pugh, M.R.; Morgan, M.; Dojcinov, S.D. Ipilimumab induced colitis: Is the Epstein Barr virus (EBV) implicated? J. Pathol. 2016, 240, 23. [Google Scholar]
- Iuchi, K.; Ichimiya, A.; Akashi, A.; Mizuta, T.; Lee, Y.E.; Tada, H.; Mori, T.; Sawamura, K.; Lee, Y.S.; Furuse, K.; et al. Non-hodgkins-lymphoma of the pleural cavity developing from long-standing pyothorax. Cancer 1987, 60, 1771–1775. [Google Scholar] [CrossRef]
- Ascani, S.; Piccioli, M.; Poggi, S.; Briskomatis, A.; Bolis, G.B.; Liberati, F.; Frongillo, R.; Caramatti, C.; Fraternali-Orcioni, G.; Gamberi, B.; et al. Pyothorax-associated lymphoma: Description of the first two cases detected in italy. Ann. Oncol. 1997, 8, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Capron, F.; Liguorybrunaud, M.D.; Defrejacques, C.; Pluot, M.; Diebold, J. Epstein-Barr virus-associated primary malignant-lymphomas of the pleural cavity occurring in longstanding pleural chronic inflammation. Hum. Pathol. 1994, 25, 1314–1318. [Google Scholar] [CrossRef]
- Petitjean, B.; Jardin, F.; Joly, B.; Martin-Garcia, N.; Tilly, H.; Picquenot, J.M.; Briere, J.; Danel, C.; Mehaut, S.; Abd-Al-Samad, I.; et al. Pyothorax-associated lymphoma—A peculiar clinicopathologic entity derived from B cells at late stage of differentiation and with occasional aberrant dual B- and T-cell phenotype. Am. J. Surg. Pathol. 2002, 26, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, S.; Yao, M.; Hoshida, Y.; Yamamoto, S.; Iuchi, K.; Aozasa, K. Pyothorax-associated lymphoma: A review of 106 cases. J. Clin. Oncol. 2002, 20, 4255–4260. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, H.; Ota, Y.; Kami, M.; Takeuchi, K.; Suzuki, R.; Matsuo, K.; Matsumura, T.; Yuji, K.; Kishi, Y.; Hamaki, T.; et al. Clinicopathological features of pyothorax-associated lymphoma; a retrospective survey involving 98 patients. Ann. Oncol. 2007, 18, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, W.; Chan, A.C.L.; Chan, J.K.C.; Lau, G.T.C.; Chan, V.N.H.; Yiu, H.H.Y. Metallic implant-associated lymphoma—A distinct subgroup of large B-cell lymphoma related to pyothorcix-associated lymphoma? Am. J. Surg. Pathol. 2005, 29, 832–836. [Google Scholar] [CrossRef] [PubMed]
- CopieBergman, C.; Niedobitek, G.; Mangham, D.C.; Selves, J.; Baloch, K.; Diss, T.C.; Knowles, D.N.; Delsol, G.; Isaacson, P.G. Epstein-Barr virus in B-cell lymphomas associated with chronic suppurative inflammation. J. Pathol. 1997, 183, 287–292. [Google Scholar] [CrossRef]
- Fujimoto, M.; Haga, H.; Okamoto, M.; Obara, E.; Ishihara, M.; Mizuta, N.; Nishimura, K.; Manabe, T. EBV-associated diffuse large B-cell lymphoma arising in the chest wall with surgical mesh implant. Pathol. Int. 2008, 58, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Aozasa, K.; Takakuwa, T.; Nakatsuka, S. Pyothorax-associated lymphoma—A lymphoma developing in chronic inflammation. Adv. Anat. Pathol. 2005, 12, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Mori, K.L.; Sakajiri, S.; Oshimi, K. B-cell marker negative (CD7(+), CD19(−)) Epstein-Barr virus-related pyothorax-associated lymphoma with rearrangement in the JH gene. Leuk. Lymphoma 2003, 44, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Yasunaga, Y.; Iuchi, K.; Yamauchi, S.; Takekawa, T.; Sugiyama, H.; Aozasa, K. Interleukin-6-mediated growth enhancement of cell lines derived from pyothorax-associated lymphoma. Lab. Investig. 1996, 75, 167–173. [Google Scholar] [PubMed]
- Miwa, H.; Takakuwa, T.; Nakatsuka, S.; Tomita, Y.; Iuchi, K.; Aozasa, K. DNA sequences of the immunoglobulin heavy chain variable region gene in pyothorax-associated lymphoma. Oncology 2002, 62, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Takakuwa, T.; Luo, W.J.; Ham, M.F.; Mizuki, M.; Iuchi, K.; Aozasa, K. Establishment and characterization of unique cell lines derived from pyothorax-associated lymphoma which develops in long-standing pyothorax and is strongly associated with Epstein-Barr virus infection. Cancer Sci. 2003, 94, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Yamato, H.; Ohshima, K.; Suzumiya, J.; Kikuchi, M. Evidence for local immunosuppression and demonstration of c-myc amplification in pyothorax-associated lymphoma. Histopathology 2001, 39, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Hongyo, T.; Kuraoka, M.; Taniguchi, E.; Iuchi, K.; Nakajima, Y.; Aozasa, K.; Nomura, T. Frequent p53 mutations at dipyrimidine sites in patients with pyothorax-associated lymphoma. Cancer Res. 1998, 58, 1105–1107. [Google Scholar] [PubMed]
- Ando, M.; Sato, Y.; Takata, K.; Nomoto, J.; Nakamura, S.; Ohshima, K.; Takeuchi, T.; Orita, Y.; Kobayashi, Y.; Yoshino, T. A20 (TNFAIP3) deletion in Epstein-Barr virus-associated lymphoproliferative disorders/lymphomas. PLoS ONE 2013, 8, e56741. [Google Scholar] [CrossRef] [PubMed]
- Nishiu, M.; Tomita, Y.; Nakatsuka, S.; Takakuwa, T.; Iizuka, N.; Hoshida, Y.; Ikeda, J.; Iuchi, K.; Yanagawa, R.; Nakamura, Y.; et al. Distinct pattern of gene expression in pyothorax-associated lymphoma (PAL), a lymphoma developing in long-standing inflammation. Cancer Sci. 2004, 95, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.F.; McKelvie, P.A.; de Leval, L.; Edlefsen, K.L.; Ko, Y.H.; Aberman, Z.A.; Kovach, A.E.; Masih, A.; Nishino, H.T.; Weiss, L.M.; et al. Fibrin-associated EBV-positive large B-cell lymphoma an indolent neoplasm with features distinct from diffuse large B-cell lymphoma associated with chronic inflammation. Am. J. Surg. Pathol. 2017, 41, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.V.; Firchau, D.J.; McClure, R.F.; Kurtin, P.J.; Feldman, A.L. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am. J. Surg. Pathol. 2010, 34, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, C.; Beltran, B.; Quinones, P.; Carbajal, T.; Vilcapaza, J.; Yabar, A.; Segura, P.; Quintanilla-Martinez, L.; Miranda, R.N.; Castillo, J.J. Large B-cell lymphoma arising in cardiac myxoma or intracardiac fibrinous mass: A localized lymphoma usually associated with Epstein-Barr virus? Cardiovasc. Pathol. 2015, 24, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Gruver, A.M.; Huba, M.A.; Dogan, A.; Hsi, E.D. Fibrin-associated large B-cell lymphoma part of the spectrum of cardiac lymphomas. Am. J. Surg. Pathol. 2012, 36, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Boroumand, N.; Ly, T.L.; Sonstein, J.; Medeiros, L.J. Microscopic diffuse large B-cell lymphoma (DLBCL) occurring in pseudocysts do these tumors belong to the category of DLBCLassociated with chronic inflammation? Am. J. Surg. Pathol. 2012, 36, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Liebow, A.A.; Carrington, C.R.; Friedman, P.J. Lymphomatoid granulomatosis. Hum. Pathol. 1972, 3, 457–558. [Google Scholar] [CrossRef]
- Guinee, D.; Jaffe, E.; Kingma, D.; Fishback, N.; Wallberg, K.; Krishnan, J.; Frizzera, G.; Travis, W.; Koss, M. Pulmonary lymphomatoid granulomatosis—Evidence for a proliferation of Epstein-Barr-virus infected B-lymphocytes with a prominent T-cell component and vasculitis. Am. J. Surg. Pathol. 1994, 18, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Y.; Pittaluga, S.; Dunleavy, K.; Grant, N.; White, T.; Jiang, L.Y.; Davies-Hill, T.; Raffeld, M.; Wilson, W.H.; Jaffe, E.S. Lymphomatoid granulomatosis—A single institute experience pathologic findings and clinical correlations. Am. J. Surg. Pathol. 2015, 39, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Kingma, D.W.; Raffeld, M.; Wittes, R.E.; Jaffe, E.S. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2B. Blood 1996, 87, 4531–4537. [Google Scholar] [PubMed]
- Beaty, M.W.; Toro, J.; Sorbara, L.; Stern, J.B.; Pittaluga, S.; Raffeld, M.; Wilson, W.H.; Jaffe, E.S. Cutaneous lymphomatoid granulomatosis—Correlation of clinical and biologic features. Am. J. Surg. Pathol. 2001, 25, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Katzenstein, A.L.A.; Carrington, C.B.; Liebow, A.A. Lymphomatoid granulomatosis—Clinicopathologic study of 152 cases. Cancer 1979, 43, 360–373. [Google Scholar] [CrossRef]
- Dunleavy, K.; Roschewski, M.; Wilson, W.H. Lymphomatoid granulomatosis and other Epstein-Barr virus associated lymphoproliferative processes. Curr. Hematol. Malig. Rep. 2012, 7, 208–215. [Google Scholar] [CrossRef] [PubMed]
- TeruyaFeldstein, J.; Jaffe, E.S.; Burd, P.R.; Kanegane, H.; Kingma, D.W.; Wilson, W.H.; Longo, D.L.; Tosato, G. The role of Mig, the monokine induced by interferon-gamma, and IP-10, the interferon-gamma-inducible protein-10, in tissue necrosis and vascular damage associated with Epstein-Barr virus-positive lymphoproliferative disease. Blood 1997, 90, 4099–4105. [Google Scholar]
- Delecluse, H.J.; Anagnostopoulos, I.; Dallenbach, F.; Hummel, M.; Marafioti, T.; Schneider, U.; Huhn, D.; SchmidtWesthausen, A.; Reichart, P.A.; Gross, U.; et al. Plasmablastic lymphomas of the oral cavity: A new entity associated with the human immunodeficiency virus infection. Blood 1997, 89, 1413–1420. [Google Scholar] [PubMed]
- Teruya-Feldstein, J.; Chiao, E.; Filippa, D.A.; Lin, O.; Comenzo, R.; Coleman, M.; Portlock, C.; Noy, A. CD20-negative large-cell lymphoma with plasmablastic features: A clinically heterogenous spectrum in both HIV-positive and -negative patients. Ann. Oncol. 2004, 15, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Morscio, J.; Dierickx, D.; Nijs, J.; Verhoef, G.; Bittoun, E.; Vanoeteren, X.; Wlodarska, I.; Sagaert, X.; Tousseyn, T. Clinicopathologic comparison of plasmablastic lymphoma in HIV-positive, immunocompetent, and posttransplant patients single-center series of 25 cases and meta-analysis of 277 reported cases. Am. J. Surg. Pathol. 2014, 38, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.J.; Bibas, M.; Miranda, R.N. The biology and treatment of plasmablastic lymphoma. Blood 2015, 125, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Colomo, L.; Loong, F.; Rives, S.; Pittaluga, S.; Martinez, A.; Lopez-Guillermo, A.; Ojanguren, J.; Romagosa, V.; Jaffe, E.S.; Campo, E. Diffuse large B-cell lymphomas with plasmablastic differentiation represent a heterogeneous group of disease entities. Am. J. Surg. Pathol. 2004, 28, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Montes-Moreno, S.; Gonzalez-Medina, A.R.; Rodriguez-Pinilla, S.M.; Maestre, L.; Sanchez-Verde, L.; Roncador, G.; Mollejo, M.; Garcia, J.F.; Menarguez, J.; Montalban, C.; et al. Aggressive large B-cell lymphoma with plasma cell differentiation: Immunohistochemical characterization of plasmablastic lymphoma and diffuse large B-cell lymphoma with partial plasmablastic phenotype. Haematologica 2010, 95, 1342–1349. [Google Scholar] [CrossRef] [PubMed]
- Montes-Moreno, S.; Martinez-Magunacelaya, N.; Zecchini-Barrese, T.; de Villambrosia, S.G.; Linares, E.; Ranchal, T.; Rodriguez-Pinilla, M.; Batlle, A.; Cereceda-Company, L.; Revert-Arce, J.B.; et al. Plasmablastic lymphoma phenotype is determined by genetic alterations in MYC and PRDM1. Mod. Pathol. 2017, 30, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Pantanowitz, L.; Dezube, B.J. HIV-associated plasmablastic lymphoma: Lessons learned from 112 published cases. Am. J. Hematol. 2008, 83, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Harmon, C.M.; Smith, L.B. Plasmablastic lymphoma a review of clinicopathologic features and differential diagnosis. Arch. Pathol. Lab. Med. 2016, 140, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A. Plasmablastic lymphoma: One or more entities? Am. J. Hematol. 2008, 83, 763–764. [Google Scholar] [CrossRef] [PubMed]
- Valera, A.; Balague, O.; Colomo, L.; Martinez, A.; Delabie, J.; Taddesse-Heath, L.; Jaffe, E.S.; Campo, E. IG/MYC rearrangements are the main cytogenetic alteration in plasmablastic lymphomas. Am. J. Surg. Pathol. 2010, 34, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.; Gentles, A.J.; Sujoy, V.; Vega, F.; Dumur, C.I.; Blevins, T.L.; Bernal-Mizrachi, L.; Mosunjac, M.; Pimentel, A.; Zhu, D.; et al. Gene expression analysis of plasmablastic lymphoma identifies downregulation of B-cell receptor signaling and additional unique transcriptional programs. Leukemia 2015, 29, 2270–2273. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D. A sarcoma involving the jaws in african children. Br. J. Surg. 1958, 46, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D.; Wright, D.H. A lymphoma syndrome in tropical Africa with a note on histology, cytology and histochemistry. Int. Rev. Exp. Pathol. 1963, 2, 67–138. [Google Scholar] [PubMed]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef]
- Moormann, A.M.; Bailey, J.A. Malaria—How this parasitic infection aids and abets EBV-associated Burkitt lymphomagenesis. Curr. Opin. Virol. 2016, 20, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Queiroga, E.M.; Gualco, G.; Weiss, L.M.; Dittmer, D.P.; Araujo, I.; Klumb, C.E.N.; Harrington, W.J.; Bacchi, C.E. Burkitt lymphoma in Brazil is characterized by geographically distinct clinicopathologic features. Am. J. Clin. Pathol. 2008, 130, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Carroll, V.; Garzino-Demo, A. HIV-associated lymphoma in the era of combination antiretroviral therapy: Shifting the immunological landscape. Pathog. Dis. 2015, 73. [Google Scholar] [CrossRef] [PubMed]
- Rimsza, L.; Pittaluga, S.; Dirnhofer, S.; Copie-Bergman, C.; de Leval, L.; Facchetti, F.; Pileri, S.; Rosenwald, A.; Wotherspoon, A.; Fend, F. The clinicopathologic spectrum of mature aggressive B cell lymphomas. Virchows Arch. 2017, 471, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Bentink, S.; Berger, H.; Klapper, W.; Wessendorf, S.; Barth, T.F.E.; Bernd, H.W.; Cogliatti, S.B.; Dierlamm, J.; Feller, A.C.; et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N. Engl. J. Med. 2006, 354, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Dallafavera, R.; Martinotti, S.; Gallo, R.C.; Erikson, J.; Croce, C.M. Translocation and rearrangements of the c-MYC oncogene locus in human undifferentiated B-cell lymphomas. Science 1983, 219, 963–967. [Google Scholar] [CrossRef]
- Zeller, K.I.; Zhao, X.D.; Lee, C.W.H.; Chiu, K.P.; Yao, F.; Yustein, J.T.; Ooi, H.S.; Orlov, Y.L.; Shahab, A.; Yong, H.C.; et al. Global mapping of c-MYC binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17834–17839. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, M.S.; Wiman, K.G. Role of genetic and epigenetic changes in Burkitt lymphoma. Semin. Cancer Biol. 2002, 12, 381–387. [Google Scholar] [CrossRef]
- Komano, J.; Sugiura, M.; Takada, K. Epstein-Barr virus contributes to the malignant phenotype and to apoptosis resistance in Burkitt’s lymphoma cell line akata. J. Virol. 1998, 72, 9150–9156. [Google Scholar] [PubMed]
- Nanbo, A.; Takada, K. The role of Epstein-Barr virus-encoded small RNAs (EBERs) in oncogenesis. Rev. Med. Virol. 2002, 12, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Murakami, M.; Verma, S.C.; Cai, Q.L.; Haldar, S.; Kaul, R.; Wasik, M.A.; Middeldorp, J.; Robertson, E.S. Epstein-Barr virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology 2011, 410, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Dheekollu, J.; Malecka, K.; Wiedmer, A.; Delecluse, H.J.; Chiang, A.K.S.; Altieri, D.C.; Messick, T.E.; Lieberman, P.M. Carcinoma-risk variant of EBNA1 deregulates Epstein-Barr virus episomal latency. Oncotarget 2017, 8, 7248–7264. [Google Scholar] [CrossRef] [PubMed]
- Fish, K.; Chen, J.; Longnecker, R. Epstein-Barr virus latent membrane protein 2A enhances MYC-driven cell cycle progression in a mouse model of B lymphoma. Blood 2014, 123, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.; Calado, D.P.; Srinivasan, L.; Kochert, K.; Zhang, B.C.; Rosolowski, M.; Rodig, S.J.; Holzmann, K.; Stilgenbauer, S.; Siebert, R.; et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 2012, 22, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.; Rajewsky, K. Burkitt lymphomagenesis linked to MYC plus PI3K in germinal center B cells. Oncotarget 2012, 3, 1066–1067. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Robbiani, D.F.; Bothmer, A.; Callen, E.; Reina-San-Martin, B.; Dorsett, Y.; Difilippantonio, S.; Bolland, D.J.; Chen, H.T.; Corcoran, A.E.; Nussenzweig, A.; et al. Aid is required for the chromosomal breaks in c-MYC that lead to c-MYC/IGH translocations. Cell 2008, 135, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Kalchschmidt, J.S.; Bashford-Rogers, R.; Paschos, K.; Gillman, A.C.T.; Styles, C.T.; Kellam, P.; Allday, M.J. Epstein-Barr virus nuclear protein EBNA3C directly induces expression of aid and somatic mutations in B cells. J. Exp. Med. 2016, 213, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Deroubaix, S.; Feldhahn, N.; Oliveira, T.Y.; Callen, E.; Wang, Q.; Jankovic, M.; Silva, I.T.; Rommel, P.C.; Bosque, D.; et al. Plasmodium infection promotes genomic instability and aid-dependent B cell lymphoma. Cell 2015, 162, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Abate, F.; Ambrosio, M.R.; Mundo, L.; Laginestra, M.A.; Fuligni, F.; Rossi, M.; Zairis, S.; Gazaneo, S.; De Falco, G.; Lazzi, S.; et al. Distinct viral and mutational spectrum of endemic Burkitt lymphoma. PLoS Pathog. 2015, 11, e1005158. [Google Scholar] [CrossRef] [PubMed]
- Tierney, R.J.; Shannon-Lowe, C.D.; Fitzsimmons, L.; Bell, A.I.; Rowe, M. Unexpected patterns of Epstein-Barr virus transcription revealed by a high throughput PCR array for absolute quantification of viral mRNA. Virology 2015, 474, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Peliccci, P.G.; Knowles, D.M.; Magrath, I.; Dalla-Favera, R. Chromosomal breakpoints and structural alterations of the c-MYC locus differ in endemic and sporadic forms of Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 1986, 83, 2984–2988. [Google Scholar] [CrossRef]
- Amato, T.; Abate, F.; Piccaluga, P.; Iacono, M.; Fallerini, C.; Renieri, A.; De Falco, G.; Ambrosio, M.R.; Mourmouras, V.; Ogwang, M.; et al. Clonality analysis of immunoglobulin gene rearrangement by next-generation sequencing in endemic Burkitt lymphoma suggests antigen drive activation of BCR as opposed to sporadic Burkitt lymphoma. Am. J. Clin. Pathol. 2016, 145, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.M.; Zhang, M.Z.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Love, C.; Sun, Z.; Jima, D.; Li, G.J.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.L.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Giulino-Roth, L.; Wang, K.; MacDonald, T.Y.; Mathew, S.; Tam, Y.; Cronin, M.T.; Palmer, G.; Lucena-Silva, N.; Pedrosa, F.; Pedrosa, M.; et al. Targeted genomic sequencing of pediatric Burkitt lymphoma identifies recurrent alterations in antiapoptotic and chromatin-remodeling genes. Blood 2012, 120, 5181–5184. [Google Scholar] [CrossRef] [PubMed]
- Mundo, L.; Ambrosio, M.R.; Picciolini, M.; Lo Bello, G.; Gazaneo, S.; Del Porro, L.; Lazzi, S.; Navari, M.; Onyango, N.; Granai, M.; et al. Unveiling another missing piece in EBV-driven lymphomagenesis: EBV-encoded microRNAs expression in EBER-negative Burkitt lymphoma cases. Front. Microbiol. 2017, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Leucci, E.; Cocco, M.; Onnis, A.; De Falco, G.; van Cleef, P.; Bellan, C.; van Rijk, A.; Nyagol, J.; Byakika, B.; Lazzi, S.; et al. MYC translocation-negative classical Burkitt lymphoma cases: An alternative pathogenetic mechanism involving miRNA deregulation. J. Pathol. 2008, 216, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Salaverria, I.; Martin-Guerrero, I.; Wagener, R.; Kreuz, M.; Kohler, C.W.; Richter, J.; Pienkowska-Grela, B.; Adam, P.; Burkhardt, B.; Claviez, A.; et al. A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood 2014, 123, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.M.; Movahed, L.A.; Warnke, R.A.; Sklar, J. Detection of Epstein-Barr viral genomes in Reed-Sternberg cells of Hodgkins-disease. N. Engl. J. Med. 1989, 320, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.K.; Re, D.; Zander, T.; Wolf, J.; Diehl, V. Epidemiology and etiology of Hodgkin’s lymphoma. Ann. Oncol. 2002, 13, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, H.; Kuppers, R.; Hansmann, M.L.; Rajewsky, K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin disease—Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B-cells at various stages of development. Proc. Natl. Acad. Sci. USA 1994, 91, 10962–10966. [Google Scholar] [CrossRef]
- Schwering, I.; Brauninger, A.; Klein, U.; Jungnickel, B.; Tinguely, M.; Diehl, V.; Hansmann, M.L.; Dalla-Favera, R.; Rajewsky, K.; Kuppers, R. Loss of the B-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003, 101, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Stein, H.; Marafioti, T.; Foss, H.D.; Laumen, H.; Hummel, M.; Anagnostopoulos, I.; Wirth, T.; Demel, G.; Falini, B. Down-regulation of BOB.1/OBF.1 and Oct2 in classical Hodgkin disease but not in lymphocyte predominant hodgkin disease correlates with immunoglobulin transcription. Blood 2001, 97, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Krenacs, L.; Himmelmann, A.W.; Quintanilla-Martinez, L.; Fest, T.; Riva, A.; Wellmann, A.; Bagdi, E.; Kehrl, J.H.; Jaffe, E.S.; Raffeld, M. Transcription factor B-cell-specific activator protein (BSAP) is differentially expressed in B cells and in subsets of B-cell lymphomas. Blood 1998, 92, 1308–1316. [Google Scholar] [PubMed]
- Pallesen, G.; Hamiltondutoit, S.J.; Rowe, M.; Young, L.S. Expression of Epstein-Barr-virus latent gene-products in tumor-cells of Hodgkins-disease. Lancet 1991, 337, 320–322. [Google Scholar] [CrossRef]
- Herbst, H.; Dallenbach, F.; Hummel, M.; Niedobitek, G.; Pileri, S.; Mullerlantzsch, N.; Stein, H. Epstein-Barr-virus latent membrane-protein expression in Hodgkin and Reed-Sternberg cells. Proc. Natl. Acad. Sci. USA 1991, 88, 4766–4770. [Google Scholar] [CrossRef]
- Anagnostopoulos, I.; Herbst, H.; Niedobitek, G.; Stein, H. Demonstration of monoclonal EBV genomes in Hodgkins-disease and Ki-1-positive anaplastic large cell lymphoma by combined Southern blot and in situ hybridization. Blood 1989, 74, 810–816. [Google Scholar] [PubMed]
- Kuppers, R. The biology of hodgkin’s lymphoma. Nat. Rev. Cancer 2009, 9, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Brauninger, A.; Schmitz, R.; Bechtel, D.; Renne, C.; Hausmann, M.L.; Kuppers, R. Molecular biology of Hodgkin’s and Reed/Sternberg cells in Hodgkin’s lymphoma. Int. J. Cancer 2006, 118, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Niedobitek, G.; Kremmer, E.; Herbst, H.; Whitehead, L.; Dawson, C.W.; Niedobitek, E.; vonOstau, C.; Rooney, N.; Grasser, F.A.; Young, L.S. Immunohistochemical detection of the Epstein-Barr virus-encoded latent membrane protein 2A in Hodgkin’s disease and infectious mononucleosis. Blood 1997, 90, 1664–1672. [Google Scholar] [PubMed]
- Mancao, C.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Connors, J.M.; Gascoyne, R.D. Molecular pathogenesis of Hodgkin’s lymphoma: Increasing evidence of the importance of the microenvironment. J. Clin. Oncol. 2011, 29, 1812–1826. [Google Scholar] [CrossRef] [PubMed]
- Vardhana, S.; Younes, A. The immune microenvironment in Hodgkin lymphoma: T cells, B cells, and immune checkpoints. Haematologica 2016, 101, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.K.; Moll, G.; Smith, C.; Dua, U.; Lambley, E.; Ramuz, O.; Gill, D.; Marlton, P.; Seymour, J.F.; Khanna, R. Galectin-1 mediated suppression of Epstein-Barr virus-specific T-cell immunity in classic Hodgkin lymphoma. Blood 2007, 110, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Nishikori, M.; Kitawaki, T.; Sakai, T.; Hishizawa, M.; Tashima, M.; Kondo, T.; Ohmori, K.; Kurata, M.; Hayashi, T.; et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood 2008, 111, 3220–3224. [Google Scholar] [CrossRef] [PubMed]
- Renne, C.; Hinsch, N.; Willenbrock, K.; Fuchs, M.; Klapper, W.; Engert, A.; Kuppers, R.; Hansmann, M.L.; Brauninger, A. The aberrant coexpression of several receptor tyrosine kinases is largely restricted to EBV-negative cases of classical Hodgkin’s lymphoma. Int. J. Cancer 2007, 120, 2504–2509. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Hansmann, M.L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. TNAFIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.R.; Quintanilla-Martinez, L.; Raffeld, M.; Richter, M.; Krugmann, J.; Burek, C.; Hartmann, E.; Rudiger, T.; Jaffe, E.S.; Muller-Hermelink, H.K.; et al. IGVH mutational status and clonality analysis of Richter’s transformation—Diffuse large B-cell lymphoma and Hodgkin lymphoma in association with B-cell chronic lymphocytic leukemia (B-CLL) represent 2 different pathways of disease evolution. Am. J. Surg. Pathol. 2007, 31, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.T.; Liao, Y.L.; Lu, C.L.; Chuang, S.S.; Li, C.Y. Plasmablastic cytomorphologic features in plasma cell neoplasms in immunocompetent patients are significantly associated with EBV. Am. J. Clin. Pathol. 2007, 128, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, N.S.; Kapadia, S.B.; Nalesnik, M.A.; Swerdlow, S.H. Extramedullary plasmacytoma of the head and neck—Use of paraffin sections to assess clonality with in-situ hybridization, growth fraction, and the presence of Epstein-Barr-virus. Mod. Pathol. 1995, 8, 503–508. [Google Scholar] [PubMed]
- Loghavi, S.; Khoury, J.D.; Medeiros, L.J. Epstein-Barr virus-positive plasmacytoma in immunocompetent patients. Histopathology 2015, 67, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Q.; Wang, J.C.; Zhang, W.Y.; Chen, M.; Chen, J.; Liu, W.P. Solitary plasmacytoma associated with Epstein-Barr virus: A clinicopathologic, cytogenetic study and literature review. Ann. Diagn. Pathol. 2017, 27, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla-Martinez, L.; Ko, Y.-H.; Kimura, H.; Jaffe, E.S. EBV-positive T-cell and NK-cell lymphoproliferative diseases of childhood. In Who Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D.A., Hasserjian, R.P., Le Beau, M.M., et al., Eds.; IARC: Lyon, France, 2017; pp. 355–363. [Google Scholar]
- Kimura, H.; Ito, Y.; Kawabe, S.; Gotoh, K.; Takahashi, Y.; Kojima, S.; Naoe, T.; Esaki, S.; Kikuta, A.; Sawada, A.; et al. EBV-associated T/NK-cell lymphoproliferative diseases in nonimmunocompromised hosts: Prospective analysis of 108 cases. Blood 2012, 119, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Ko, Y.H.; Yoo, K.H.; Koo, H.H.; Kim, S.J.; Kim, W.S.; Park, H. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J. Pathol. 2013, 47, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ohshima, K.; Karube, K.; Suzumiya, J.; Ohga, S.; Ishihara, S.; Tamura, K.; Kikuchi, M. Clinicopathological states of Epstein-Barr virus-associated T/NK-cell lymphoproliferative disorders (severe chronic active EBV infection) of children and young adults. Int. J. Oncol. 2004, 24, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Chang, S.T.; Hsieh, Y.C.; Huang, W.T.; Hsu, J.D.; Tseng, C.E.; Wang, M.C.; Hwang, W.S.; Wang, J.; Chuang, S.S. Spectrum of Epstein-Barr virus-associated T-cell lymphoproliferative disorder in adolescents and young adults in taiwan. Int. J. Clin. Exp. Pathol. 2014, 7, 2430–2437. [Google Scholar] [PubMed]
- Jones, J.F.; Straus, S.E. Chronic Epstein-Barr virus-infection. Annu. Rev. Med. 1987, 38, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Straus, S.E. The chronic mononucleosis syndrome. J. Infect. Dis. 1988, 157, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Hoshino, Y.; Kanegane, H.; Tsuge, I.; Okamura, T.; Kawa, K.; Morishima, T. Clinical and virologic characteristics of chronic active Epstein-Barr virus infection. Blood 2001, 98, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Pathogenesis of chronic active Epstein-Barr virus infection: Is this an infectious disease, lymphoproliferative disorder, or immunodeficiency? Rev. Med. Virol. 2006, 16, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.E.; Jones, A.; Smith, L.; Lai, R.; Preiksaitis, J.; Robinson, J. Severe chronic active Epstein-Barr virus infection mimicking steroid-dependent inflammatory bowel disease. Pediatr. Infect. Dis. J. 2005, 24, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Quintanilla-Martinez, L.; Bienemann, K.; Zeus, T.; Germing, U.; Sander, O.; Kandolf, R.; Haussinger, D.; Klingel, K. An unusual presentation of a common infection. Infection 2013, 41, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Isobe, Y.; Aritaka, N.; Setoguchi, Y.; Ito, Y.; Kimura, H.; Hamano, Y.; Sugimoto, K.; Komatsu, N. T/NK cell type chronic active Epstein-Barr virus disease in adults: An underlying condition for Epstein-Barr virus-associated T/NK-cell lymphoma. J. Clin. Pathol. 2012, 65, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Arai, A.; Imadome, K.I.; Watanabe, Y.; Yoshimori, M.; Koyama, T.; Kawaguchi, T.; Nakaseko, C.; Fujiwara, S.; Miura, O. Clinical features of adult-onset chronic active Epstein-Barr virus infection: A retrospective analysis. Int. J. Hematol. 2011, 93, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Morishima, T.; Kanegane, H.; Ohga, S.; Hoshino, Y.; Maeda, A.; Imai, S.; Okano, M.; Morio, T.; Yokota, S.; et al. Prognostic factors for chronic active Epstein-Barr virus infection. J. Infect. Dis. 2003, 187, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Hoshino, Y.; Hara, S.; Sugaya, N.; Kawada, J.; Shibata, Y.; Kojima, S.; Nagasaka, T.; Kuzushima, K.; Morishima, T. Differences between T cell-type and natural killer cell-type chronic active Epstein-Barr virus infection. J. Infect. Dis. 2005, 191, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Kimura, H.; Yoshino, T.; Kim, C.W.; Ko, Y.H.; Lee, S.S.; Peh, S.C.; Chan, J.K.C.; Grp, C.S. Proposed categorization of pathological states of EBV-associated T/natural killer-cell lymphoproliferative disorder (LPD) in children and young adults: Overlap with chronic active EBV infection and infantile fulminant EBV T-LPD. Pathol. Int. 2008, 58, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Tosato, G.; Straus, S.; Henle, W.; Pike, S.E.; Blaese, R.M. Characteristic T-cell dysfunction in patients with chronic active Epstein-Barr virus-infection (chronic infectious-mononucleosis). J. Immunol. 1985, 134, 3082–3088. [Google Scholar] [PubMed]
- Cohen, J.I.; Jaffe, E.S.; Dale, J.K.; Pittaluga, S.; Heslop, H.E.; Rooney, C.M.; Gottschalk, S.; Bollard, C.M.; Rao, V.K.; Marques, A.; et al. Characterization and treatment of chronic active Epstein-Barr virus disease: A 28-year experience in the united states. Blood 2011, 117, 5835–5849. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, I.; Morishima, T.; Kimura, H.; Kuzushima, K.; Matsuoka, H. Impaired cytotoxic T lymphocyte response to Epstein-Barr virus-infected NK cells in patients with severe chronic active EBV infection. J. Med. Virol. 2001, 64, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, N.; Kimura, H.; Hara, S.; Hoshino, Y.; Kojima, S.; Morishima, T.; Tsurumi, T.; Kuzushima, K. Quantitative analysis of Epstein-Barr virus (EBV)-specific CD8(+) T cells in patients with chronic active EBV infection. J. Infect. Dis. 2004, 190, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Sugiura, M.; Oikawa, O.; Koizumi, S.; Hirao, M.; Kimura, H.; Hayashibara, H.; Terai, N.; Tsutsumi, H.; Oda, T.; et al. Epstein-Barr virus (EBV)-carrying and -expressing T-cell lines established from severe chronic active EBV infection. Blood 1996, 87, 1446–1457. [Google Scholar] [PubMed]
- Quintanilla-Martinez, L.; Ridaura, C.; Nagl, F.; Saez-de-Ocariz, M.; Duran-McKinster, C.; Ruiz-Maldonado, R.; Alderete, G.; Grube, P.; Lome-Maldonado, C.; Bonzheim, I.; et al. Hydroa vacciniforme-like lymphoma: A chronic EBV+ lymphoproliferative disorder with risk to develop a systemic lymphoma. Blood 2013, 122, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Ruizmaldonado, R.; Parrilla, F.M.; Orozcocovarrubias, M.D.; Ridaura, C.; Sanchez, L.T.; Mckinster, C.D. Edematous, scarring vasculitic panniculitis—A new multisystemic disease with malignant potential. J. Am. Acad. Dermatol. 1995, 32, 37–44. [Google Scholar] [CrossRef]
- Barrionuevo, C.; Anderson, V.M.; Zevallos-Giampietri, E.; Zaharia, M.; Misad, O.; Bravo, J.; Caceres, H.; Taxa, L.; Martinez, M.T.; Wachtel, A.; et al. Hydroa-like cutaneous T-cell lymphoma: A clinicopathologic and molecular genetic study of 16 pediatric cases from Peru. Appl Immunohistochem. Mol. Morphol. 2002, 10, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, K.; Xu, Z.; Takata, M.; Iguchi, M.; Ohtsuka, M.; Akiba, H.; Mitsuhashi, Y.; Takenoshita, H.; Sugiuchi, R.; Tagami, H.; et al. The association of latent Epstein-Barr virus infection with hydroa vacciniforme. Br. J. Dermatol. 1999, 140, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, K.; Ohtsuka, M.; Akiba, H.; Kaneko, F. Atypical hydroa vacciniforme in childhood: From a smoldering stage to Epstein-Barr virus-associated lymphoid malignancy. J. Am. Acad. Dermatol. 1999, 40, 283–284. [Google Scholar] [CrossRef]
- Magana, M.; Sangueza, P.; Gil-Beristain, J.; Sanchez-Sosa, S.; Salgado, A.; Ramon, G.; Sangueza, O.P. Angiocentric cutaneous T-cell lymphoma of childhood (hydroa-like lymphoma): A distinctive type of cutaneous T-cell lymphoma. J. Am. Acad. Dermatol. 1998, 38, 574–579. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L.; Kimura, H.; Jaffe, E.S. EBV-positive T-cell lymphoproliferative disorders of childhood. In Who Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Vardiman, J.W., Eds.; IARC: Lyon, France, 2008; pp. 278–280. [Google Scholar]
- Cho, K.H.; Lee, S.H.; Kim, C.W.; Jeon, Y.K.; Kwon, I.H.; Cho, Y.J.; Lee, S.K.; Suh, D.H.; Chung, J.H.; Yoon, T.Y.; et al. Epstein-Barr virus-associated lymphoproliferative lesions presenting as a hydroa vacciniforme-like eruption: An analysis of six cases. Br. J. Dermatol. 2004, 151, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, K.; Satoh, M.; Yamamoto, T.; Oono, T.; Morizane, S.; Ohtsuka, M.; Xu, Z.G.; Suzuki, D.; Tsuji, K. Pathogenic link between hydroa vacciniforme and Epstein-Barr virus-associated hematologic disorders. Arch. Dermatol. 2006, 142, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Sangueza, M.; Plaza, J.A. Hydroa vacciniforme-like cutaneous T-cell lymphoma: Clinicopathologic and immunohistochemical study of 12 cases. J. Am. Acad. Dermatol. 2013, 69, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pinilla, S.M.; Barrionuevo, C.; Garcia, J.; Martinez, M.T.; Pajares, R.; Montes-Moreno, S.; Casavilca, S.; Montes, J.; Bravo, F.; Zaharia, M.; et al. EBV-associated cutaneous NK/T-cell lymphoma review of a series of 14 cases from Peru in children and young adults. Am. J. Surg. Pathol. 2010, 34, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Hirai, Y.; Yamamoto, T.; Kimura, H.; Ito, Y.; Tsuji, K.; Miyake, T.; Morizane, S.; Suzuki, D.; Fujii, K.; Iwatsuki, K. Hydroa vacciniforme is associated with increased numbers of Epstein-Barr virus-infected gamma delta T cells. J. Investig. Dermatol. 2012, 132, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Toga, A.; Sakakibara, Y.; Toma, T.; Hasegawa, M.; Takehara, K.; Shigemura, T.; Agematsu, K.; Yachie, A. Clonal expansion of Epstein-Barr virus (EBV)-infected gamma delta T cells in patients with chronic active EBV disease and hydroa vacciniforme-like eruptions. Int. J. Hematol. 2012, 96, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Massone, C.; Magana, P.; Cerroni, L. Clinicopathologic features of hydroa vacciniforme-like lymphoma: A series of 9 patients. Am. J. Dermatopathol. 2016, 38, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Wada, K.; Tobita, S.; Gotoh, K.; Ito, Y.; Demachi-Okamura, A.; Shimizu, N.; Nishiyama, Y.; Kimura, H. Quantitative analysis of Epstein-Barr virus (EBV)-related gene expression in patients with chronic active EBV infection. J. Gen. Virol. 2010, 91, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Okada, S.; Wakiguchi, H.; Kurashige, T.; Hirai, K.; KawaHa, K. Clonal lymphoproliferation following chronic active Epstein-Barr virus infection and hypersensitivity to mosquito bites. Am. J. Hematol. 1997, 54, 276–281. [Google Scholar] [CrossRef]
- Ishihara, S.; Ohshima, K.; Tokura, Y.; Yabuta, R.; Imaishi, H.; Wakiguchi, H.; Kurashige, T.; Kishimoto, H.; Katayama, I.; Okada, S.; et al. Hypersensitivity to mosquito bites conceals clonal lymphoproliferation of Epstein-Barr viral DNA-positive natural killer cells. Jpn. J. Cancer Res. 1997, 88, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Tokura, Y.; Tamura, Y.; Takigawa, M.; Koide, M.; Satoh, T.; Sakamoto, T.; Horiguchi, D.; Yamada, M. Severe hypersensitivity to mosquito bites associated with natural-killer-cell lymphocytosis. Arch. Dermatol. 1990, 126, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Asada, H. Hypersensitivity to mosquito bites: A unique pathogenic mechanism linking Epstein-Barr virus infection, allergy and oncogenesis. J. Dermatol. Sci. 2007, 45, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Asada, H.; Saito-Katsuragi, M.; Niizeki, H.; Yoshioka, A.; Suguri, S.; Isonokami, M.; Aoki, T.; Ishihara, S.; Tokura, Y.; Iwatsuki, K.; et al. Mosquito salivary gland extracts induce EBV-infected NK cell oncogenesis via CD4+ T cells in patients with hypersensitivity to mosquito bites. J. Investig. Dermatol. 2005, 125, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Tokura, Y.; Matsuoka, H.; Koga, C.; Asada, H.; Seo, N.; Ishihara, S.; Adachi, A.; Ibe, M. Enhanced T-cell response to mosquito extracts by NK cells in hypersensitivity to mosquito bites associated with EBV infection and NK cell lymphocytosis. Cancer Sci. 2005, 96, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Asada, H.; Miyagawa, S.; Sumikawa, Y.; Yamaguchi, Y.; Itami, S.; Suguri, S.; Harada, M.; Tokura, Y.; Ishihara, S.; Ohshima, S.; et al. CD4(+) T-lymphocyte-induced Epstein-Barr virus reactivation in a patient with severe hypersensitivity to mosquito bites and Epstein-Barr virus-infected NK cell lymphocytosis. Arch. Dermatol. 2003, 139, 1601–1607. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Quintanilla-Martinez, L.; Kumar, S.; Fend, F.; Reyes, E.; Teruya-Feldstein, J.; Kingma, D.W.; Sorbara, L.; Raffeld, M.; Straus, S.E.; Jaffe, E.S. Fulminant EBV+ T-cell lymphoproliferative disorder following acute/chronic EBV infection: A distinct clinicopathologic syndrome. Blood 2000, 96, 443–451. [Google Scholar] [PubMed]
- Henter, J.I.; Horne, A.; Arico, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.B.; Allen, C.E.; Weitzman, S.; Filipovich, A.H.; McClain, K.L. How I treat hemophagocytic lymphohistiocytosis. Blood 2011, 118, 4041–4052. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Cohen, D.N.; Greig, B.; Yenamandra, S.; Vnencak-Jones, C.; Thompson, M.A.; Kim, A.S. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int. J. Clin. Exp. Pathol. 2014, 7, 5738–5749. [Google Scholar] [PubMed]
- Jones, J.F.; Shurin, S.; Abramowsky, C.; Tubbs, R.R.; Sciotto, C.G.; Wahl, R.; Sands, J.; Gottman, D.; Katz, B.Z.; Sklar, J. T-cell lymphomas containing Epstein-Barr viral-DNA in patients with chronic Epstein-Barr virus-infections. N. Engl. J. Med. 1988, 318, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Kanegane, H.; Bhatia, K.; Gutierrez, M.; Kaneda, H.; Wada, T.; Yachie, A.; Seki, H.; Arai, T.; Kagimoto, S.; Okazaki, M.; et al. Syndrome of peripheral blood T-cell infection with Epstein-Barr virus (EBV) followed by EBV-positive T-cell lymphoma. Blood 1998, 91, 2085–2091. [Google Scholar] [PubMed]
- Kikuta, H.; Sakiyama, Y.; Matsumoto, S.; Ohishi, T.; Nakano, T.; Nagashima, T.; Oka, T.; Hironaka, T.; Hirai, K. Fatal Epstein-Barr virus-associated hemophagocytic syndrome. Blood 1993, 82, 3259–3264. [Google Scholar] [PubMed]
- Chen, R.L.; Su, I.J.; Lin, K.H.; Lee, S.H.; Lin, D.T.; Chuu, W.M.; Lin, K.S.; Huang, L.M.; Lee, C.Y. Fulminant childhood hemophagocytic syndrome mimicking histiocytic medullary reticulosis—An atypical form of Epstein-Barr-virus infection. Am. J. Clin. Pathol. 1991, 96, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Imamura, N.; Kusunoki, Y.; Kawaha, K.; Yumura, K.; Hara, J.; Oda, K.; Abe, K.; Dohy, H.; Inada, T.; Kajihara, H.; et al. Aggressive natural-killer-cell leukemia lymphoma—Report of 4 cases and review of the literature—Possible existence of a new clinical entity originating from the 3rd lineage of lymphoid-cells. Br. J. Haematol. 1990, 75, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla-Martinez, L.; Jaffe, E.S. Aggressive NK cell lymphomas: Insights into the spectrum of NK cell derived malignancies. Histopathology 2000, 37, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.C.; Jaffe, E.S.; Ko, Y.-H. Aggressive NK-cell leukemia. In Who Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D.A., Hasserjian, R.P., Le Beau, M.M., et al., Eds.; IARC: Lyon, France, 2017; pp. 353–354. [Google Scholar]
- Chan, J.K.C.; Sin, V.C.; Wong, K.F.; Ng, C.S.; Tsang, W.Y.W.; Chan, C.H.; Cheung, M.M.C.; Lau, W.H. Nonnasal lymphoma expressing the natural killer cell marker CD56: A clinicopathologic study of 49 cases of an uncommon aggressive neoplasm. Blood 1997, 89, 4501–4513. [Google Scholar] [PubMed]
- Li, C.R.; Tan, Y.; Wang, J.; Zhu, L.; Huang, L.; Wang, N.; Xu, D.M.; Cao, Y.; Li, J.Y.; Zhou, J.F. Abnormal immunophenotype provides a key diagnostic marker: A report of 29 cases of de novo aggressive natural killer cell leukemia. Transl. Res. 2014, 163, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Yamashita, Y.; Tsuzuki, T.; Nakayama, A.; Nakazawa, M.; Hasegawa, Y.; Kojima, H.; Nagasawa, T. Lymphomatous features of aggressive NK cell leukaemia/lymphoma with massive necrosis, haemophagocytosis and EB virus infection. Histopathology 2000, 37, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Song, S.Y.; Kim, W.S.; Ko, Y.H.; Kim, K.; Lee, M.H.; Park, K. Aggressive natural killer cell leukemia: Clinical features and treatment outcome. Haematologica 2002, 87, 1343–1345. [Google Scholar] [PubMed]
- Suzuki, R.; Suzumiya, J.; Nakamura, S.; Aoki, S.; Notoya, A.; Ozaki, S.; Gondo, H.; Hino, N.; Mori, H.; Sugimori, H.; et al. Aggressive natural killer-cell leukemia revisited: Large granular lymphocyte leukemia of cytotoxic NK cells. Leukemia 2004, 18, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Ohshima, K.; Ishihara, S.; Anzai, K.; Suzumiya, J.; Kikuchi, M. Elevated serum soluble fas ligand in natural killer cell proliferative disorders. Br. J. Haematol. 1998, 103, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Ito, T.; Momose, K.; Nakazawa, H.; Shimodaira, S.; Kamijo, Y.; Nakazawa, Y.; Ichikawa, N.; Ueno, M.; Kobayashi, H.; et al. Chemokine system and tissue infiltration in aggressive NK-cell leukemia. Leuk. Res. 2007, 31, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.H.; Behdad, A.; Ji, P.; Wolniak, K.L.; Frankfurt, O.; Chen, Y.H. EBV-negative aggressive NK-cell leukemia/lymphoma: A clinical and pathological study from a single institution. Mod. Pathol. 2017, 30, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Nicolae, A.; Ganapathi, K.A.; Pham, T.H.T.; Xi, L.Q.; Torres-Cabala, C.A.; Nanaji, N.M.; Zha, H.D.; Fan, Z.; Irwin, S.; Pittaluga, S.; et al. EBV-negative aggressive NK-cell leukemia/lymphoma clinical, pathologic, and genetic features. Am. J. Surg. Pathol. 2017, 41, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Ko, Y.H. Epstein-Barr virus-associated T/natural killer-cell lymphoproliferative disorders. J. Dermatol. 2014, 41, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Ryder, J.; Wang, X.; Bao, L.; Gross, S.A.; Hua, F.; Irons, R.D. Aggressive natural killer cell leukemia: Report of a chinese series and review of the literature. Int. J. Hematol. 2007, 85, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Tagawa, H.; Suzuki, R.; Karnan, S.; Karube, K.; Ohshima, K.; Muta, K.; Nawata, H.; Morishima, Y.; Nakamura, S.; et al. Genome-wide array-based comparative genomic hybridization of natural killer cell lymphoma/leukemia: Different genomic alteration patterns of aggressive NK-cell leukemia and extranodal NK/T-cell lymphoma, nasal type. Genes Chromosome Cancer 2005, 44, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Suda, T.; Haze, K.; Nakamura, N.; Sato, K.; Kimura, F.; Motoyoshi, K.; Mizuki, M.; Tagawa, S.; Ohga, S.; et al. Fas ligand in human serum. Nat. Med. 1996, 2, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.M.; Zhao, S.; Liu, W.P.; Zhang, W.Y.; Li, G.D.; Kucuk, C.; Hu, X.Z.; Chan, W.C.; Tang, Y.; Ding, W.S.; et al. Clinicopathologic characterization of aggressive natural killer cell leukemia involving different tissue sites. Am. J. Surg. Pathol. 2016, 40, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Lanier, L.L. NK cell development, homeostasis and function: Parallels with CD8(+) T cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.C.; Quintanilla-Martinez, L.; Ferry, J.A. Extranodal NK/T-cell lymphoma, nasal type. In Who Classification of Tumours of Haematopoietic and Lymphoid Tissue; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D.A., Hasserjian, R.P., Le Beau, M.M., et al., Eds.; IARC: Lyon, France, 2017; pp. 368–371. [Google Scholar]
- Quintanilla-Martinez, L.; Franklin, J.L.; Guerrero, I.; Krenacs, L.; Naresh, K.N.; Rama-Rao, C.; Bhatia, K.; Raffeld, M.; Magrath, I.T. Histological and immunophenotypic profile of nasal NK T cell lymphomas from Peru: High prevalence of p53 overexpression. Hum. Pathol. 1999, 30, 849–855. [Google Scholar] [CrossRef]
- Au, W.Y.; Weisenburger, D.D.; Intragumtornchai, T.; Nakamura, S.; Kim, W.S.; Sng, I.; Vose, J.; Armitage, J.O.; Liang, R.; International Peripheral T-Cell Lymphoma Project. Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: A study of 136 cases from the international peripheral T-cell lymphoma project. Blood 2009, 113, 3931–3937. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Kato, S.; Nakamura, S. Epstein–Barr virus-associated natural killer/T-cell lymphomas. Best Pract. Res. Clin. Haematol. 2013, 26, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.F.; Chan, J.K.C.; Cheung, M.M.C.; So, J.C.C. Bone marrow involvement by nasal NK cell lymphoma at diagnosis is uncommon. Am. J. Clin. Pathol. 2001, 115, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Kwong, Y.L.; Chan, A.C.L.; Liang, R.; Chiang, A.K.S.; Chim, C.S.; Chan, T.K.; Todd, D.; Ho, F.C.S. CD56(+) NK lymphomas: Clinicopathological features and prognosis. Br. J. Haematol. 1997, 97, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Kern, W.F.; Spier, C.M.; Hanneman, E.H.; Miller, T.P.; Matzner, M.; Grogan, T.M. Neural cell-adhesion molecule-positive peripheral T-cell lymphoma—A rare variant with a propensity for unusual sites of involvement. Blood 1992, 79, 2432–2437. [Google Scholar] [PubMed]
- Wong, K.F.; Chan, J.K.C.; Ng, C.S.; Lee, K.C.; Tsang, W.Y.W.; Cheung, M.M.C. CD56 (NKH1)-positive hematolymphoid malignancies—An aggressive neoplasm featuring frequent cutaneous mucosal involvement, cytoplasmic azurophilic granules, and angiocentricity. Hum. Pathol. 1992, 23, 798–804. [Google Scholar] [CrossRef]
- Hasserjian, R.P.; Harris, N.L. NK-cell lymphomas and leukemias—A spectrum of tumors with variable manifestations and immunophenotype. Am. J. Clin. Pathol. 2007, 127, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S.; Chan, J.K.C.; Su, I.J.; Frizzera, G.; Mori, S.; Feller, A.C.; Ho, F.C.S. Report of the workshop on nasal and related extranodal angiocentric T natural killer cell lymphomas—Definitions, differential diagnosis, and epidemiology. Am. J. Surg. Pathol. 1996, 20, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Jhuang, J.Y.; Chang, S.T.; Weng, S.F.; Pan, S.T.; Chu, P.Y.; Hsieh, P.P.; Wei, C.H.; Chou, S.C.; Koo, C.L.; Chen, C.J.; et al. Extranodal natural killer/T-cell lymphoma, nasal type in taiwan: A relatively higher frequency of T-cell lineage and poor survival for extranasal tumors. Hum. Pathol. 2015, 46, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Nam, S.J.; Kim, S.; Kim, T.M.; Heo, D.S.; Kim, C.W.; Jeon, Y.K. Prognostic implications of CD30 expression in extranodal natural killer/T-cell lymphoma according to treatment modalities. Leuk. Lymphoma 2015, 56, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.K.; Kim, H.; Park, S.O.; Choi, H.Y.; Kim, Y.A.; Park, S.S.; Kim, J.E.; Kim, Y.N.; Kim, C.W. Resistance to FAS-mediated apoptosis is restored by cycloheximide through the downregulation of cellular FLIPl in NK/T-cell lymphoma. Lab. Investig. 2005, 85, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Suzumiya, J.; Shimazaki, K.; Kato, A.; Tanaka, T.; Kanda, M.; Kikuchi, M. Nasal T/NK cell lymphomas commonly express perforin and fas ligand: Important mediators of tissue damage. Histopathology 1997, 31, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Chiang, A.K.S.; Wong, K.Y.; Liang, A.C.T.; Srivastava, G. Comparative analysis of Epstein-Barr virus gene polymorphisms in nasal T/NK-cell lymphomas and normal nasal tissues: Implications on virus strain selection in malignancy. Int. J. Cancer 1999, 80, 356–364. [Google Scholar] [CrossRef]
- Dirnhofer, S.; Angeles-Angeles, A.; Ortiz-Hidalgo, C.; Reyes, E.; Gredler, E.; Krugmann, J.; Fend, F.; Quintanilla-Martinez, L. High prevalence of a 30-base pair deletion in the Epstein-Barr virus (EBV) latent membrane protein 1 gene and of strain type B EBV in Mexican classical Hodgkin’s disease and reactive lymphoid tissue. Hum. Pathol. 1999, 30, 781–787. [Google Scholar] [CrossRef]
- Kwong, Y.L.; Lam, C.C.K.; Chan, T.M. Post-transplantation lymphoproliferative disease of natural killer cell lineage: A clinicopathological and molecular analysis. Br. J. Haematol. 2000, 110, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Lee, T.; Kang, S.Y.; Kim, S.J.; Kim, W.; Ko, Y.H. Nasal-type NK/T-cell lymphomas are more frequently T rather than NK lineage based on T-cell receptor gene, RNA, and protein studies: Lineage does not predict clinical behavior. Mod. Pathol. 2016, 29, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Weisenburger, D.D.; Chowdhury, A.; Tsai, M.Y.; Srivastava, G.; Greiner, T.C.; Kucuk, C.; Deffenbacher, K.; Vose, J.; Smith, L.; et al. Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic gamma delta T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro. Leukemia 2011, 25, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.L.P.; Chan, V.; Chan, J.K.C.; Wong, K.F.; Liang, R.; Kwong, Y.L. Consistent patterns of allelic loss in natural killer cell lymphoma. Am. J. Pathol. 2000, 157, 1803–1809. [Google Scholar] [CrossRef]
- Jiang, L.; Gu, Z.H.; Yan, Z.X.; Zhao, X.; Xie, Y.Y.; Zhang, Z.G.; Pan, C.M.; Hu, Y.; Cai, C.P.; Dong, Y.; et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat. Genet. 2015, 47, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, H.Y.; Kang, S.Y.; Kim, S.J.; Hwang, J.; Lee, S.; Kwak, S.H.; Park, K.S.; Yoo, H.Y.; Kim, W.S.; et al. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget 2015, 6, 17764–17776. [Google Scholar] [CrossRef] [PubMed]
- Dobashi, A.; Tsuyama, N.; Asaka, R.; Togashi, Y.; Ueda, K.; Sakata, S.; Baba, S.; Sakamoto, K.; Hatake, K.; Takeuchi, K. Frequent BCOR aberrations in extranodal NK/T-cell lymphoma, nasal type. Genes Chromosome Cancer 2016, 55, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Kucuk, C.; Jiang, B.; Hu, X.Z.; Zhang, W.Y.; Chan, J.K.C.; Xiao, W.M.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gamma delta-T or NK cells. Nat. Commun. 2015, 6, 6025. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla-Martinez, L.; Kremer, M.; Keller, G.; Nathrath, M.; Gamboa-Dominguez, A.; Meneses, A.; Luna-Contreras, L.; Cabras, A.; Hoefler, H.; Mohar, A.; et al. P53 mutations in nasal natural killer/T-cell lymphoma from Mexico—Association with large cell morphology and advanced disease. Am. J. Pathol. 2001, 159, 2095–2105. [Google Scholar] [CrossRef]
- Attygalle, A.D.; Cabecadas, J.; Gaulard, P.; Jaffe, E.S.; de Jong, D.; Ko, Y.H.; Said, J.; Klapper, W. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward—Report on the lymphoma workshop of the XVIth meeting of the European Association for Haematopathology and the Society for Hematopathology. Histopathology 2014, 64, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.K.; Kim, J.H.; Sung, J.Y.; Han, J.H.; Ko, Y.H.; Pathologists, K.S. Epstein-Barr virus-positive nodal T/NK-cell lymphoma: An analysis of 15 cases with distinct clinicopathological features. Hum. Pathol. 2015, 46, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Asano, N.; Miyata-Takata, T.; Takata, K.; Elsayed, A.A.; Satou, A.; Takahashi, E.; Kinoshita, T.; Nakamura, S. T-cell receptor (TCR) phenotype of nodal Epstein-Barr virus (EBV)-positive cytotoxic T-cell lymphoma (CTL) a clinicopathologic study of 39 cases. Am. J. Surg. Pathol. 2015, 39, 462–471. [Google Scholar] [CrossRef] [PubMed]
- NG, S.B.; Chung, T.H.; Nakamura, S.; Takahashi, E.; Ko, Y.-H.; Khoury, J.D.; Yin, C.C.; Soong, R.; Jeyasekharan, A.D.; Hoppe, M.M.; et al. EBV-associated primary nodal T/NK-cell lymphoma shows distinct molecular signature and copy number changes. Haematologica 2017. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. B-cell lymphoproliferations EBV-associated
|
2. B-cell lymphomas that might be EBV-associated
|
3. B-cell lymphomas rarely EBV-associated
|
4. Immunodeficiency-associated lymphoproliferative disorders
|
|
| Anatomic Sites and Distribution | EBV (%) | LMP1 | EBNA2 | Morphologic Features | Immunophenotypic Features and Clonality | Pathogenesis | Main Genetic Alterations | |
|---|---|---|---|---|---|---|---|---|
| EBV+ DLBCL, NOS | LN or extranodal sites | 100 | +/− | −/+ | Large cells; centroblastic or HRS-like morphology, TCRBL-like; angioinvasion and necrosis | Mainly post GC phenotype: CD45+/−, Pan-B cell markers+, CD138−, CD10−, BCL6+/−. IRF4/MUM1+; 68%, CD30+ and CD15+. IGH monoclonal. | Immunosenescence, Immune evasion (PDL2 upregulated) | NFkB and JAK/STAT pathways activated. GEP: “host immune response” |
| EBV+ mucocutaneous ulcer | Oral cavity, skin and GI tract | 100 | + | −/+ | Circumscribed ulcer with large cells immunoblastic or Hodgkin-like features | Post GC phenotype: CD45+, Pan-B cell markers+, CD138−. CD10−; variable positivity for BCL6, IRF4/MUM1+. ~50% co-express CD30 and CD15. IGH monoclonal | Immuno-senescence or immune-suppression. Postulated immune sequestration | Not known common T-cell oligoclonality or monoclonality |
| DLBCL-associated with chronic inflammation | Pleural cavity, cysts and other confined spaces | 100 | + | + | Morphology similar to conventional DLBCL | Post GC phenotype: Pan-B cell markers+, CD10−, BCL6+/−, CD30+, MUM1/IRF4+; plasmablastic phenotype possible (pan-Bcell markers−, CD138/MUM1+). IGH monoclonal | Immune sequestration in confined spaces due to prolonged inflammation | High genetic complexity; common TP53 mutation, MYC amplification and TNFAIP3 deletion |
| Fibrin-associated DLBCL | Cardiac myxoma, cardiac fibrin thrombi, implants | 100 | + | + | Large cells centroblastic, immunoblastic or plasmablastic features | Post GC phenotype: CD45+, CD20+, PAX5+, CD79a+, BCL6+/−, MUM1/IRF4+, CD30+, MYC (<50%), p53 (<30%). IGH monoclonal | Immune sequestration in avascular fibrin masses | Low complexity of genetic abnormalities |
| Lymphomatoid granulomatosis | Lung, CNS, skin, liver or kidney | 100 | −/+ | −/+ | Large cells with centroblastic, immunoblastic or HRS-like features in a T-cell reactive background; Angioinvasion and necrosis | Post GC phenotype: CD45+, Pan-B cell markers+, CD30+, CD15−; IGH monoclonal. | Underlying inherent immunosuppression | Alterations of oncogenes not detected |
| Plasmablastic lymphoma | Solid extranodal masses, GI tract, LN | 70–80 | −/+ | − | Plasmablastic, immunoblasticor anaplastic | Terminally differentiated B-cell: CD45−, CD20−, PAX5−, CD79a−/+, CD138+, CD38+, CD10−/+, CD56−/+, BCL6−, MUM1/IRF4+, BLIMP1+, XBP1+, cIgG; IGH monoclonal | EBV driven B-cell proliferation in an immunosuppressed setting | Complex karyotypes; MYC rearrangement (>50%); PRDM1 mutations (49%) |
| Burkitt lymphoma -Endemic -Sporadic -HIV+ | LN or extranodal sites | 100 5–80 30–40 | − | − | monotonous medium-sized blasts without prominent nucleoli “Starry sky” appearance; | GC phenotype: CD45+, Pan-B cell markers+, CD10+, BCL6+, BCL2−, sIgM+, Ki67 100%, MYC 100% IGH monoclonal | Synergistic effect of EBV and MYC; EBV may not be essential. | MYC/IG translocation (>90%); TP53 (30%), TCF3 (70% sBL) mutations. |
| Classic Hodgkin lymphoma | LN | 20–100 | + | − | HRS cells in a typical inflammatory background | CD45−, CD20−/+, CD79a−/+, PAX5+ (weak), OCT2−, BOB1−, Ig−, CD30+, CD15+, CD10−, BCL6−/+, MUM1+ | EBV pathogenetic role likely in some cases “Crippling” mutations of the IGH genes. Aberrant Ig transcription | NFkB and JAK/STAT pathways activated. GEP: “Host immune response” Altered PD1-PD-L1 signalling |
| Anatomic Sites and Distribution | EBV (%) | LMP1 | Cell of Origin | Morphologic Features | Immunophenotypic Features and Clonality | Pathogenesis and Main Genetic Alterations | |
|---|---|---|---|---|---|---|---|
| CAEBV, systemic form | spleen, liver, LN, extranodal sites | 100 | +/− | T cell (59%), NK cell (41%) | There are no changes suggestive of a malignant LPD | CD4 > CD8 CD56+ (41%) TCR monoclonal in 84% | Unknown. Racial predisposition, defective EBV immune response |
| Hydroa vacciniforme-like LPD | skin | 100 | +/− | T cell (85%) αβ and γδ rarely NK cell (15%) | Intraepidermal spongiotic vesicles. Periadnexal and perivascular infiltrate with angiodestruction | CD8 > CD4 CD56+ (15%) CD30 often express TCR often monoclonal | Unknown. Racial predisposition, defective EBV immune response |
| Severe mosquito bite allergy | skin | 100 | − | NK cell | Small reactive looking lymphocytes to large atypical neoplastic cells | CD3ε+, CD56+, TIA1+, granzyme B+, CD30+ TCR polyclonal | CD4+ T cells response to mosquito salivary gland secretion that reactivates EBV infection |
| Systemic EBV+ T-cell lymphoma of childhood | Spleen, liver, LN | 100 | +/− | T cell (CD8 > CD4) | Hemophagocytosis with T cell infiltrates in LN, liver, spleen | CD8+, CD3+, CD2+, CD56−; rare cases CD4+ TCR monoclonal | Unknown. Racial predisposition, defective EBV immune response |
| Aggressive NK-cell leukaemia | BM, PB, spleen, liver | 95 | − | NK cell | Hemophagocytosis. PB and BM infiltrated with atypical granular lymphocytes with broad cytological spectrum | CD3ε+, CD56+, CD2+, TIA1+, granzyme B+, CD16+ (75%), CD95+ (FAS), FASL+, CD3−, CD5, CD57−. TCR polyclonal | Unknown. Racial predisposition, defective EBV response. Complex karyotype with gains of 1q23.1–1q24.2 and 1q31.3–q44 and losses of 7p15.1–p22.3 and 17p13.1. STAT3, STAT5B and TP53 mutations |
| Extranodal NK/T-cell lymphoma, nasal type | Nasal cavity, nasopharynx, palate, skin, GI, testis, CNS | 100 | +/− | NK cell rarely T cell | Ulceration, coagulative necrosis, karyorrhexis, angiocentricity and angioinvasion. Cytological spectrum broad | CD3ε+, CD2+, CD56+, TIA1+, granzyme B+, CD30+, CD3−, CD5−, CD16−, CD57−, CD4/CD8−. TCR polyclonal | Racial predisposition, defective EBV immune response. Cytogenetic alterations with del (6)(q21q25) or i(6)(p10). Recurrent mutations in STAT3, STAT5B, DDX3X, TP53, MGA, PRDM1, FOXO3, HACE1, BCOR |
| Primary EBV+ nodal T- or NK-cell lymphoma | LN | 100 | +/− | T cell TCRαβ (58%), TCRγδ (15%), TCR silent (29%), rarely NK cell | LN with diffuse infiltration of pleomorphic, anaplastic or HRS-like cells | cytotoxic T-cell phenotype: CD3+, CD8+, TIA1+, granzyme B+, CD56−, CD4−, CD30+/−. TCR monoclonal | Racial predisposition, defective EBV immune response. Cytogenetic alterations with frequent losses of 14q11.2. GEP: enrichment of immune response genes and genes associated with cytotoxic activation. PD-L1 upregulation |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dojcinov, S.D.; Fend, F.; Quintanilla-Martinez, L. EBV-Positive Lymphoproliferations of B- T- and NK-Cell Derivation in Non-Immunocompromised Hosts. Pathogens 2018, 7, 28. https://doi.org/10.3390/pathogens7010028
Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-Positive Lymphoproliferations of B- T- and NK-Cell Derivation in Non-Immunocompromised Hosts. Pathogens. 2018; 7(1):28. https://doi.org/10.3390/pathogens7010028
Chicago/Turabian StyleDojcinov, Stefan D., Falko Fend, and Leticia Quintanilla-Martinez. 2018. "EBV-Positive Lymphoproliferations of B- T- and NK-Cell Derivation in Non-Immunocompromised Hosts" Pathogens 7, no. 1: 28. https://doi.org/10.3390/pathogens7010028
APA StyleDojcinov, S. D., Fend, F., & Quintanilla-Martinez, L. (2018). EBV-Positive Lymphoproliferations of B- T- and NK-Cell Derivation in Non-Immunocompromised Hosts. Pathogens, 7(1), 28. https://doi.org/10.3390/pathogens7010028