
Viral Interactions with PDZ Domain-Containing Proteins—An Oncogenic Trait?

Abstract
:1. Introduction

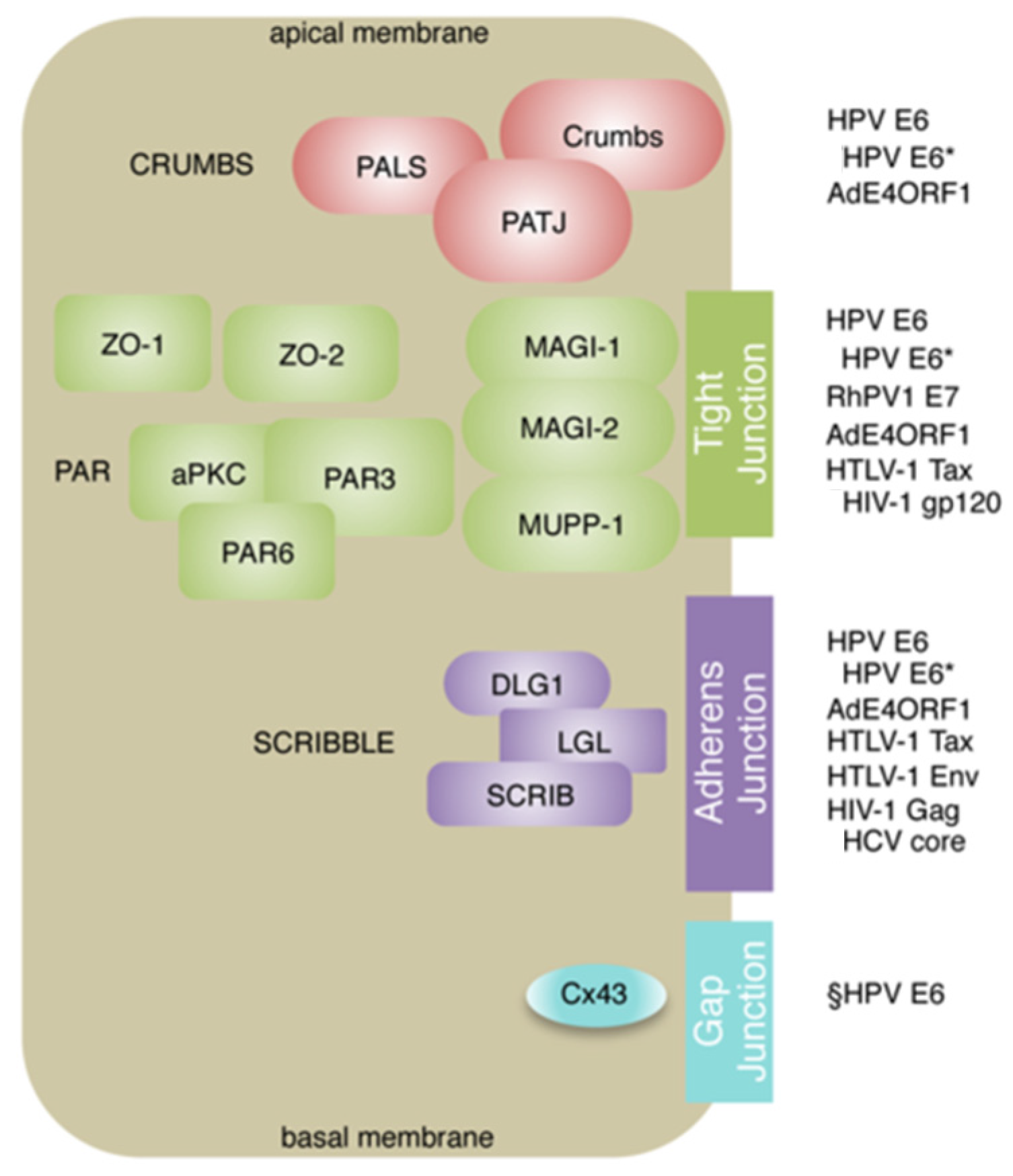{kind=link}
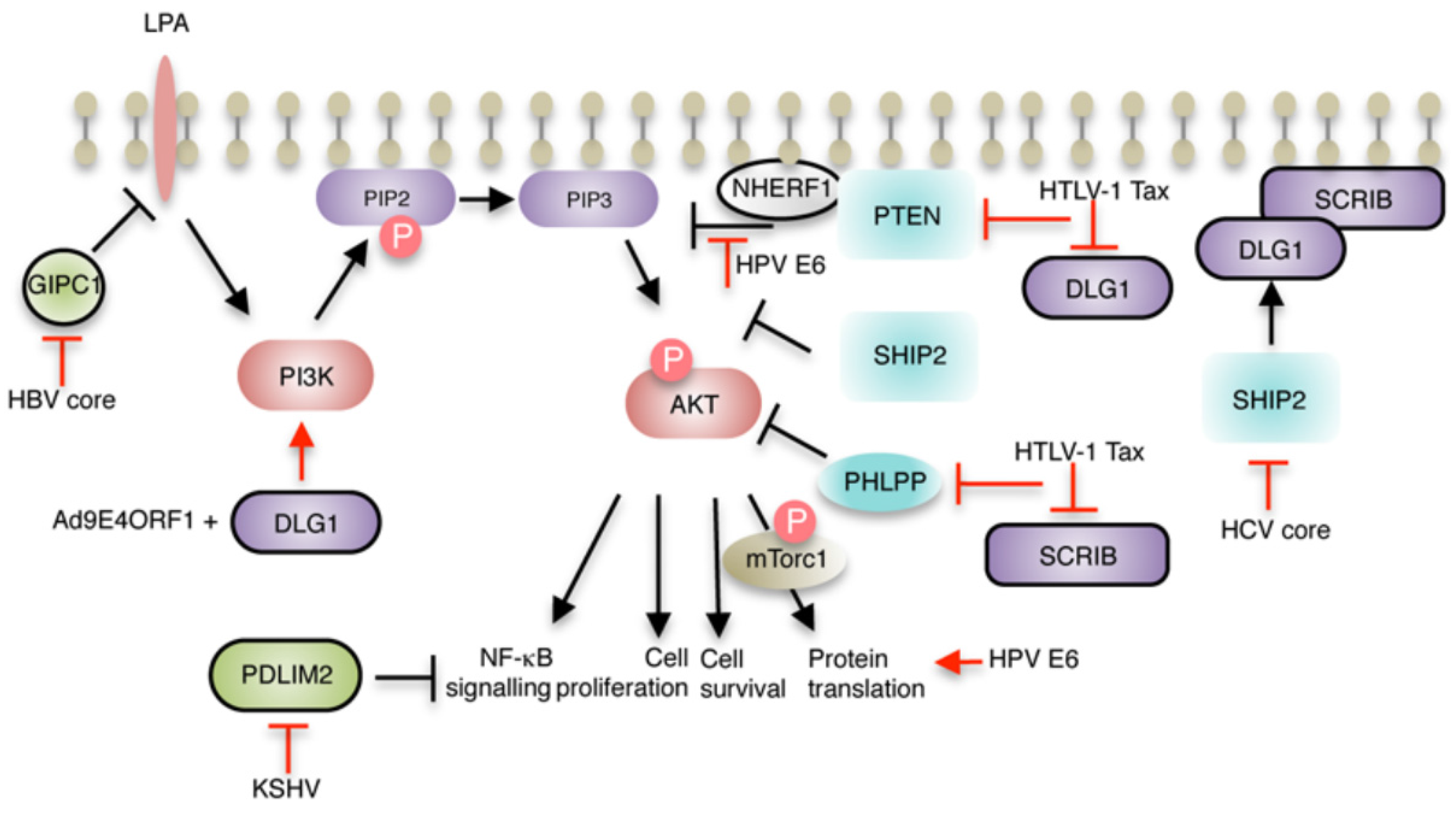{kind=link}
| Virus/Viral Protein | PDZ Target | PBM Mediated Interaction (Type, Sequence) | Virus Mediated Effect upon PDZ Target (If Known) | References |
|---|---|---|---|---|
| HCV Core protein | DLG1, SCRIB | No | Decreased expression (DLG1); mislocalization (SCRIB) | [25] |
| HBV Core protein | GIPC1 (also known as TIP-2), PTPN3 | Yes (non-typical, SQC) | Disruption of GIPC1 interaction with LPA signalling molecule (?) | [26,27] |
| KSHV | PDLIM2 | No | Suppresses PDLIM2 transcription by increased promoter methylation | [28] |
| HIV-1 Gag | PDZD8, DLG1 | No | Stabilizes viral capsid (PDZD8 [?]); colocalization in plasma membrane, antagonistic to virus infectivity (DLG1) | [29,30] |
| HIV-1 Env | SDCBP1 (Syntenin-1) | No | Recruited to the plasma membrane during virus attachment, antagonistic to virus infectivity (?) | [31] |
| HIV-1 Glycoprotein 120 | ZO-1 | No | Displacement from tight junctions, reduced ZO-1 transcription | [32] |
| HTLV-1 Env | DLG1 | Yes (class I, SSL) | Colocalization in plasma membrane, at possible virological synapse, also colocalization with major HTLV-1 receptor GLUT-1 | [33] |
| HTLV-1 Tax | DLG1, SCRIB, MAGI-1, MAGI-3, Pro-IL-16, TIP-1 | Yes (class I, TEV) | Altered subcellular localization (DLG1, SCRIB, MAGI-1) | [34,35,36,37] |
| Ad E4ORF1 | DLG1, MAGI-1, MUPP1,PATJ, ZO-2 | Yes (class I, TLV) | Altered subcellular localization (MAGI-1, MUPP1, PATJ, ZO-2), associated with tight junction disruption, accumulate at plasma membrane (DLG1) where activates PI3K signalling | [38,39,40,41,42] |
| Beta-HPV8 E6 | SDCBP2 (Syntenin-2) | No | Repression of SDCBP2 transcription | [43] |
| RhPV1 (MmPV1) E7 | PARD3 (PAR3) | Yes (class I, SRV) | Degradation via the proteasome | [44] |
| High-risk alpha-HPV E6 | Includes DLG1, SCRIB, MAGI-1, MAGI-2, MAGI-3, PTPN3, PTPN13, PATJ, PAR3, SDCBP2 (see Table 2 for full list) | Yes (class I, S/TXV/L) | Degradation via the proteasome, transcriptional downregulation, altered subcellular localization, complex formation to alter function | See Table 2 |
| 2High-risk alpha-HPV E6* | DLG1, MAGI-1, PATJ, SCRIB | No | Degradation via the proteasome | [45,46] |
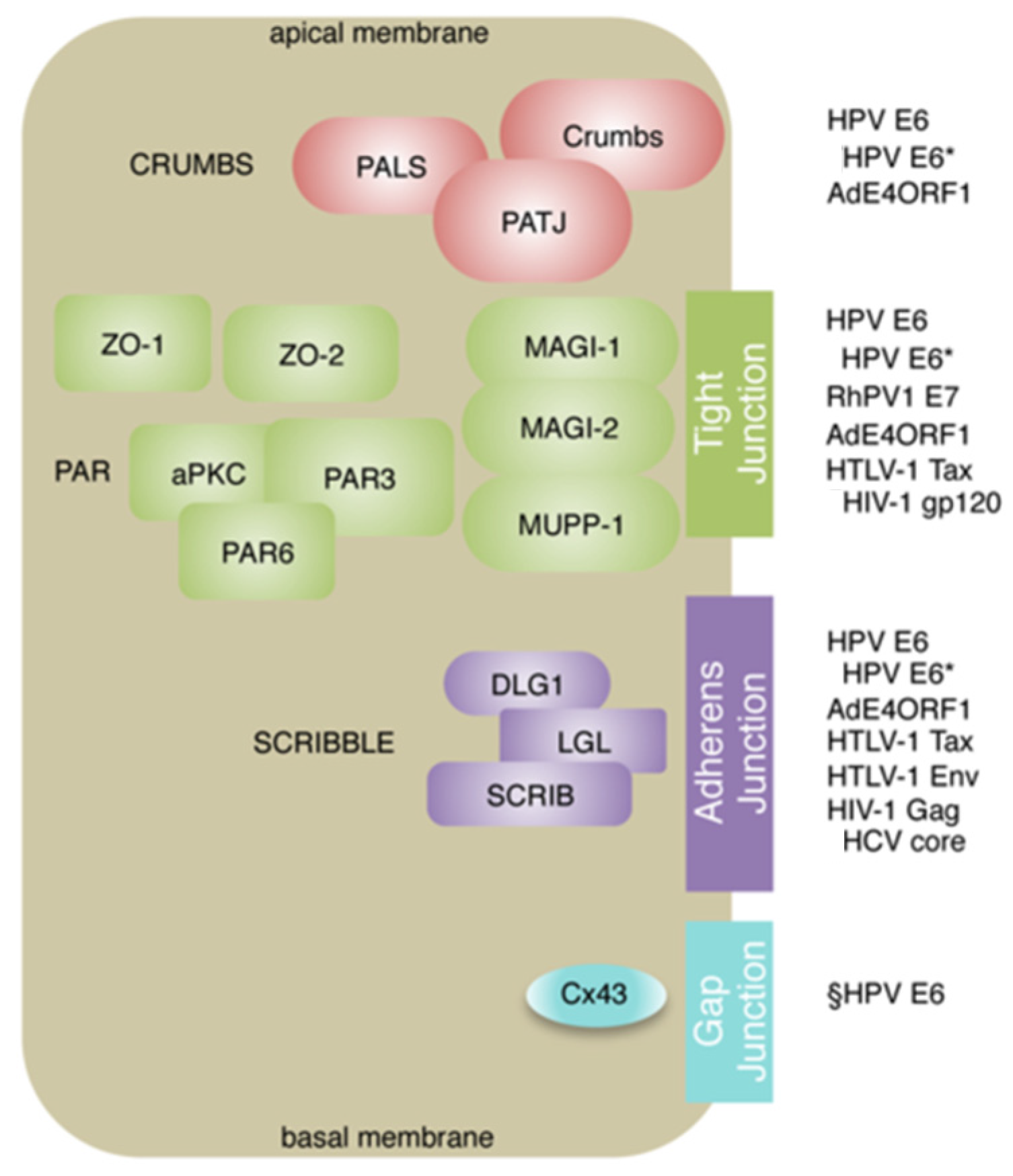
2. Oncogenic Viruses and PDZ Proteins
2.1. Hepatitis C virus (HCV)
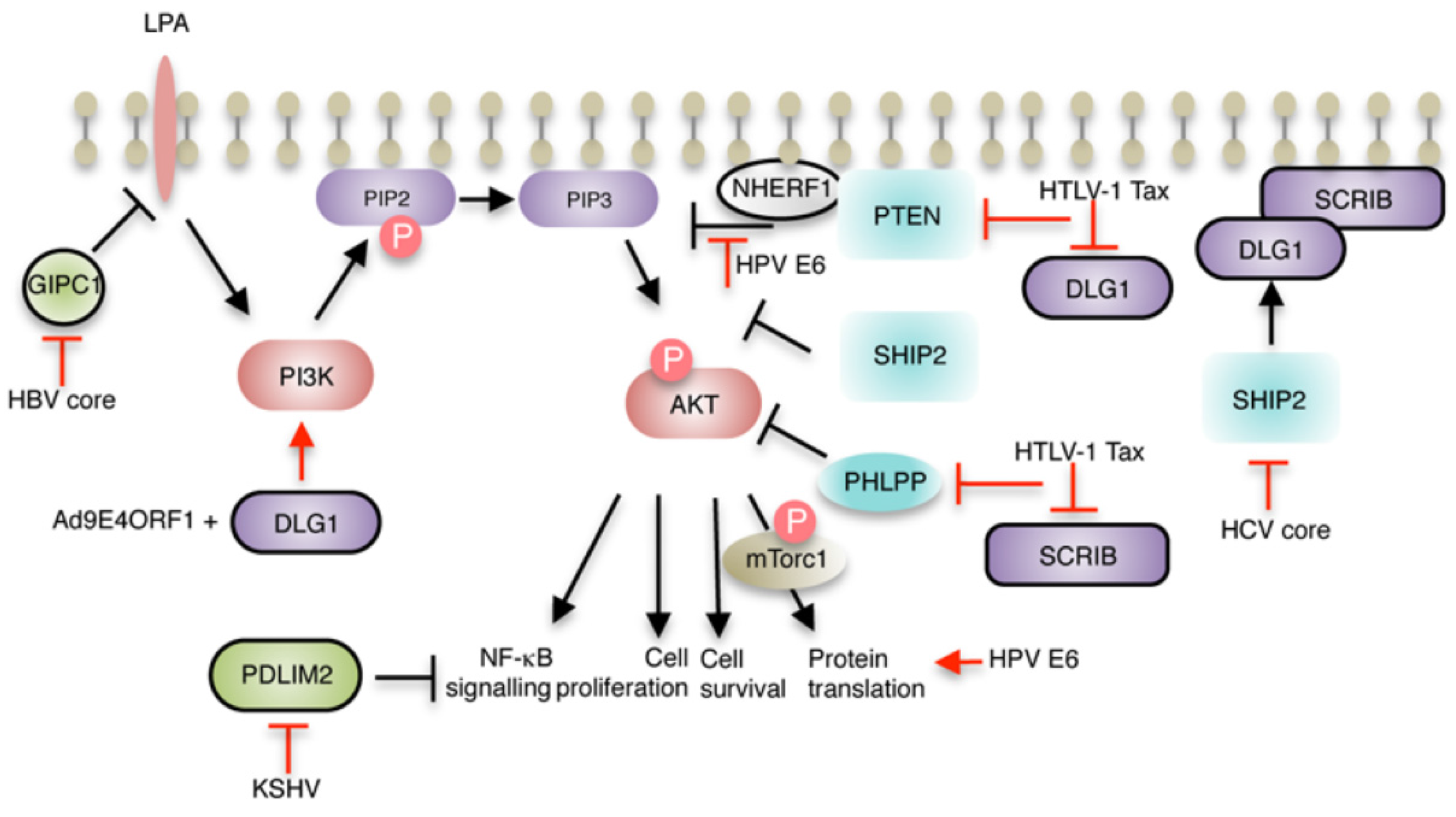
2.2. Hepatitis B virus (HBV)
2.3. Kaposi Sarcoma Herpesvirus (KSHV)
2.4. Human Immunodeficiency Virus Type 1 (HIV-1)
2.5. Human T Cell Leukaemia Virus (HTLV-1)
2.6. Adenoviruses (Ad)
2.7. High-Risk Human Papillomaviruses (HPV)
| PDZ Protein (Gene Symbol) | Alternative Name(s) | Cellular Functions | Effect of E6 on PDZ Protein | References |
|---|---|---|---|---|
| DLG1 | SAP97 | Cell polarity, proliferation, asymmetric cell division, migration and adhesion | Degradation, complex formation with subcellular pools of DLG1 | [104,109,110,111] |
| SCRIB | Scribble, Vartul, LAP4 | Cell polarity, proliferation, apoptotic signalling | Degradation | [112] |
| MPDZ | MUPP1 | Tight junction localization | Degradation | [40] |
| MAGI-1 | AIP3, WWP3 | Tight junction integrity, cell growth. | Degradation | [38] |
| MAGI-2 | AIP1 | Tight junction function, planar cell polarity, proliferation and motility | Degradation | [113] |
| MAGI-3 | Cell polarity, receptor trafficking, cell survival | Degradation | [113] | |
| TAX1BP3 | TIP-1 | Cell polarity, cell motility, ion transport | Complex formation | [114] |
| GIPC1 | Synectin, TIP-2 | Cell surface receptor expression and trafficking, TGFβ signalling, | Degradation | [115] |
| INADL | PATJ | Tight junction formation, cell polarity | Degradation | [41,46] |
| DLG4 | PSD95, SAP90, TIP15 | Receptor clustering in neuronal cells, epithelial cell polarization | Degradation | [116] |
| PTPN3 | PTPH1 | Tyrosine phosphatase, cell growth regulator | Degradation | [103,117] |
| GOPC | CAL | Intracellular trafficking | Degradation | [118] |
| PTPN13 | FAP-1, PTPL-1, PTP-BAS, PTPBL | Fas-associated phosphatase, cell growth signalling, apoptotic signalling | Degradation | [119] |
| SLC9A3R1 | NHERF1, EBP50 | G-protein couple receptor regulation, cytoskeleton anchoring | Degradation of phosphorylated forms | [120] |
| SDCBP2 | Syntenin-2 | Phosphoinositol mediated cell signalling, cell division and survival | mRNA downregulation | [43] |
| PARD3 | PAR3 | Cell polarity, asymmetrical cell division | Altered subcellular localisation | [121] |
| PDZRN3 | LNX3 | E3 ubiquitin ligase, planar cell polarity signalling | Degradation | [122] |
3. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, H.J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell. Commun. Signal. 2010, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Subbaiah, V.K.; Kranjec, C.; Thomas, M.; Banks, L. PDZ domains: The building blocks regulating tumorigenesis. Biochem. J. 2011, 439, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.; Delury, C.; Marsh, E. The PDZ protein discs-large (dlg): The “Jekyll and Hyde” of the epithelial polarity proteins. FEBS J. 2012, 279, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Zori, P.; Roberts, S.; Banks, L. Differential regulation of cell-cell contact, invasion and anoikis by hScrib and hDlg in keratinocytes. PLoS ONE 2012, 7, e40279. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, E.D.; Oliaro, J.; Ramsbottom, K.M.; Newbold, A.; Humbert, P.O.; Johnstone, R.W.; Russell, S.M. Scribble acts as an oncogene in emu-myc-driven lymphoma. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Feigin, M.E.; Akshinthala, S.D.; Araki, K.; Rosenberg, A.Z.; Muthuswamy, L.B.; Martin, B.; Lehmann, B.D.; Berman, H.K.; Pietenpol, J.A.; Cardiff, R.D.; et al. Mislocalization of the cell polarity protein scribble promotes mammary tumorigenesis and is associated with basal breast cancer. Cancer Res. 2014, 74, 3180–3194. [Google Scholar] [CrossRef] [PubMed]
- Tonikian, R.; Zhang, Y.; Sazinsky, S.L.; Currell, B.; Yeh, J.H.; Reva, B.; Held, H.A.; Appleton, B.A.; Evangelista, M.; Wu, Y.; et al. A specificity map for the PDZ domain family. PLoS Biol. 2008, 6, e239. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Guardeno, J.M.; Nieto-Torres, J.L.; DeDiego, M.L.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castano-Rodriguez, C.; Enjuanes, L. The PDZ-binding motif of severe acute respiratory syndrome coronavirus envelope protein is a determinant of viral pathogenesis. PLoS Pathog. 2014, 10, e1004320. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castano-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef] [PubMed]
- Teoh, K.T.; Siu, Y.L.; Chan, W.L.; Schluter, M.A.; Liu, C.J.; Peiris, J.S.; Bruzzone, R.; Margolis, B.; Nal, B. The SARS coronavirus E protein interacts with pals1 and alters tight junction formation and epithelial morphogenesis. Mol. Biol. Cell. 2010, 21, 3838–3852. [Google Scholar] [CrossRef] [PubMed]
- Terrien, E.; Chaffotte, A.; Lafage, M.; Khan, Z.; Prehaud, C.; Cordier, F.; Simenel, C.; Delepierre, M.; Buc, H.; Lafon, M.; et al. Interference with the Pten-Mast2 interaction by a viral protein leads to cellular relocalization of Pten. Sci. Signal. 2012, 5, ra58. [Google Scholar] [CrossRef] [PubMed]
- Prehaud, C.; Wolff, N.; Terrien, E.; Lafage, M.; Megret, F.; Babault, N.; Cordier, F.; Tan, G.S.; Maitrepierre, E.; Menager, P.; et al. Attenuation of rabies virulence: Takeover by the cytoplasmic domain of its envelope protein. Sci. Signal. 2010, 3, ra5. [Google Scholar] [CrossRef] [PubMed]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.; Mukatira, S.; Finkelstein, D.B.; Xu, X.; Wang, J.; Ma, J.; Fan, Y.; et al. Large-scale sequence analysis of avian influenza isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef] [PubMed]
- Soubies, S.M.; Volmer, C.; Croville, G.; Loupias, J.; Peralta, B.; Costes, P.; Lacroux, C.; Guerin, J.L.; Volmer, R. Species-specific contribution of the four C-terminal amino acids of Influenza A virus NS1 protein to virulence. J. Virol. 2010, 84, 6733–6747. [Google Scholar] [CrossRef] [PubMed]
- Cauthen, A.N.; Swayne, D.E.; Sekellick, M.J.; Marcus, P.I.; Suarez, D.L. Amelioration of influenza virus pathogenesis in chickens attributed to the enhanced interferon-inducing capacity of a virus with a truncated NS1 gene. J. Virol. 2007, 81, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, Y.; Jiao, P.; Wang, A.; Zhao, F.; Tian, G.; Wang, X.; Yu, K.; Bu, Z.; Chen, H. The NS1 gene contributes to the virulence of H5N1 avian influenza viruses. J. Virol. 2006, 80, 11115–11123. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Hwang, M.W.; Lee, I.; Park, S.; Lee, S.; Bae, J.Y.; Heo, J.; Kim, D.; Jang, S.I.; Park, M.S.; et al. The PDZ-binding motif of the avian NS1 protein affects transmission of the 2009 Influenza A(H1N1) virus. Biochem. Biophys. Res. Commun. 2014, 449, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Soubies, S.M.; Volmer, C.; Guerin, J.L.; Volmer, R. Truncation of the NS1 protein converts a low pathogenic avian influenza virus into a strong interferon inducer in duck cells. Avian Dis. 2010, 54, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Hossain, M.J.; Hickman, D.; Perez, D.R.; Lamb, R.A. A new influenza virus virulence determinant: The NS1 protein four c-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 2008, 105, 4381–4386. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Kranjec, C.; Nagasaka, K.; Matlashewski, G.; Banks, L. Analysis of the PDZ binding specificities of influenza a virus NS1 proteins. Virol. J. 2011, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Golebiewski, L.; Liu, H.; Javier, R.T.; Rice, A.P. The avian influenza virus NS1 ESEV PDZ binding motif associates with Dlg1 and scribble to disrupt cellular tight junctions. J. Virol. 2011, 85, 10639–10648. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, X.; Wang, Y.; Li, B.; Li, H.; Li, Y.; Zhou, W.; Zhang, C.; Rao, Z.; Bartlam, M.; et al. PDLIM2 selectively interacts with the PDZ binding motif of highly pathogenic avian H5N1 influenza A virus NS1. PLoS ONE 2011, 6, e19511. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Golebiewski, L.; Dow, E.C.; Krug, R.M.; Javier, R.T.; Rice, A.P. The ESEV PDZ-binding motif of the avian influenza a virus NS1 protein protects infected cells from apoptosis by directly targeting scribble. J. Virol. 2010, 84, 11164–11174. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Awad, A.; Sar, S.; Barre, R.; Cariven, C.; Marin, M.; Salles, J.P.; Erneux, C.; Samuel, D.; Gassama-Diagne, A. Ship2 regulates epithelial cell polarity through its lipid product, which binds to Dlg1, a pathway subverted by hepatitis C virus core protein. Mol. Biol. Cell. 2013, 24, 2171–2185. [Google Scholar] [CrossRef] [PubMed]
- Razanskas, R.; Sasnauskas, K. Interaction of hepatitis B virus core protein with human GIPC1. Arch. Virol. 2010, 155, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.C.; Lin, Y.C.; Hung, C.S.; Huang, C.J.; Lee, M.Y.; Yang, S.C.; Ting, L.P. Suppression of hepatitis B viral gene expression by protein-tyrosine phosphatase PTPN3. J. Biomed. Sci. 2007, 14, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Xiao, Y.; Qu, Z. Oncovirus kaposi sarcoma herpesvirus (KSHV) represses tumor suppressor pdlim2 to persistently activate nuclear factor kappaB (NF-kappaB) and Stat3 transcription factors for tumorigenesis and tumor maintenance. J. Biol. Chem. 2015, 290, 7362–7368. [Google Scholar] [CrossRef] [PubMed]
- Henning, M.S.; Morham, S.G.; Goff, S.P.; Naghavi, M.H. PDZD8 is a novel Gag-interacting factor that promotes retroviral infection. J. Virol. 2010, 84, 8990–8995. [Google Scholar] [CrossRef] [PubMed]
- Perugi, F.; Muriaux, D.; Ramirez, B.C.; Chabani, S.; Decroly, E.; Darlix, J.L.; Blot, V.; Pique, C. Human discs large is a new negative regulator of human immunodeficiency virus-1 infectivity. Mol. Biol. Cell 2009, 20, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Alonso, M.; Rocha-Perugini, V.; Alvarez, S.; Moreno-Gonzalo, O.; Ursa, A.; Lopez-Martin, S.; Izquierdo-Useros, N.; Martinez-Picado, J.; Munoz-Fernandez, M.A.; Yanez-Mo, M.; et al. The PDZ-adaptor protein syntenin-1 regulates HIV-1 entry. Mol. Biol. Cell 2012, 23, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Nazli, A.; Chan, O.; Dobson-Belaire, W.N.; Ouellet, M.; Tremblay, M.J.; Gray-Owen, S.D.; Arsenault, A.L.; Kaushic, C. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog. 2010, 6, e1000852. [Google Scholar] [CrossRef] [PubMed]
- Blot, V.; Delamarre, L.; Perugi, F.; Pham, D.; Benichou, S.; Benarous, R.; Hanada, T.; Chishti, A.H.; Dokhelar, M.C.; Pique, C. Human DLG protein binds to the envelope glycoproteins of human T-cell leukemia virus type 1 and regulates envelope mediated cell-cell fusion in T lymphocytes. J. Cell Sci. 2004, 117, 3983–3993. [Google Scholar] [CrossRef] [PubMed]
- Rousset, R.; Fabre, S.; Desbois, C.; Bantignies, F.; Jalinot, P. The C-terminus of the HTLV-1 Tax oncoprotein mediates interaction with the PDZ domain of cellular proteins. Oncogene 1998, 16, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ohsugi, Y.; Uchida-Toita, M.; Akiyama, T.; Yoshida, M. Tax oncoprotein of HTLV-1 binds to the human homologue of drosophila discs large tumor suppressor protein, hDlg, and perturbs its function in cell growth control. Oncogene 1999, 18, 5967–5972. [Google Scholar] [CrossRef] [PubMed]
- Makokha, G.N.; Takahashi, M.; Higuchi, M.; Saito, S.; Tanaka, Y.; Fujii, M. Human T-cell leukemia virus type 1 Tax protein interacts with and mislocalizes the PDZ domain protein Magi-1. Cancer Sci. 2013, 104, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Sakurai, M.; Higuchi, M.; Mori, N.; Fukushi, M.; Oie, M.; Coffey, R.J.; Yoshiura, K.; Tanaka, Y.; Uchiyama, M.; et al. Human T-cell leukemia virus type 1 Tax oncoprotein induces and interacts with a multi-PDZ domain protein, Magi-3. Virology 2004, 320, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.A.; Lee, S.S.; Thomas, M.; Banks, L.; Javier, R. Interactions of the PDZ-protein Magi-1 with adenovirus E4-orf1 and high-risk papillomavirus E6 oncoproteins. Oncogene 2000, 19, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.A.; Weiss, R.S.; Lee, S.S.; Javier, R. Link of the unique oncogenic properties of adenovirus type 9 E4-Orf1 to a select interaction with the candidate tumor suppressor protein Zo-2. Embo J. 2001, 20, 5578–5586. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Glaunsinger, B.; Mantovani, F.; Banks, L.; Javier, R.T. Multi-PDZ domain protein Mupp1 is a cellular target for both adenovirus E4-Orf1 and high-risk papillomavirus type 18 E6 oncoproteins. J. Virol. 2000, 74, 9680–9693. [Google Scholar] [CrossRef] [PubMed]
- Latorre, I.J.; Roh, M.H.; Frese, K.K.; Weiss, R.S.; Margolis, B.; Javier, R.T. Viral oncoprotein-induced mislocalization of select PDZ proteins disrupts tight junctions and causes polarity defects in epithelial cells. J. Cell Sci. 2005, 118, 4283–4293. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.K.; Latorre, I.J.; Chung, S.H.; Caruana, G.; Bernstein, A.; Jones, S.N.; Donehower, L.A.; Justice, M.J.; Garner, C.C.; Javier, R.T. Oncogenic function for the Dlg1 mammalian homolog of the Drosophila discs-large tumor suppressor. Embo J. 2006, 25, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Lazic, D.; Hufbauer, M.; Zigrino, P.; Buchholz, S.; Kazem, S.; Feltkamp, M.C.; Mauch, C.; Steger, G.; Pfister, H.; Akgul, B. Human papillomavirus type 8 E6 oncoprotein inhibits transcription of the PDZ protein syntenin-2. J. Virol. 2012, 86, 7943–7952. [Google Scholar] [CrossRef] [PubMed]
- Tomaic, V.; Gardiol, D.; Massimi, P.; Ozbun, M.; Myers, M.; Banks, L. Human and primate tumour viruses use PDZ binding as an evolutionarily conserved mechanism of targeting cell polarity regulators. Oncogene 2009, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Tomaic, V.; Banks, L. The human papillomavirus (hpv) E6* proteins from high-risk, mucosal hpvs can direct degradation of cellular proteins in the absence of full-length E6 protein. J. Virol. 2009, 83, 9863–9874. [Google Scholar] [CrossRef] [PubMed]
- Storrs, C.H.; Silverstein, S.J. Patj, a tight junction-associated pdz protein, is a novel degradation target of high-risk human papillomavirus E6 and the alternatively spliced isoform 18 E6. J. Virol. 2007, 81, 4080–4090. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, Y.; Arnold, R.; McLaughlin, M.; Nim, S.; Joshi, R.; Ray, D.; Liu, B.; Teyra, J.; Pawson, T.; Moffat, J.; et al. Large-scale interaction profiling of PDZ domains through proteomic peptide-phage display using human and viral phage peptidomes. Proc. Natl. Acad. Sci. USA 2014, 111, 2542–2547. [Google Scholar] [CrossRef] [PubMed]
- Belotti, E.; Polanowska, J.; Daulat, A.M.; Audebert, S.; Thome, V.; Lissitzky, J.C.; Lembo, F.; Blibek, K.; Omi, S.; Lenfant, N.; et al. The human PDZome: A gateway to PSD95-disc large-zonula occludens (PDZ)-mediated functions. Mol. Cell. Proteom. 2013, 12, 2587–2603. [Google Scholar] [CrossRef] [PubMed]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Mee, C.J.; Farquhar, M.J.; Harris, H.J.; Hu, K.; Ramma, W.; Ahmed, A.; Maurel, P.; Bicknell, R.; Balfe, P.; McKeating, J.A. Hepatitis C virus infection reduces hepatocellular polarity in a vascular endothelial growth factor-dependent manner. Gastroenterology 2010, 138, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Eyre, N.S.; Drummer, H.E.; Beard, M.R. The SR-BI partner PDZK1 facilitates hepatitis C virus entry. PLoS Pathog. 2010, 6, e1001130. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yu, X.; Guo, Y.; Kong, L. Interaction networks of hepatitis C virus NS4B: Implications for antiviral therapy. Cell. Microbiol. 2012, 14, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Melik, W.; Ellencrona, K.; Wigerius, M.; Hedstrom, C.; Elvang, A.; Johansson, M. Two PDZ binding motifs within NS5 have roles in tick-borne encephalitis virus replication. Virus Res. 2012, 169, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Ellencrona, K.; Syed, A.; Johansson, M. Flavivirus NS5 associates with host-cell proteins zonula occludens-1 (Zo-1) and regulating synaptic membrane exocytosis-2 (RIMS2) via an internal PDZ binding mechanism. Biol. Chem. 2009, 390, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Javier, R.T.; Rice, A.P. Emerging theme: Cellular PDZ proteins as common targets of pathogenic viruses. J. Virol. 2011, 85, 11544–11556. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, T.; Galet, C.; Kishi, M.; Ascoli, M. Gipc binds to the human lutropin receptor (hLHR) through an unusual PDZ domain binding motif, and it regulates the sorting of the internalized human choriogonadotropin and the density of cell surface hLHR. J. Biol. Chem. 2003, 278, 49348–49357. [Google Scholar] [CrossRef] [PubMed]
- Varsano, T.; Taupin, V.; Guo, L.; Baterina, O.Y., Jr.; Farquhar, M.G. The PDZ protein Gipc regulates trafficking of the LPA1 receptor from APPl signaling endosomes and attenuates the cell's response to LPA. PLoS ONE 2012, 7, e49227. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.; Bouchard, M.J. The hepatitis B virus (HBV) hbx protein activates Akt to simultaneously regulate HBV replication and hepatocyte survival. J. Virol. 2015, 89, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Grusby, M.J.; Kaisho, T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 2007, 8, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Guth, C.A.; Sodroski, J. Contribution of PDXD8 to stabilization of the human immunodeficiency virus type 1 capsid. J. Virol. 2014, 88, 4612–4623. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernandez, A.; Alvarez, S.; Gordon-Alonso, M.; Barrero, M.; Ursa, A.; Cabrero, J.R.; Fernandez, G.; Naranjo-Suarez, S.; Yanez-Mo, M.; Serrador, J.M.; et al. Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol. Biol. Cell 2005, 16, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- Henning, M.S.; Stiedl, P.; Barry, D.S.; McMahon, R.; Morham, S.G.; Walsh, D.; Naghavi, M.H. PDZD8 is a novel moesin-interacting cytoskeletal regulatory protein that suppresses infection by herpes simplex virus type 1. Virology 2011, 415, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sodroski, J. Efficient human immunodeficiency virus (hiv-1) infection of cells lacking PDZD8. Virology 2015, 481, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Guo, S.; Ye, D.; Ma, T.Y. TNF-alpha modulation of intestinal epithelial tight junction barrier is regulated by Erk1/2 activation of ELK-1. Am. J. Pathol. 2013, 183, 1871–1884. [Google Scholar] [CrossRef] [PubMed]
- Ilinskaya, A.; Heidecker, G.; Derse, D. Opposing effects of a tyrosine-based sorting motif and a PDZ-binding motif regulate human t-lymphotropic virus type 1 envelope trafficking. J. Virol. 2010, 84, 6995–7004. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Higuchi, M.; Shoji, T.; Yoshita, M.; Ishioka, K.; Takahashi, M.; Oie, M.; Tanaka, Y.; Uchiyama, M.; Fujii, M. Knockdown of synapse-associated protein Dlg1 reduces syncytium formation induced by human t-cell leukemia virus type 1. Virus Genes 2008, 37, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Higuchi, M.; Niinuma, A.; Ohashi, M.; Fukushi, M.; Oie, M.; Akiyama, T.; Tanaka, Y.; Gejyo, F.; Fujii, M. PDZ domain-binding motif of human T-cell leukemia virus type 1 Tax oncoprotein augments the transforming activity in a rat fibroblast cell line. Virology 2004, 318, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Takahashi, C.; Yamaoka, S.; Nosaka, T.; Maki, M.; Hatanaka, M. Oncogenic transformation by the Tax gene of humanT-cell leukemia virus type 1 in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Grossman, W.J.; Kimata, J.T.; Wong, F.H.; Zutter, M.; Ley, T.J.; Ratner, L. Development of leukemia in mice transgenic for the Tax gene of human T-cell leukemia virus type 1. Proc. Natl. Acad. Sci. USA 1995, 92, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Yamamoto, B.; Haoudi, A.; Semmes, O.J.; Green, P.L. PDZ binding motif of HTLV-1 Tax promotes virus-mediated t-cell proliferation in vitro and persistence in vivo. Blood 2006, 107, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Humphries, L.A.; Tomassian, T.; Mittelstadt, P.; Zhang, M.; Miceli, M.C. Scaffold protein Dlgh1 coordinates alternative p38 kinase activation, directing T cell receptor signals toward NFAT but not NF-kappaB transcription factors. Nat. Immunol. 2007, 8, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Bidoia, C.; Mazzorana, M.; Pagano, M.A.; Arrigoni, G.; Meggio, F.; Pinna, L.A.; Bertazzoni, U. The pleiotropic protein kinase CK2 phosphorylates HTLV-1 Tax protein in vitro, targeting its PDZ-binding motif. Virus Genes 2010, 41, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.; Rabizadeh, S.; Ishiguro, K.; Andre, N.; Ortiz, J.B.; Wachtel, H.; Morris, D.G.; Lopez-Ilasaca, M.; Shaw, A.C.; Swat, W.; et al. Discs large (dlg1) complexes in lymphocyte activation. J. Cell Biol. 2004, 166, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Ludford-Menting, M.J.; Oliaro, J.; Sacirbegovic, F.; Cheah, E.T.; Pedersen, N.; Thomas, S.J.; Pasam, A.; Iazzolino, R.; Dow, L.E.; Waterhouse, N.J.; et al. A network of PDZ-containing proteins regulates T cell polarity and morphology during migration and immunological synapse formation. Immunity 2005, 22, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Arpin-Andre, C.; Mesnard, J.M. The PDZ domain-binding motif of the human T cell leukemia virus type 1 Tax protein induces mislocalization of the tumor suppressor hscrib in T cells. J. Biol. Chem. 2007, 282, 33132–33141. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Weiss, R.S.; Javier, R.T. Binding of human virus oncoproteins to hdlg/SAP97, a mammalian homolog of the drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6670–6675. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, L.M.; Sammut, B.; Graham, D.B.; Chan-Wang, J.; Brim, K.L.; Huett, A.S.; Miletic, A.V.; Kloeppel, T.; Landry, A.; Xavier, R.; et al. Dlgh1 is a negative regulator of T-lymphocyte proliferation. Mol. Cell. Biol. 2007, 27, 7574–7581. [Google Scholar] [CrossRef] [PubMed]
- Cherian, M.A.; Baydoun, H.H.; Al-Saleem, J.; Shkriabai, N.; Kvaratskhelia, M.; Green, P.; Ratner, L. Akt pathway activation by human T-cell leukemia virus type 1 Tax oncoprotein. J. Biol. Chem. 2015, 290, 26270–26281. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Takahashi, M.; Higuchi, M.; Oie, M.; Tanaka, Y.; Kiyono, T.; Aoyagi, Y.; Fujii, M. The PDZ domain binding motif (PBM) of human T-cell leukemia virus type 1 Tax can be substituted by heterologous pbms from viral oncoproteins during T-cell transformation. Virus Genes 2010, 40, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Narayan, N.; Massimi, P.; Banks, L. Cdk phosphorylation of the discs large tumour suppressor controls its localisation and stability. J. Cell Sci. 2009, 122, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Ishioka, K.; Higuchi, M.; Takahashi, M.; Yoshida, S.; Oie, M.; Tanaka, Y.; Takahashi, S.; Xie, L.; Green, P.L.; Fujii, M. Inactivation of tumor suppressor dlg1 augments transformation of a T-cell line induced by humanT-cell leukemia virus type 1 Tax protein. Retrovirology 2006, 3, 71. [Google Scholar] [CrossRef] [PubMed]
- Zanin-Zhorov, A.; Lin, J.; Scher, J.; Kumari, S.; Blair, D.; Hippen, K.L.; Blazar, B.R.; Abramson, S.B.; Lafaille, J.J.; Dustin, M.L. Scaffold protein disc large homolog 1 is required for T-cell receptor-induced activation of regulatory T-cell function. Proc. Natl. Acad. Sci. USA 2012, 109, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.; Liu, J.; Schmidt, M.D.; Gao, T. Scribble-mediated membrane targeting of PHLPP1 is required for its negative regulation of Akt. EMBO Rep. 2011, 12, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Adey, N.B.; Huang, L.; Ormonde, P.A.; Baumgard, M.L.; Pero, R.; Byreddy, D.V.; Tavtigian, S.V.; Bartel, P.L. Threonine phosphorylation of the MMAC1/Pten PDZ binding domain both inhibits and stimulates PDZ binding. Cancer Res. 2000, 60, 35–37. [Google Scholar] [PubMed]
- Sotelo, N.S.; Valiente, M.; Gil, A.; Pulido, R. A functional network of the tumor suppressors APC, hdlg, and Pten, that relies on recognition of specific PDZ-domains. J. Cell. Biochem. 2012, 113, 2661–2670. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.C.; Center, D.M.; Cruikshank, W.W.; Zhang, Y. Binding of HTLV-1 tax oncoprotein to the precursor of interleukin-16, a T cell PDZ domain-containing protein. Virology 2003, 306, 60–67. [Google Scholar] [CrossRef]
- Javier, R.; Raska, K., Jr.; Macdonald, G.J.; Shenk, T. Human adenovirus type 9-induced rat mammary tumors. J. Virol. 1991, 65, 3192–3202. [Google Scholar] [PubMed]
- Javier, R.; Raska, K., Jr.; Shenk, T. Requirement for the adenovirus type 9 E4 region in production of mammary tumors. Science 1992, 257, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.K.; Lee, S.S.; Thomas, D.L.; Latorre, I.J.; Weiss, R.S.; Glaunsinger, B.A.; Javier, R.T. Selective pdz protein-dependent stimulation of phosphatidylinositol 3-kinase by the adenovirus E4-Orf1 oncoprotein. Oncogene 2003, 22, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Frese, K.K.; Weiss, R.S.; Prasad, B.V.; Javier, R.T. A new crucial protein interaction element that targets the adenovirus E4-Orf1 oncoprotein to membrane vesicles. J. Virol. 2007, 81, 4787–4797. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.; Kumar, M.; Taruishi, M.; Javier, R.T. The human adenovirus E4-Orf1 protein subverts discs large 1 to mediate membrane recruitment and dysregulation of phosphatidylinositol 3-kinase. PLoS Pathog. 2014, 10, e1004102. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.; Kumar, M.; Taruishi, M.; Javier, R.T. Adenovirus E4-Orf1 dysregulates epidermal growth factor and insulin/insulin-like growth factor receptors to mediate constitutive myc expression. J. Virol. 2015, 89, 10774–10785. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kong, K.; Javier, R.T. Hijacking Dlg1 for oncogenic phosphatidylinositol 3-kinase activation in human epithelial cells is a conserved mechanism of human adenovirus E4-Orf1 proteins. J. Virol. 2014, 88, 14268–14277. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; DeSalle, R.; Einstein, M.H.; Burk, R.D. Degradation of human PDZ-proteins by human alphapapillomaviruses represents an evolutionary adaptation to a novel cellular niche. PLoS Pathog. 2015, 11, e1004980. [Google Scholar] [CrossRef] [PubMed]
- Delury, C.P.; Marsh, E.K.; James, C.D.; Boon, S.S.; Banks, L.; Knight, G.L.; Roberts, S. The role of protein kinase A regulation of the E6 PDZ-binding domain during the differentiation-dependent life cycle of human papillomavirus type 18. J. Virol. 2013, 87, 9463–9472. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Laimins, L.A. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 2004, 78, 12366–12377. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, L.; Davy, C.; Raj, K.; Kranjec, C.; Banks, L.; Doorbar, J. Stabilization of HPV16 E6 protein by PDZ proteins, and potential implications for genome maintenance. Virology 2011, 414, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Brimer, N.; Vande Pol, S.B. Papillomavirus E6 PDZ interactions can be replaced by repression of p53 to promote episomal human papillomavirus genome maintenance. J. Virol. 2014, 88, 3027–3030. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.M.; Nguyen, M.L.; Caruana, G.; Bernstein, A.; Lambert, P.F.; Griep, A.E. Requirement of PDZ-containing proteins for cell cycle regulation and differentiation in the mouse lens epithelium. Mol. Cell. Biol. 2003, 23, 8970–8981. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Nguyen, M.M.; Lee, D.; Griep, A.E.; Lambert, P.F. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6's induction of epithelial hyperplasia in vivo. J. Virol. 2003, 77, 6957–6964. [Google Scholar] [CrossRef] [PubMed]
- Spanos, W.C.; Geiger, J.; Anderson, M.E.; Harris, G.F.; Bossler, A.D.; Smith, R.B.; Klingelhutz, A.J.; Lee, J.H. Deletion of the PDZ motif of HPV16E preventing immortalization and anchorage-independent growth in human tonsil epithelial cells. Head Neck 2008, 30, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.A.; Thomas, M.; Banks, L.; Roberts, S. Activity of the human papillomavirus E6 PDZ-binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J. Cell Sci. 2003, 116, 4925–4934. [Google Scholar] [CrossRef] [PubMed]
- Jing, M.; Bohl, J.; Brimer, N.; Kinter, M.; Vande Pol, S.B. Degradation of tyrosine phosphatase Ptpn3 (PTPH1) by association with oncogenic human papillomavirus E6 proteins. J. Virol. 2007, 81, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef] [PubMed]
- Shai, A.; Brake, T.; Somoza, C.; Lambert, P.F. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res. 2007, 67, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Muench, P.; Hiller, T.; Probst, S.; Florea, A.M.; Stubenrauch, F.; Iftner, T. Binding of PDZ proteins to HPV E6 proteins does neither correlate with epidemiological risk classification nor with the immortalization of foreskin keratinocytes. Virology 2009, 387, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Shai, A.; Lambert, P.; Banks, L. Hpv E6 degradation of p53 and PDZ containing substrates in an E6AP null background. Oncogene 2008, 27, 1800–1804. [Google Scholar] [CrossRef] [PubMed]
- Kuballa, P.; Matentzoglu, K.; Scheffner, M. The role of the ubiquitin ligase E6-AP in human papillomavirus E6-mediated degradation of PDZ domain-containing proteins. J. Biol. Chem. 2007, 282, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Gardiol, D.; Kuhne, C.; Glaunsinger, B.; Lee, S.S.; Javier, R.; Banks, L. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene 1999, 18, 5487–5496. [Google Scholar] [CrossRef] [PubMed]
- Krishna Subbaiah, V.; Massimi, P.; Boon, S.S.; Myers, M.P.; Sharek, L.; Garcia-Mata, R.; Banks, L. The invasive capacity of HPV transformed cells requires the hDlg-dependent enhancement of SGEF/RHOG activity. PLoS Pathog. 2012, 8, e1002543. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Dong, L.; MacDonald, A.I.; Akbari, S.; Edward, M.; Hodgins, M.B.; Johnstone, S.R.; Graham, S.V. HPV16 E6 controls the gap junction protein Cx43 in cervical tumour cells. Viruses 2015, 7, 5243–5256. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Huibregtse, J.M. Human scribble (vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus e6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 2000, 20, 8244–8253. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Laura, R.; Hepner, K.; Guccione, E.; Sawyers, C.; Lasky, L.; Banks, L. Oncogenic human papillomavirus E6 proteins target the Magi-2 and Magi-3 proteins for degradation. Oncogene 2002, 21, 5088–5096. [Google Scholar] [CrossRef] [PubMed]
- Hampson, L.; Li, C.; Oliver, A.W.; Kitchener, H.C.; Hampson, I.N. The PDZ protein Tip-1 is a gain of function target of the HPV16 E6 oncoprotein. Int. J. Oncol. 2004, 25, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Favre-Bonvin, A.; Reynaud, C.; Kretz-Remy, C.; Jalinot, P. Human papillomavirus type 18 E6 protein binds the cellular PDZ protein Tip-2/GIPC, which is involved in transforming growth factor beta signaling and triggers its degradation by the proteasome. J. Virol. 2005, 79, 4229–4237. [Google Scholar] [CrossRef] [PubMed]
- Handa, K.; Yugawa, T.; Narisawa-Saito, M.; Ohno, S.; Fujita, M.; Kiyono, T. E6AP-dependent degradation of Dlg4/PSD95 by high-risk human papillomavirus type 18 E6 protein. J. Virol. 2007, 81, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Topffer, S.; Muller-Schiffmann, A.; Matentzoglu, K.; Scheffner, M.; Steger, G. Protein tyrosine phosphatase H1 is a target of the E6 oncoprotein of high-risk genital human papillomaviruses. J. Gen. Virol. 2007, 88, 2956–2965. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.W.; Kim, H.Z.; Kim, S.; Kim, Y.S.; Choe, J. Human papillomavirus type 16 E6 protein interacts with cystic fibrosis transmembrane regulator-associated ligand and promotes E6-associated protein-mediated ubiquitination and proteasomal degradation. Oncogene 2007, 26, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Spanos, W.C.; Hoover, A.; Harris, G.F.; Wu, S.; Strand, G.L.; Anderson, M.E.; Klingelhutz, A.J.; Hendriks, W.; Bossler, A.D.; Lee, J.H. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. J. Virol. 2008, 82, 2493–2500. [Google Scholar] [CrossRef] [PubMed]
- Accardi, R.; Rubino, R.; Scalise, M.; Gheit, T.; Shahzad, N.; Thomas, M.; Banks, L.; Indiveri, C.; Sylla, B.S.; Cardone, R.A.; et al. E6 and E7 from human papillomavirus type 16 cooperate to target the PDZ protein Na/H exchange regulatory factor 1. J. Virol. 2011, 85, 8208–8216. [Google Scholar] [CrossRef] [PubMed]
- Facciuto, F.; Bugnon Valdano, M.; Marziali, F.; Massimi, P.; Banks, L.; Cavatorta, A.L.; Gardiol, D. Human papillomavirus (HPV)-18 E6 oncoprotein interferes with the epithelial cell polarity PAR3 protein. Mol. Oncol. 2014, 8, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Banks, L. PDZRN3/LNX3 is a novel target of human papillomavirus type 16 (HPV-16) and hpv-18 E6. J. Virol. 2015, 89, 1439–1444. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roberts, S.; Calautti, E.; Vanderweil, S.; Nguyen, H.O.; Foley, A.; Baden, H.P.; Viel, A. Changes in localization of human discs large (hdlg) during keratinocyte differentiation are [corrected] associated with expression of alternatively spliced hdlg variants. Exp. Cell Res. 2007, 313, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Gammoh, N.; Thomas, M.; Banks, L. Hpv E6 specifically targets different cellular pools of its PDZ domain-containing tumour suppressor substrates for proteasome-mediated degradation. Oncogene 2004, 23, 8033–8039. [Google Scholar] [CrossRef] [PubMed]
- Narayan, N.; Subbaiah, V.K.; Banks, L. The high-risk HPV E6 oncoprotein preferentially targets phosphorylated nuclear forms of hdlg. Virology 2009, 387, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, A.I.; Sun, P.; Hernandez-Lopez, H.; Aasen, T.; Hodgins, M.B.; Edward, M.; Roberts, S.; Massimi, P.; Thomas, M.; Banks, L.; et al. A functional interaction between the MAGUK protein hdlg and the gap junction protein connexin 43 in cervical tumour cells. Biochem. J. 2012, 446, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.A.; Rollason, T.P.; Reynolds, G.M.; Murray, P.G.; Banks, L.; Roberts, S. Changes in expression of the human homologue of the drosophila discs large tumour suppressor protein in high-grade premalignant cervical neoplasias. Carcinogenesis 2002, 23, 1791–1796. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Massimi, P.; Banks, L. Restoration of Magi-1 expression in human papillomavirus-positive tumor cells induces cell growth arrest and apoptosis. J. Virol. 2014, 88, 7155–7169. [Google Scholar] [CrossRef] [PubMed]
- James, M.A.; Lee, J.H.; Klingelhutz, A.J. Human papillomavirus type 16 E6 activates NF-kappaB, induces ciap-2 expression, and protects against apoptosis in a PDZ binding motif-dependent manner. J. Virol. 2006, 80, 5301–5307. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Ghosh-Choudhury, N.; Munger, K. Activation of cap-dependent translation by mucosal human papillomavirus E6 proteins is dependent on the integrity of the LXXLL binding motif. J. Virol. 2012, 86, 7466–7472. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cavatorta, A.L.; Fumero, G.; Chouhy, D.; Aguirre, R.; Nocito, A.L.; Giri, A.A.; Banks, L.; Gardiol, D. Differential expression of the human homologue of drosophila discs large oncosuppressor in histologic samples from human papillomavirus-associated lesions as a marker for progression to malignancy. Int. J. Cancer 2004, 111, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Yano, T.; Nakagawa, K.; Takizawa, S.; Suzuki, Y.; Yasugi, T.; Huibregtse, J.M.; Taketani, Y. Analysis of the expression and localisation of a LAP protein, human scribble, in the normal and neoplastic epithelium of uterine cervix. Br. J. Cancer 2004, 90, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Banks, L. A systematic analysis of human papillomavirus (HPV) E6 PDZ substrates identifies Magi-1 as a major target of HPV type 16 (HPV-16) and HPV-18 whose loss accompanies disruption of tight junctions. J. Virol. 2011, 85, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Lee, S.; Choi, T.; Lee, C. Roles of the PDZ domain-binding motif of the human papillomavirus type 16 E6 on the immortalization and differentiation of primary human foreskin keratinocytes. Virus Genes 2014, 48, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Simonson, S.J.; Difilippantonio, M.J.; Lambert, P.F. Two distinct activities contribute to human papillomavirus 16 E6’s oncogenic potential. Cancer Res. 2005, 65, 8266–8273. [Google Scholar] [CrossRef] [PubMed]
- Kuhne, C.; Gardiol, D.; Guarnaccia, C.; Amenitsch, H.; Banks, L. Differential regulation of human papillomavirus E6 by protein kinase A: Conditional degradation of human discs large protein by oncogenic E6. Oncogene 2000, 19, 5884–5891. [Google Scholar] [CrossRef] [PubMed]
- Boon, S.S.; Tomaic, V.; Thomas, M.; Roberts, S.; Banks, L. Cancer-causing human papillomavirus E6 proteins display major differences in the phospho-regulation of their PDZ interactions. J. Virol. 2015, 89, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Boon, S.S.; Banks, L. High-risk human papillomavirus e6 oncoproteins interact with 14–3-3zeta in a PDZ binding motif-dependent manner. J. Virol. 2013, 87, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Massimi, P.; Navarro, C.; Borg, J.P.; Banks, L. The hscrib/dlg apico-basal control complex is differentially targeted by HPV-16 and HPV-18 E6 proteins. Oncogene 2005, 24, 6222–6230. [Google Scholar] [CrossRef] [PubMed]
- Hoover, A.C.; Strand, G.L.; Nowicki, P.N.; Anderson, M.E.; Vermeer, P.D.; Klingelhutz, A.J.; Bossler, A.D.; Pottala, J.V.; Hendriks, W.J.; Lee, J.H. Impaired PTPN13 phosphatase activity in spontaneous or hpv-induced squamous cell carcinomas potentiates oncogene signaling through the MAP kinase pathway. Oncogene 2009, 28, 3960–3970. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. The Jak-Stat transcriptional regulator, Stat-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P. PDZ domain-phosphoinositide interactions in cell-signaling. Verh K Acad. Geneeskd Belg. 2006, 68, 271–286. [Google Scholar] [PubMed]
- Diehl, N.; Schaal, H. Make yourself at home: Viral hijacking of the PI3k/Akt signaling pathway. Viruses 2013, 5, 3192–3212. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Poirson, J.; Foltz, C.; Chebaro, Y.; Schrapp, M.; Meyer, A.; Bonetta, A.; Forster, A.; Jacob, Y.; Masson, M.; et al. Targeting the two oncogenic functional sites of the HPV E6 oncoprotein with a high-affinity bivalent ligand. Angew. Chem. Int. Ed. Engl. 2015, 54, 7958–7962. [Google Scholar] [CrossRef] [PubMed]
- Belyaeva, T.A.; Nicol, C.; Cesur, O.; Trave, G.; Blair, G.E.; Stonehouse, N.J. An RNA aptamer targets the PDZ-binding motif of the HPV16 E6 oncoprotein. Cancers (Basel) 2014, 6, 1553–1569. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
James, C.D.; Roberts, S. Viral Interactions with PDZ Domain-Containing Proteins—An Oncogenic Trait? Pathogens 2016, 5, 8. https://doi.org/10.3390/pathogens5010008
James CD, Roberts S. Viral Interactions with PDZ Domain-Containing Proteins—An Oncogenic Trait? Pathogens. 2016; 5(1):8. https://doi.org/10.3390/pathogens5010008
Chicago/Turabian StyleJames, Claire D., and Sally Roberts. 2016. "Viral Interactions with PDZ Domain-Containing Proteins—An Oncogenic Trait?" Pathogens 5, no. 1: 8. https://doi.org/10.3390/pathogens5010008
APA StyleJames, C. D., & Roberts, S. (2016). Viral Interactions with PDZ Domain-Containing Proteins—An Oncogenic Trait? Pathogens, 5(1), 8. https://doi.org/10.3390/pathogens5010008