
Full Genomic Characterization of a Saffold Virus Isolated in Peru

 , and
, and
Abstract
:1. Introduction
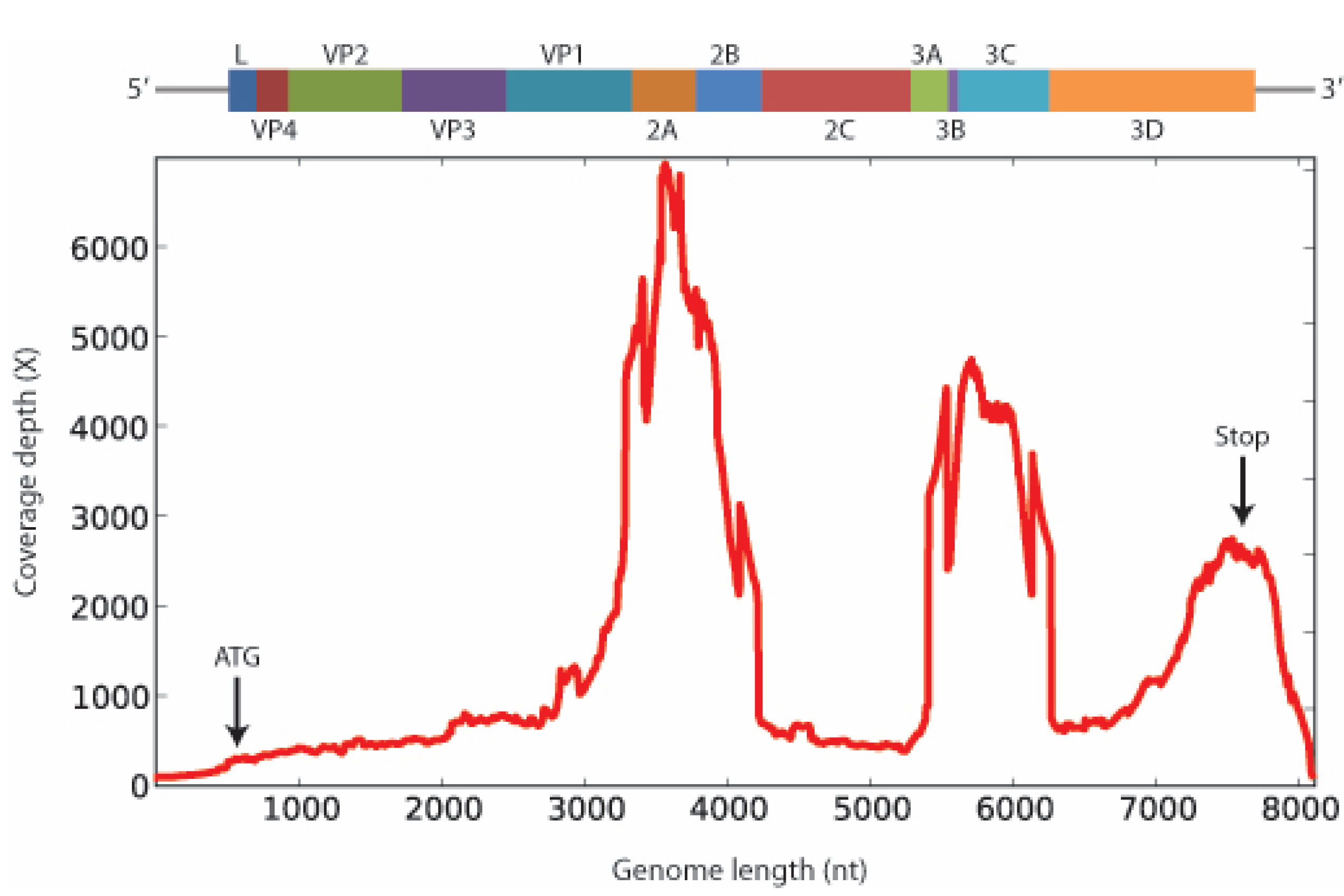
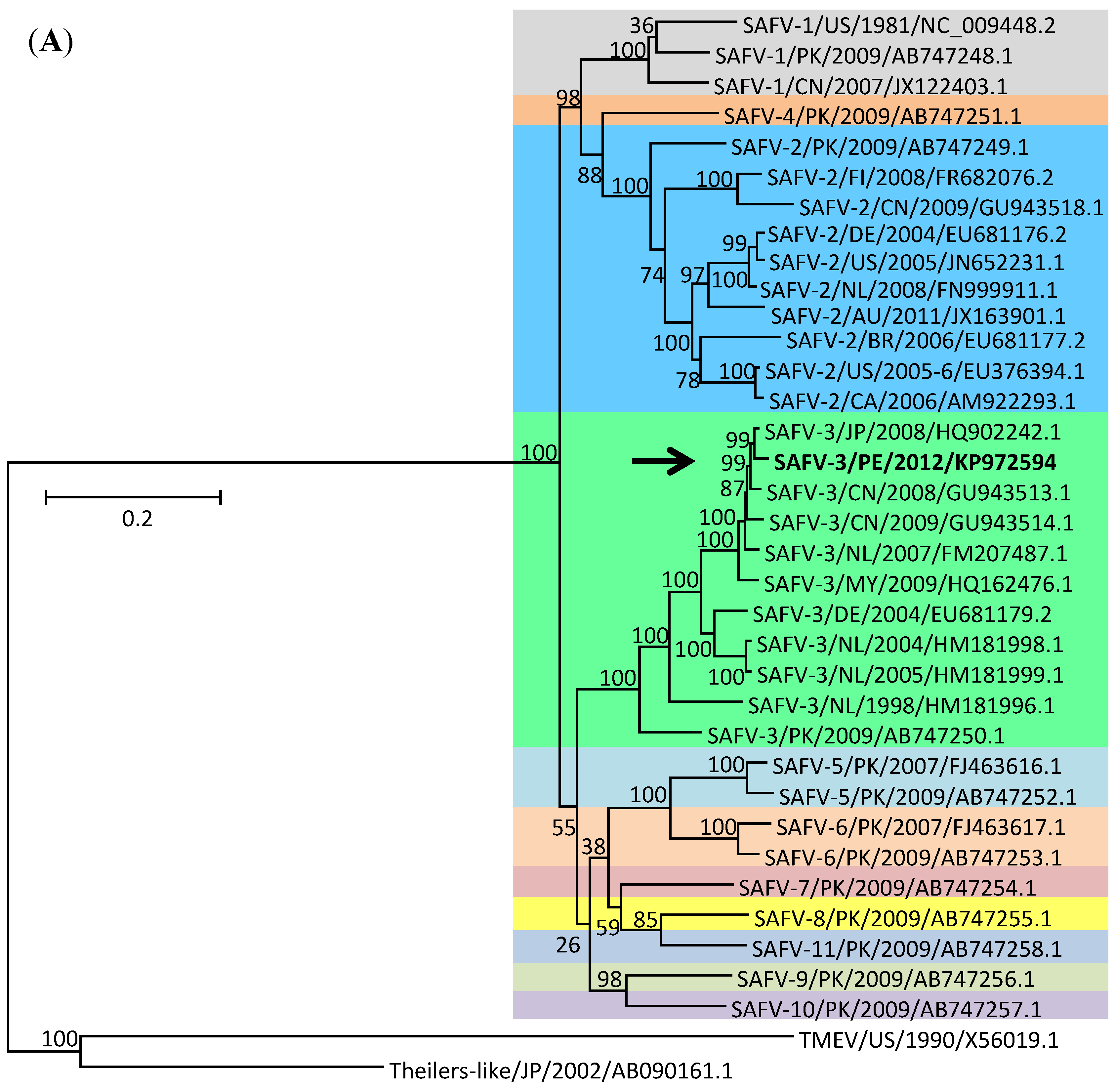
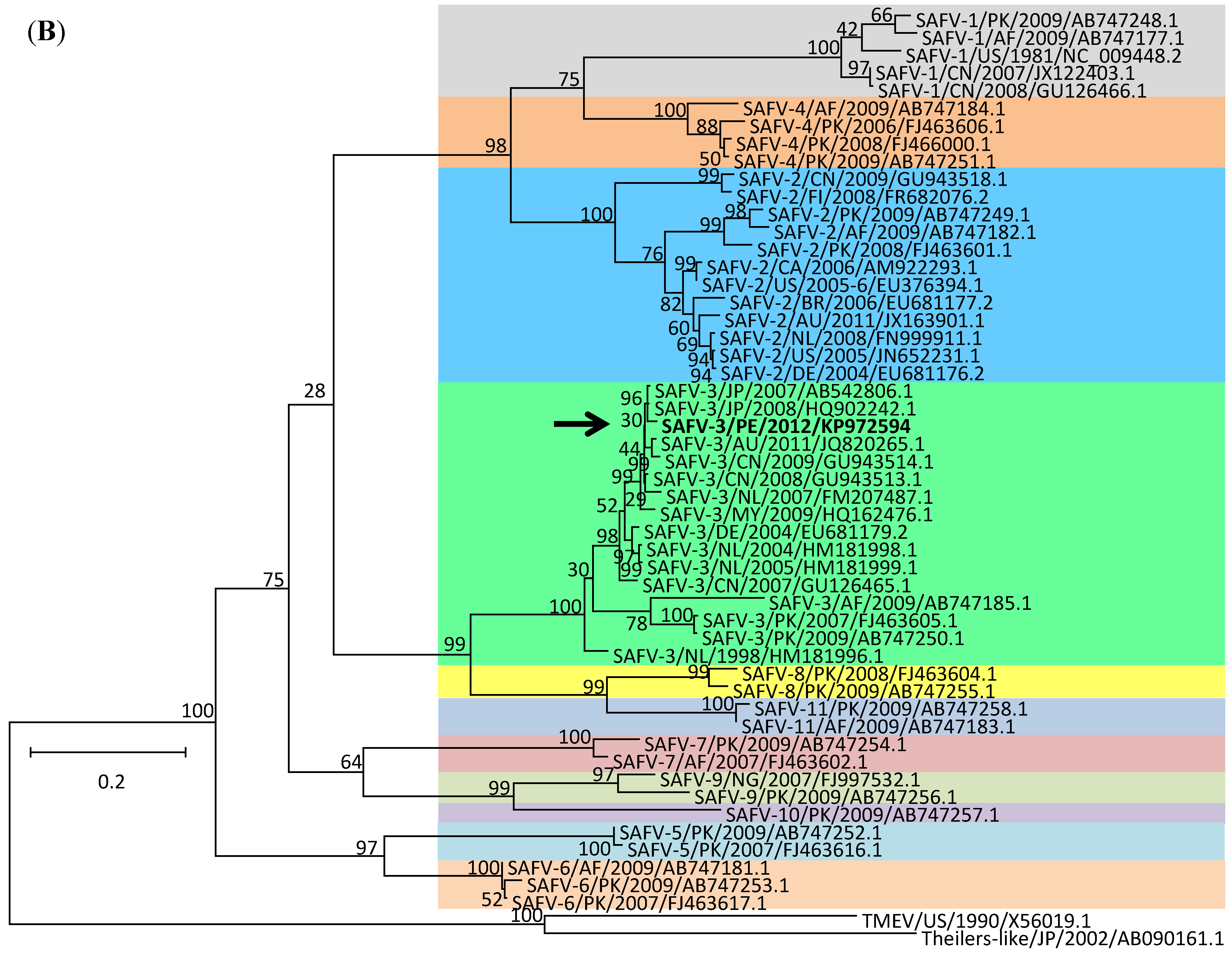
2. Results and Discussion
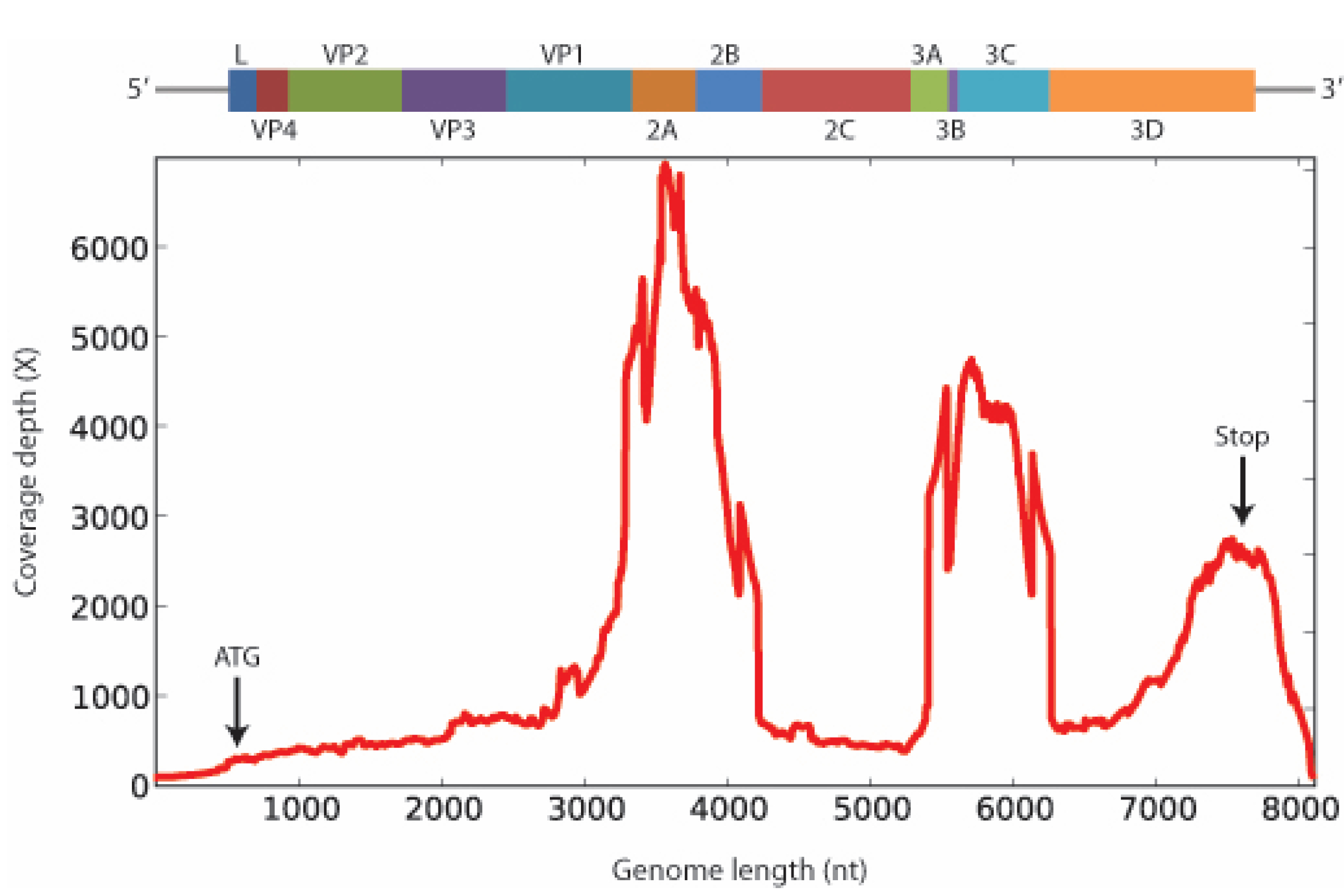
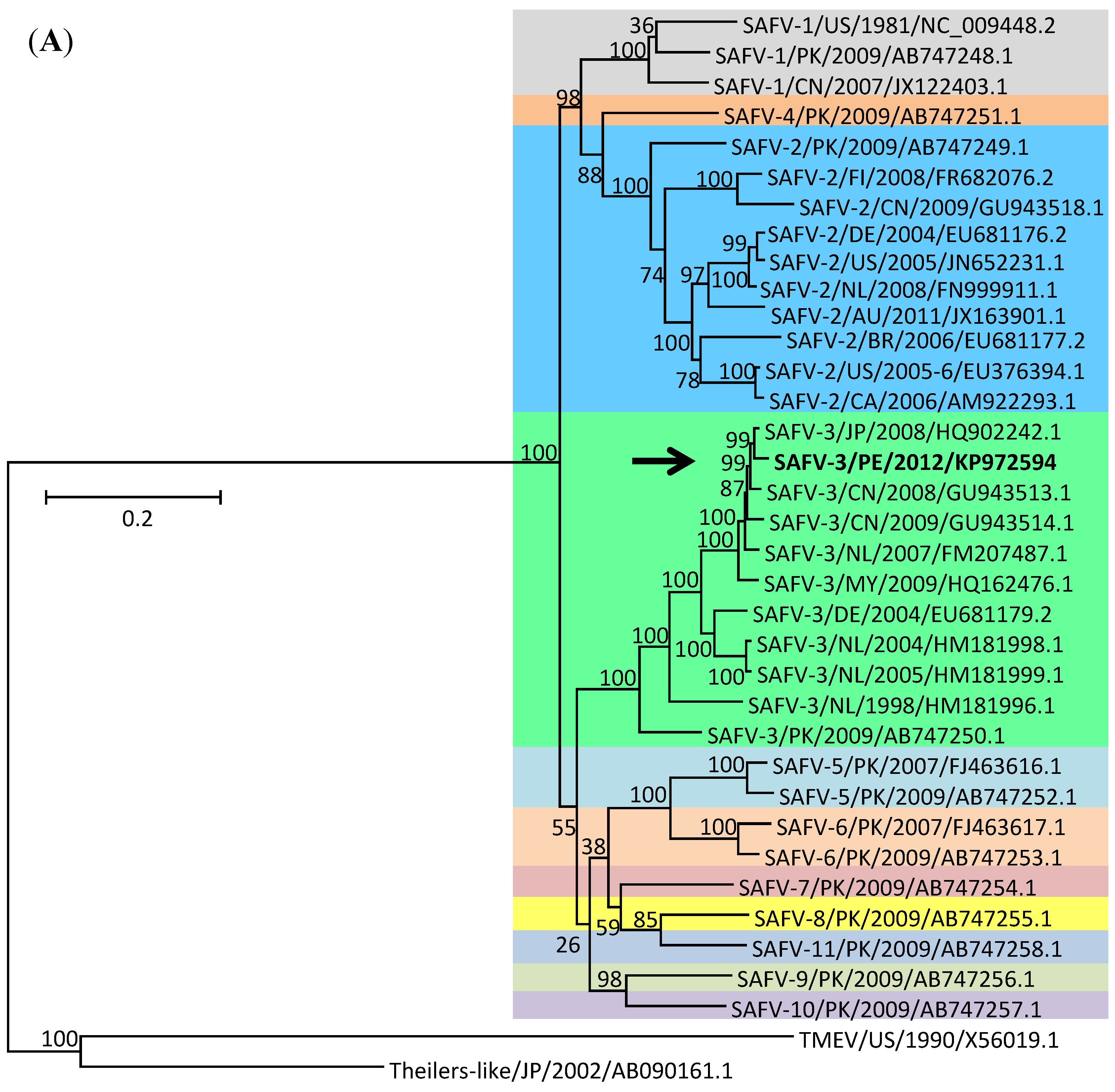
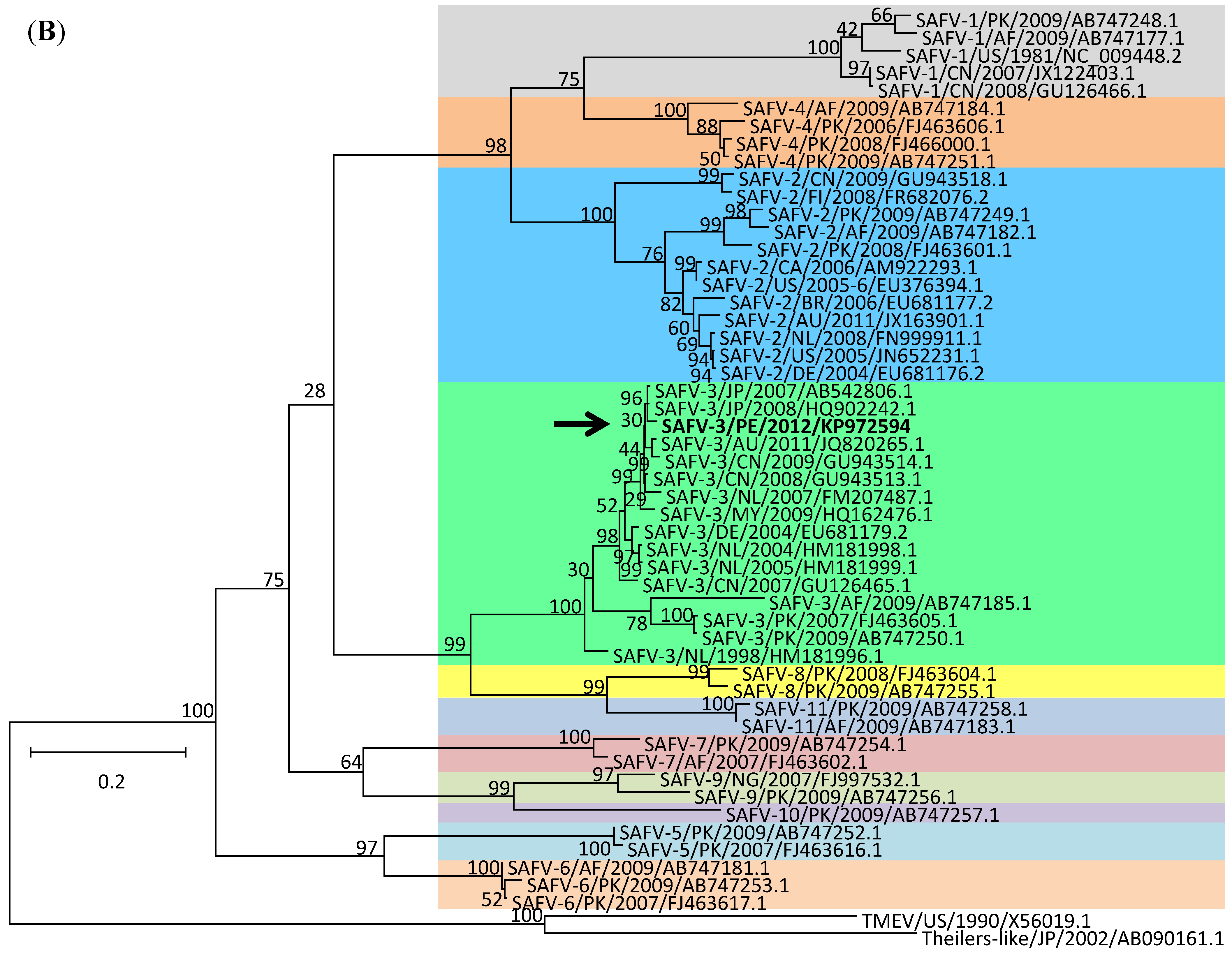
3. Experimental Section
3.1. Cell Culture
3.2. MassTag PCR
3.3. Sequencing Library Preparation
3.4. Sequencing and Bioinformatics
3.5. Phylogenetics

{kind=link}
{kind=link}
{kind=link}
| Virus | Accession # | Year | Location | Sequence Type | Length (nt) |
|---|---|---|---|---|---|
| SAFV-1 | NC_009448.2 | 1981 | USA | Complete genome | 8115 |
| SAFV-1 | JX219488.1 | 2002 | Bolivia | Partial VP1 | 321 |
| SAFV-1 | JX219490.1 | 2003 | Bolivia | Partial VP1 | 356 |
| SAFV-1 | JX122403.1 | 2007 | China | Complete genome | 7856 |
| SAFV-1 | GU126466.1 | 2008 | China | Complete VP1 | 822 |
| SAFV-1 | AB747177.1 | 2009 | Afghanistan | Complete VP1 | 847 |
| SAFV-1 | AB747248.1 | 2009 | Pakistan | Complete genome | 8078 |
| SAFV-2 | JX219484.1 | 2002 | Bolivia | Partial VP1 | 357 |
| SAFV-2 | JX219485.1 | 2003 | Bolivia | Partial VP1 | 376 |
| SAFV-2 | EU681176.2 | 2004 | Germany | Complete genome | 7842 |
| SAFV-2 | JN652231.1 | 2005 | USA | Complete genome | 7885 |
| SAFV-2 | EU376394.1 | 2005–2006 | USA | Complete genome | 7961 |
| SAFV-2 | EU681177.2 | 2006 | Brazil | Complete genome | 7809 |
| SAFV-2 | AM922293 | 2006 | Canada | Complete genome | 6882 |
| SAFV-2 | FR682076.2 | 2008 | Finland | Complete genome | 8046 |
| SAFV-2 | FN999911.1 | 2008 | Netherlands | Complete genome | 8045 |
| SAFV-2 | FJ463601.1 | 2008 | Pakistan | Complete VP1 | 816 |
| SAFV-2 | AB747182.1 | 2009 | Afghanistan | Complete VP1 | 856 |
| SAFV-2 | GU943518.1 | 2009 | China | Complete genome | 7846 |
| SAFV-2 | AB747249.1 | 2009 | Pakistan | Complete genome | 8075 |
| SAFV-2 | JX163901.1 | 2011 | Australia | Complete genome | 7781 |
| SAFV-3 | HM181996.1 | 1998 | Netherlands | Complete genome | 7984 |
| SAFV-3 | EU681179.2 | 2004 | Germany | Complete genome | 7846 |
| SAFV-3 | HM181998.1 | 2004 | Netherlands | Complete genome | 7991 |
| SAFV-3 | HM181999.1 | 2005 | Netherlands | Complete genome | 7991 |
| SAFV-3 | GU126465.1 | 2007 | China | Complete VP1 | 810 |
| SAFV-3 | AB542806.1 | 2007 | Japan | Complete VP1 | 813 |
| SAFV-3 | FM207487.1 | 2007 | Netherlands | Complete genome | 8051 |
| SAFV-3 | FJ463605.1 | 2007 | Pakistan | Complete VP1 | 810 |
| SAFV-3 | GU943513.1 | 2008 | China | Complete genome | 8054 |
| SAFV-3 | HQ902242.1 | 2008 | Japan | Complete genome | 8082 |
| SAFV-3 | AB747185.1 | 2009 | Afghanistan | Complete VP1 | 835 |
| SAFV-3 | GU943514.1 | 2009 | China | Complete genome | 7853 |
| SAFV-3 | HQ162476.1 | 2009 | Malaysia | Complete genome | 8073 |
| SAFV-3 | AB747250.1 | 2009 | Pakistan | Complete genome | 8079 |
| SAFV-3 | JQ820265.1 | 2011 | Australia | Complete VP1 | 843 |
| SAFV-3 | KP972594 | 2012 | Peru | Complete genome | 8064 |
| SAFV-4 | JX219486.1 | 2002 | Bolivia | Partial VP1 | 315 |
| SAFV-4 | JX219487.1 | 2003 | Bolivia | Partial VP1 | 312 |
| SAFV-4 | FJ463606.1 | 2006 | Pakistan | Complete VP1 | 813 |
| SAFV-4 | FJ463600.1 | 2008 | Pakistan | Complete VP1 | 813 |
| SAFV-4 | AB747184.1 | 2009 | Afghanistan | Complete VP1 | 845 |
| SAFV-4 | AB747251.1 | 2009 | Pakistan | Complete genome | 8027 |
| SAFV-5 | FJ463616.1 | 2007 | Pakistan | Complete genome | 7639 |
| SAFV-5 | AB747252.1 | 2009 | Pakistan | Complete genome | 8083 |
| SAFV-6 | FJ463617.1 | 2007 | Pakistan | Complete genome | 7410 |
| SAFV-6 | AB747181.1 | 2009 | Afghanistan | Complete VP1 | 822 |
| SAFV-6 | AB747253.1 | 2009 | Pakistan | Complete genome | 8084 |
| SAFV-7 | FJ463602.1 | 2007 | Afghanistan | Complete VP1 | 816 |
| SAFV-7 | AB747254.1 | 2009 | Pakistan | Complete genome | 8087 |
| SAFV-8 | FJ463604.1 | 2008 | Pakistan | Complete VP1 | 810 |
| SAFV-8 | AB747255.1 | 2009 | Pakistan | Complete genome | 8081 |
| SAFV-9 | JX219494.1 | 2002 | Bolivia | Partial VP1 | 343 |
| SAFV-9 | FJ997532.1 | 2007 | Nigeria | Complete VP1 | 789 |
| SAFV-9 | AB747256.1 | 2009 | Pakistan | Complete genome | 8081 |
| SAFV-10 | AB747257.1 | 2009 | Pakistan | Complete genome | 8083 |
| SAFV-11 | AB747183.1 | 2009 | Afghanistan | Complete VP1 | 829 |
| SAFV-11 | AB747258.1 | 2009 | Pakistan | Complete genome | 8079 |
| TMEV | X56019.1 | 1990 | USA | Complete genome | 8101 |
| Theilers-like virus | AB090161.1 | 2002 | Japan | Complete genome | 8021 |
4. Conclusions
Supplementary Files
Supplementary File 1Disclaimers
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jones, M.S.; Lukashov, V.V.; Ganac, R.D.; Schnurr, D.P. Discovery of a novel human picornavirus in a stool sample from a pediatric patient presenting with fever of unknown origin. J. Clin. Microbiol. 2007, 45, 2144–2150. [Google Scholar] [CrossRef] [PubMed]
- Carocci, M.; Bakkali-Kassimi, L. The encephalomyocarditis virus. Virulence 2012, 3, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Naeem, A.; Hosomi, T.; Nishimura, Y.; Alam, M.M.; Oka, T.; Zaidi, S.S.; Shimizu, H. Genetic diversity of circulating saffold viruses in Pakistan and Afghanistan. J. Gen. Virol. 2014, 95, 1945–1957. [Google Scholar] [CrossRef] [PubMed]
- Taboada, B.; Espinoza, M.A.; Isa, P.; Aponte, F.E.; Arias-Ortiz, M.A.; Monge-Martinez, J.; Rodriguez-Vazquez, R.; Diaz-Hernandez, F.; Zarate-Vidal, F.; Wong-Chew, R.M.; et al. Is there still room for novel viral pathogens in pediatric respiratory tract infections? PLoS ONE 2014, 9, e113570. [Google Scholar] [CrossRef] [PubMed]
- Yodmeeklin, A.; Khamrin, P.; Chuchaona, W.; Saikruang, W.; Malasao, R.; Chaimongkol, N.; Kongsricharoern, T.; Ukarapol, N.; Maneekarn, N. Saffold viruses in pediatric patients with diarrhea in Thailand. J. Med. Virol. 2015, 87, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Zoll, J.; Erkens Hulshof, S.; Lanke, K.; Verduyn Lunel, F.; Melchers, W.J.; Schoondermark-van de Ven, E.; Roivainen, M.; Galama, J.M.; van Kuppeveld, F.J. Saffold virus, a human theiler’s-like cardiovirus, is ubiquitous and causes infection early in life. PLoS Pathog. 2009, 5, e1000416. [Google Scholar] [CrossRef] [PubMed]
- Abed, Y.; Boivin, G. New saffold cardioviruses in 3 children, Canada. Emerg. Infect. Dis. 2008, 14, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Luna, L.K.; Stocker, A.; Almeida, P.S.; Ribeiro, T.C.; Petersen, N.; Herzog, P.; Pedroso, C.; Huppertz, H.I.; Ribeiro Hda, C., Jr.; et al. Circulation of 3 lineages of a novel saffold cardiovirus in humans. Emerg. Infect. Dis. 2008, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Nix, W.A.; Khetsuriani, N.; Penaranda, S.; Maher, K.; Venczel, L.; Cselko, Z.; Freire, M.C.; Cisterna, D.; Lema, C.L.; Rosales, P.; et al. Diversity of picornaviruses in rural bolivia. J. Gen. Virol. 2013, 94, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Lamson, D.; Renwick, N.; Kapoor, V.; Liu, Z.; Palacios, G.; Ju, J.; Dean, A.; St George, K.; Briese, T.; Lipkin, W.I. Masstag polymerase-chain-reaction detection of respiratory pathogens, including a new rhinovirus genotype, that caused influenza-like illness in New York state during 2004–2005. J. Infect. Dis. 2006, 194, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, S.R.; Briese, T.; Palacios, G.; Hui, J.; Villari, J.; Kapoor, V.; Tokarz, R.; Glode, M.P.; Anderson, M.S.; Robinson, C.C.; et al. Multiplex MassTag-PCR for respiratory pathogens in pediatric nasopharyngeal washes negative by conventional diagnostic testing shows a high prevalence of viruses belonging to a newly recognized rhinovirus clade. J. Clin. Virol. 2008, 43, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; Depasse, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. Viral genome sequencing by random priming methods. BMC Genomics 2008, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, S.; Raymond, F.; Godzaridis, E.; Laviolette, F.; Corbeil, J. Ray meta: Scalable de novo metagenome assembly and profiling. Genome Biol. 2012, 13, R122. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Czechowicz, J.; Huaman, J.L.; Forshey, B.M.; Morrison, A.C.; Castillo, R.; Huaman, A.; Caceda, R.; Eza, D.; Rocha, C.; Blair, P.J.; et al. Prevalence and risk factors for encephalomyocarditis virus infection in Peru. Vector Borne Zoonotic Dis. 2011, 11, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Gotuzzo, E.; Blair, P.; Nix, W.A.; Ksiazek, T.G.; Comer, J.A.; Rollin, P.; Goldsmith, C.S.; Olson, J.; Kochel, T.J. Human febrile illness caused by encephalomyocarditis virus infection, Peru. Emerg. Infect. Dis. 2009, 15, 640–646. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leguia, M.; Loyola, S.; Rios, J.; Juarez, D.; Guevara, C.; Silva, M.; Prieto, K.; Wiley, M.; Kasper, M.R.; Palacios, G.; et al. Full Genomic Characterization of a Saffold Virus Isolated in Peru. Pathogens 2015, 4, 816-825. https://doi.org/10.3390/pathogens4040816
Leguia M, Loyola S, Rios J, Juarez D, Guevara C, Silva M, Prieto K, Wiley M, Kasper MR, Palacios G, et al. Full Genomic Characterization of a Saffold Virus Isolated in Peru. Pathogens. 2015; 4(4):816-825. https://doi.org/10.3390/pathogens4040816
Chicago/Turabian StyleLeguia, Mariana, Steev Loyola, Jane Rios, Diana Juarez, Carolina Guevara, Maria Silva, Karla Prieto, Michael Wiley, Matthew R. Kasper, Gustavo Palacios, and et al. 2015. "Full Genomic Characterization of a Saffold Virus Isolated in Peru" Pathogens 4, no. 4: 816-825. https://doi.org/10.3390/pathogens4040816
APA StyleLeguia, M., Loyola, S., Rios, J., Juarez, D., Guevara, C., Silva, M., Prieto, K., Wiley, M., Kasper, M. R., Palacios, G., & Bausch, D. G. (2015). Full Genomic Characterization of a Saffold Virus Isolated in Peru. Pathogens, 4(4), 816-825. https://doi.org/10.3390/pathogens4040816