
Systems Level Dissection of Candida Recognition by Dectins: A Matter of Fungal Morphology and Site of Infection

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
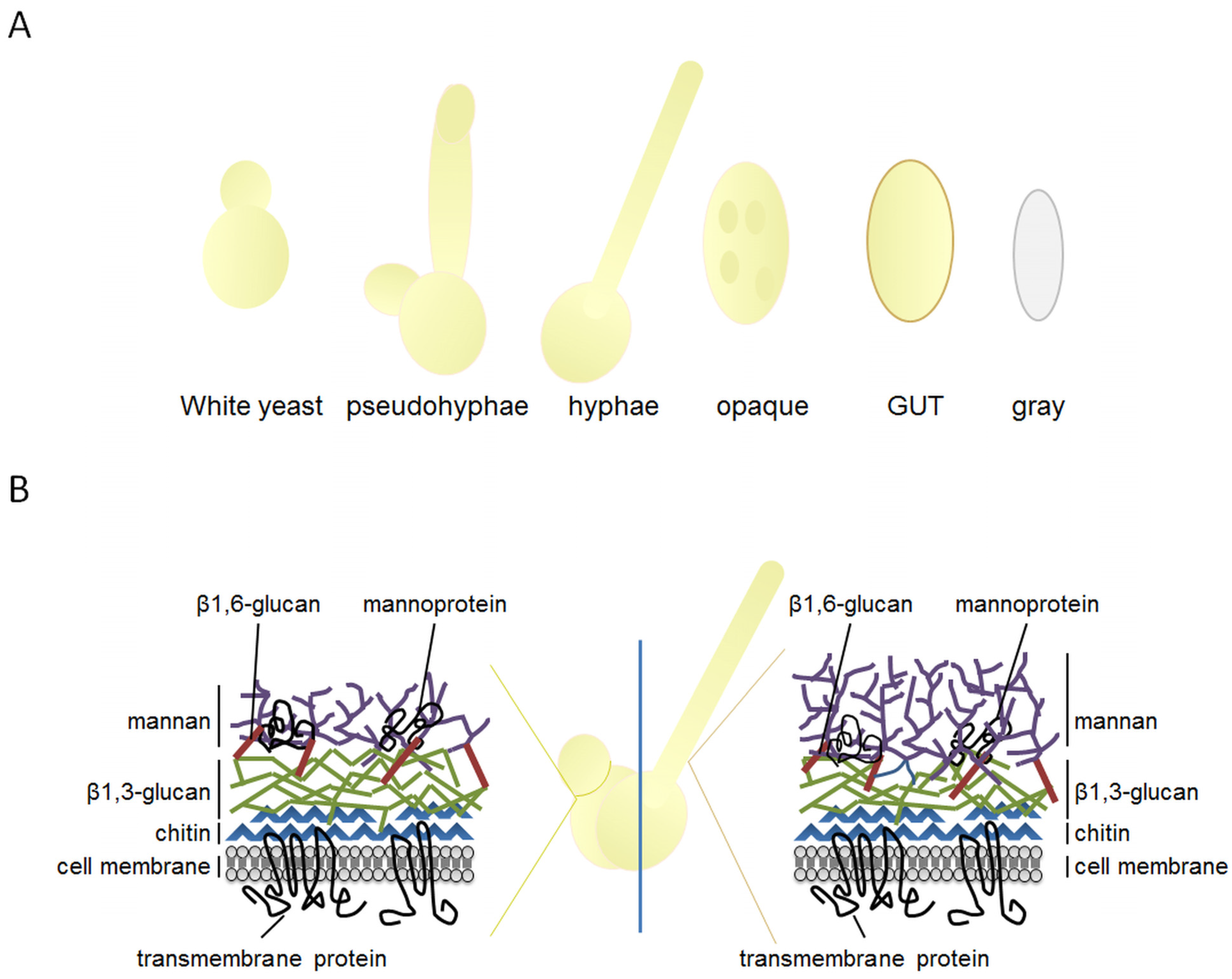
2. The Highly Plastic Nature of C. albicans Morphology and Its Implications for Pathogenicity
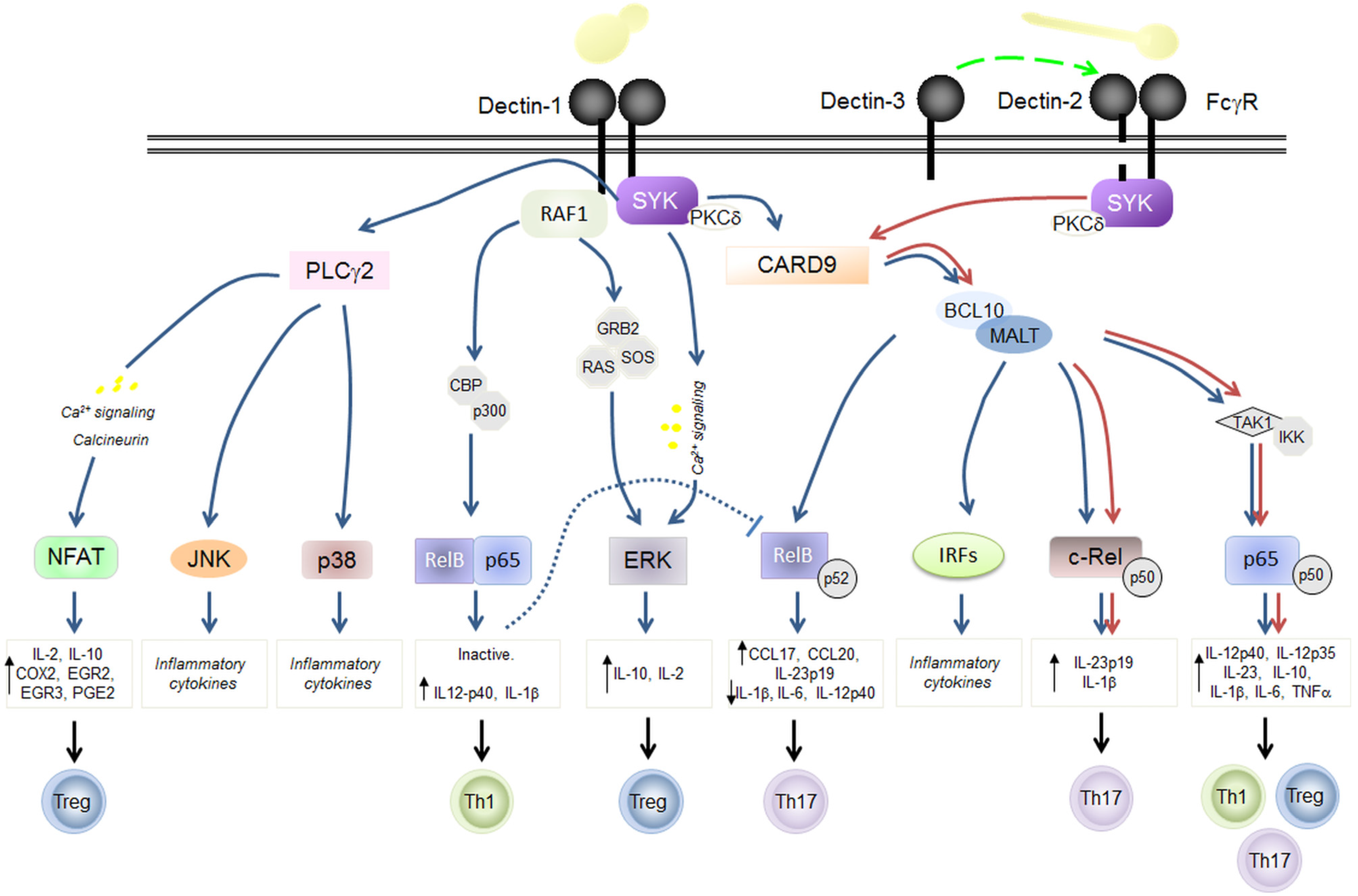
3. C-Type Lectins in Fungal Immunity: Dectin-1 and Dectin-2 Signaling
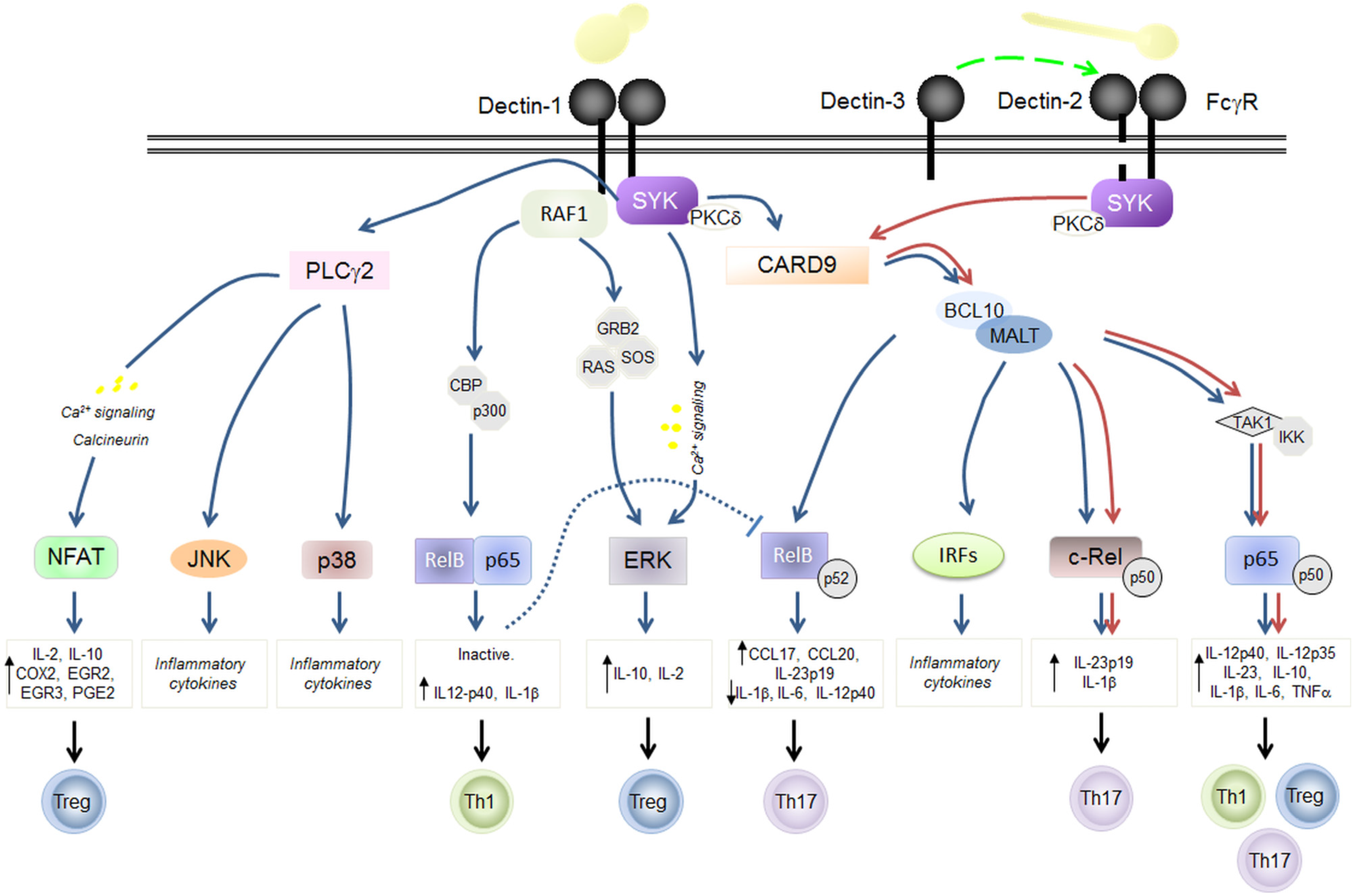
3.1. Dectin-1
3.2. Dectin-2
3.3. Dectin-3
3.4. Dectin-1 Antagonist: CLRs Antagonism
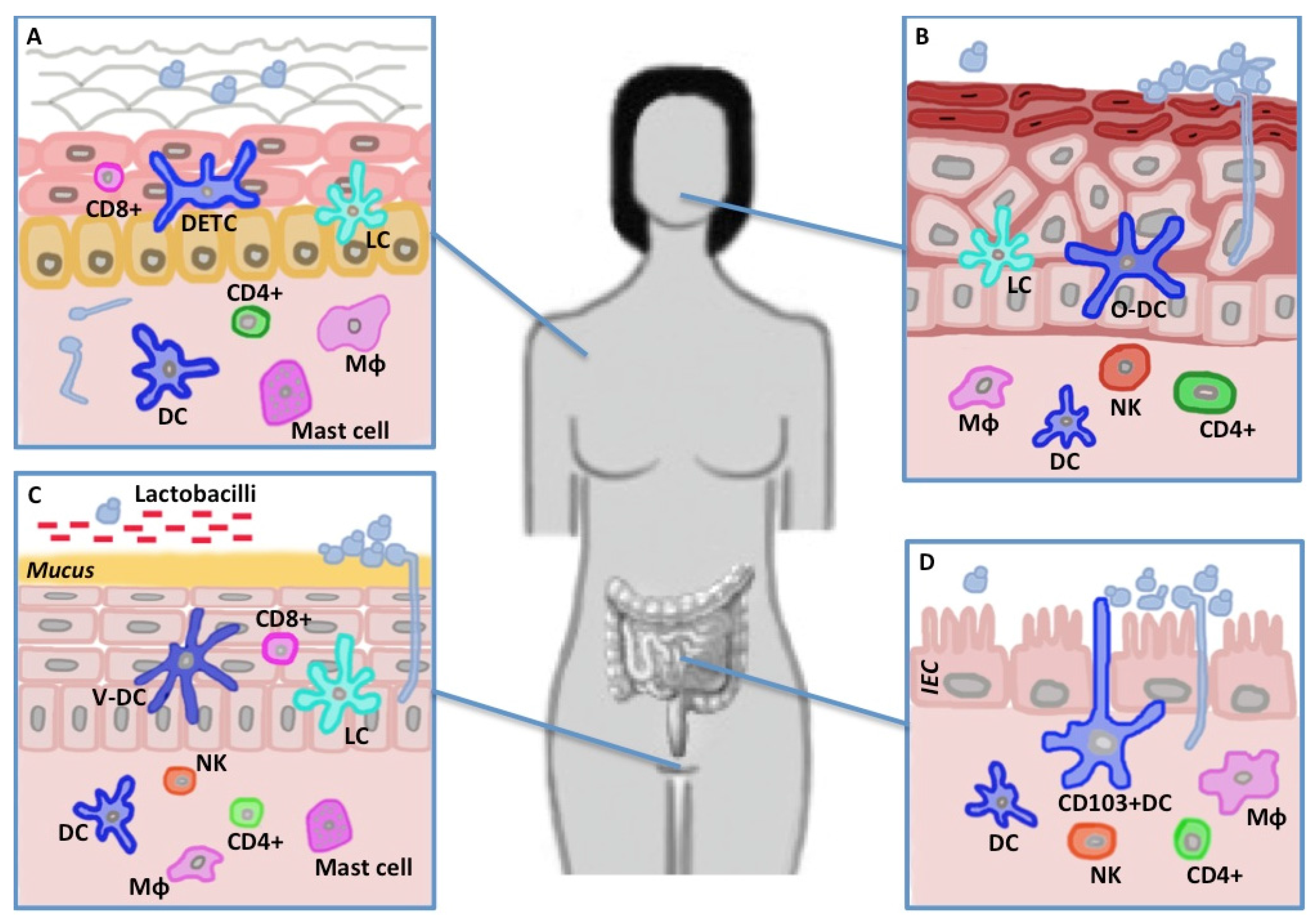
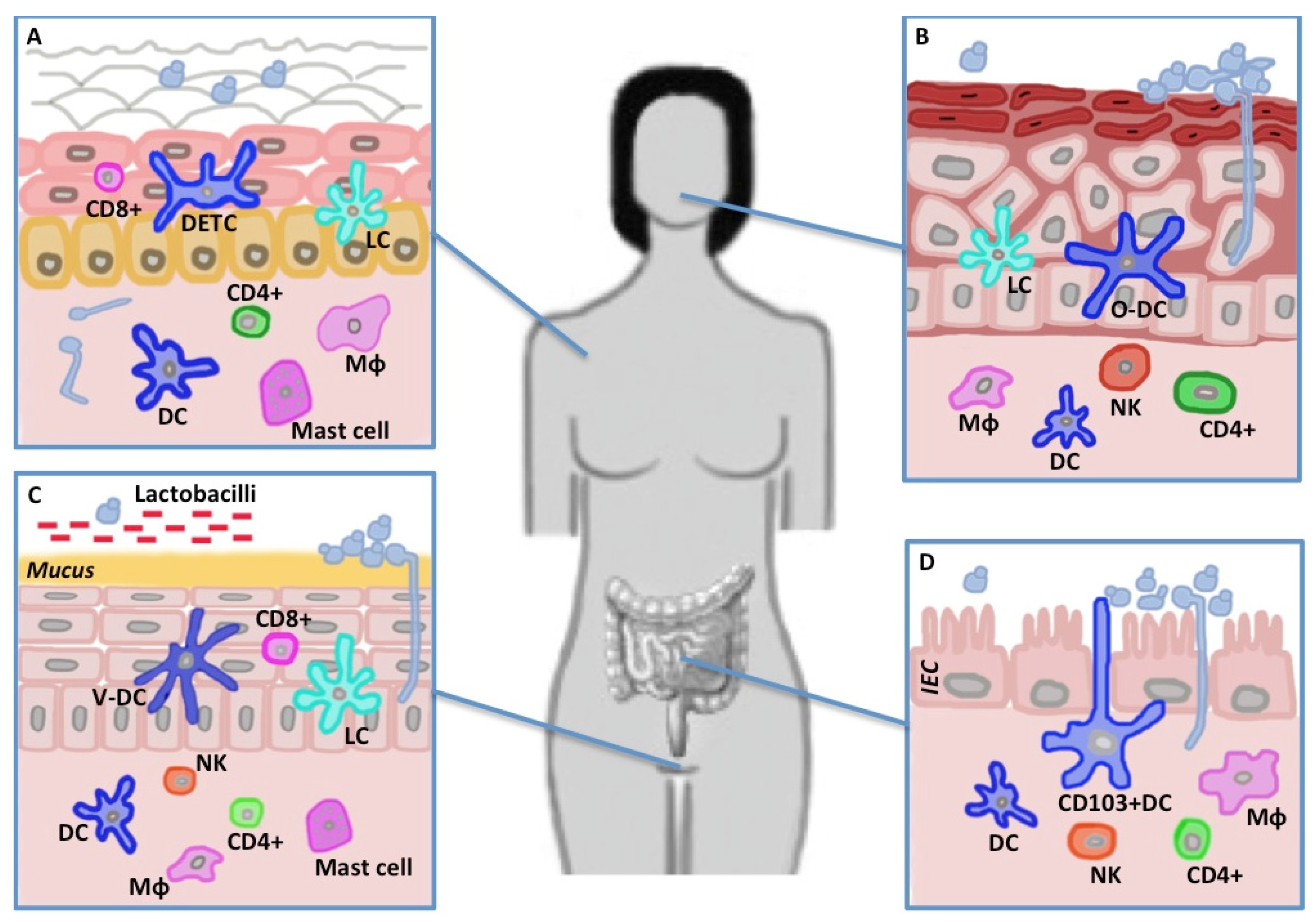
4. The Mucosal and Epithelial Barrier and C. albicans
4.1. Skin
4.2. Mouth
4.3. Gut
4.4. Vagina
5. Conclusion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Oh, J.; Freeman, A.F.; NISC Comparative Sequencing Program; Park, M.; Sokolic, R.; Candotti, F.; Holland, S.M.; Segre, J.A.; Kong, H.H. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013, 23, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Iliev, I.D.; Underhill, D.M. Striking a Balance: Fungal Commensalism versus Pathogenesis. Curr. Opin. Microbiol. 2013, 16, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; NIH Intramural Sequencing Center Comparative Sequencing Program; Kong, H.H.; Segre, J.A. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Ghannoum, M.A.; Jurevic, R.J.; Mukherjee, P.K.; Cui, F.; Sikaroodi, M.; Naqvi, A.; Gillevet, P.M. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010, 6, e1000713. [Google Scholar] [CrossRef] [PubMed]
- Drell, T.; Lillsaar, T.; Tummeleht, L.; Simm, J.; Aaspõllu, A.; Väin, E.; Saarma, I.; Salumets, A.; Donders, G.G.G.; Metsis, M. Characterization of the vaginal micro- and mycobiome in asymptomatic reproductive-age Estonian women. PLoS ONE 2013, 8, e54379. [Google Scholar] [CrossRef] [PubMed]
- Merenstein, D.; Hu, H.; Wang, C.; Hamilton, P.; Blackmon, M.; Chen, H.; Calderone, R.; Li, D. Colonization by Candida species of the oral and vaginal mucosa in HIV-infected and noninfected women. AIDS Res. Hum. Retrovir. 2013, 29, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef] [PubMed]
- Perlroth, J.; Choi, B.; Spellberg, B. Nosocomial fungal infections: Epidemiology, diagnosis, and treatment. Med. Mycol. 2007, 45, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, L.; De Filippo, C.; Cavalieri, D. Richness and diversity of mammalian fungal communities shape innate and adaptive immunity in health and disease. Eur. J. Immunol. 2014, 44, 3166–3181. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Chandra, J.; Retuerto, M.; Sikaroodi, M.; Brown, R.E.; Jurevic, R.; Salata, R.A.; Lederman, M.M.; Gillevet, P.M.; Ghannoum, M.A. Oral mycobiome analysis of HIV-infected patients: Identification of Pichia as an antagonist of opportunistic fungi. PLoS Pathog. 2014, 10, e1003996. [Google Scholar] [CrossRef] [PubMed]
- Charlson, E.S.; Diamond, J.M.; Bittinger, K.; Fitzgerald, A.S.; Yadav, A.; Haas, A.R.; Bushman, F.D.; Collman, R.G. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am. J. Respir. Crit. Care Med. 2012, 186, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Delhaes, L.; Monchy, S.; Fréalle, E.; Hubans, C.; Salleron, J.; Leroy, S.; Prevotat, A.; Wallet, F.; Wallaert, B.; Dei-Cas, E.; et al. The airway microbiota in cystic fibrosis: A complex fungal and bacterial community--implications for therapeutic management. PLoS ONE 2012, 7, e36313. [Google Scholar] [CrossRef] [PubMed]
- WHO. Antimicrobial Resistance: Global Report on Surveillance 2014. Available online: http://www.who.int/drugresistance/documents/surveillancereport/en/ (accessed on 10 July 2015).
- Mochon, A.B.; Jin, Y.; Ye, J.; Kayala, M.A.; Wingard, J.R.; Clancy, C.J.; Nguyen, M.H.; Felgner, P.; Baldi, P.; Liu, H. Serological profiling of a Candida albicans protein microarray reveals permanent host-pathogen interplay and stage-specific responses during candidemia. PLoS Pathog. 2010, 6, e1000827. [Google Scholar] [CrossRef] [PubMed]
- Sendid, B.; Jouault, T.; Vitse, A.; Fradin, C.; Colombel, J.F.; Poulain, D. Anti-glycan antibodies establish an unexpected link between C. albicans and Crohn disease. Méd. Sci. MS 2009, 25, 473–481. [Google Scholar]
- Saville, S.P.; Lazzell, A.L.; Monteagudo, C.; Lopez-Ribot, J.L. Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryot. Cell 2003, 2, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Wächtler, B.; Wilson, D.; Haedicke, K.; Dalle, F.; Hube, B. From attachment to damage: Defined genes of Candida albicans mediate adhesion, invasion and damage during interaction with oral epithelial cells. PLoS ONE 2011, 6, e17046. [Google Scholar] [CrossRef] [PubMed]
- Vautier, S.; MacCallum, D.M.; Brown, G.D. C-type lectin receptors and cytokines in fungal immunity. Cytokine 2012, 58, 89–99. [Google Scholar] [CrossRef] [PubMed]
- LeibundGut-Landmann, S.; Gross, O.; Robinson, M.J.; Osorio, F.; Slack, E.C.; Tsoni, S.V.; Schweighoffer, E.; Tybulewicz, V.; Brown, G.D.; Ruland, J.; et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 2007, 8, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.J.; Osorio, F.; Rosas, M.; Freitas, R.P.; Schweighoffer, E.; Gross, O.; Verbeek, J.S.; Ruland, J.; Tybulewicz, V.; Brown, G.D.; et al. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J. Exp. Med. 2009, 206, 2037–2051. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Den Dunnen, J.; Litjens, M.; Van der Vlist, M.; Wevers, B.; Bruijns, S.C.M.; Geijtenbeek, T.B.H. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat. Immunol. 2009, 10, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Saijo, S.; Ikeda, S.; Yamabe, K.; Kakuta, S.; Ishigame, H.; Akitsu, A.; Fujikado, N.; Kusaka, T.; Kubo, S.; Chung, S.; et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 2010, 32, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Marakalala, M.J.; Vautier, S.; Potrykus, J.; Walker, L.A.; Shepardson, K.M.; Hopke, A.; Mora-Montes, H.M.; Kerrigan, A.; Netea, M.G.; Murray, G.I.; et al. Differential adaptation of Candida albicans in vivo modulates immune recognition by dectin-1. PLoS Pathog. 2013, 9, e1003315. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, L.; Giovannini, G.; Bromley, M.; Bowyer, P.; Romani, L.; Cavalieri, D. Strain dependent variation of immune responses to A. fumigatus: Definition of pathogenic species. PLoS ONE 2013, 8, e56651. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.M.; French, S.; Kohn, L.A.; Chen, V.; Johnson, A.D. Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat. Genet. 2010, 42, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Sudbery, P.E. Growth of Candida albicans hyphae. Nat. Rev. Microbiol. 2011, 9, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; Simmons, R.M.; Underhill, D.M. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.T.; Fink, G.R. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog. 2006, 2, e35. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D. Innate antifungal immunity: The key role of phagocytes. Annu. Rev. Immunol. 2011, 29, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.M.; Louw, J.; Lewis, L.E.; Okai, B.; Walls, C.A.; Ballou, E.R.; Walker, L.A.; Reid, D.; Munro, C.A.; Brown, A.J.P.; et al. Candida albicans Hypha Formation and Mannan Masking of β-Glucan Inhibit Macrophage Phagosome Maturation. MBio 2014, 5, e01874–e01914. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, T.R.; Veri, A.O.; Ketela, T.; Jiang, B.; Roemer, T.; Cowen, L.E. Global analysis of fungal morphology exposes mechanisms of host cell escape. Nat. Commun. 2015, 6, 6741. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.E.; Bain, J.M.; Lowes, C.; Gillespie, C.; Rudkin, F.M.; Gow, N.A.R.; Erwig, L.-P. Stage specific assessment of Candida albicans phagocytosis by macrophages identifies cell wall composition and morphogenesis as key determinants. PLoS Pathog. 2012, 8, e1002578. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.; Rasmussen, M.D.; Lin, M.F.; Santos, M.A.S.; Sakthikumar, S.; Munro, C.A.; Rheinbay, E.; Grabherr, M.; Forche, A.; Reedy, J.L.; et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 2009, 459, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, A.R.; Simões, J.; Lee, W.; Rung, J.; Weil, T.; Gut, I.G.; Gut, M.; Bayés, M.; Rizzetto, L.; Cavalieri, D.; et al. Reversion of a fungal genetic code alteration links proteome instability with genomic and phenotypic diversification. Proc. Natl. Acad. Sci. USA. 2013, 110, 11079–11084. [Google Scholar] [CrossRef] [PubMed]
- Miranda, I.; Silva-Dias, A.; Rocha, R.; Teixeira-Santos, R.; Coelho, C.; Gonçalves, T.; Santos, M.A.S.; Pina-Vaz, C.; Solis, N.V.; Filler, S.G.; et al. Candida albicans CUG mistranslation is a mechanism to create cell surface variation. MBio 2013, 4, e00285–e00313. [Google Scholar] [CrossRef] [PubMed]
- Morschhäuser, J. Regulation of white-opaque switching in Candida albicans. Med. Microbiol. Immunol. 2010, 199, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Soll, D.R. Why does Candida albicans switch? FEMS Yeast Res. 2009, 9, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Du, H.; Guan, G.; Dai, Y.; Nobile, C.J.; Liang, W.; Cao, C.; Zhang, Q.; Zhong, J.; Huang, G. Discovery of a “White-Gray-Opaque” Tristable Phenotypic Switching System in Candida albicans: Roles of Non-genetic Diversity in Host Adaptation. PLoS Biol. 2014, 12, e1001830. [Google Scholar] [CrossRef] [PubMed]
- Pande, K.; Chen, C.; Noble, S.M. Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat. Genet. 2013, 45, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Soll, D.R. The evolution of alternative biofilms in an opportunistic fungal pathogen: An explanation for how new signal transduction pathways may evolve. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2014, 22, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Finkel, J.S.; Mitchell, A.P. Genetic control of Candida albicans biofilm development. Nat. Rev. Microbiol. 2011, 9, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hardison, S.E.; Brown, G.D. C-type lectin receptors orchestrate antifungal immunity. Nat. Immunol. 2012, 13, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Ishihara, C.; Takeuchi, A.; Imanishi, T.; Xue, L.; Morris, S.W.; Inui, M.; Takai, T.; Shibuya, A.; Saijo, S.; et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol. 2007, 8, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Ferwerda, B.; Ferwerda, G.; Plantinga, T.S.; Willment, J.A.; Van Spriel, A.B.; Venselaar, H.; Elbers, C.C.; Johnson, M.D.; Cambi, A.; Huysamen, C.; et al. Human Dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 2009, 361, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Ariizumi, K.; Shen, G.L.; Shikano, S.; Xu, S.; Ritter, R.; Kumamoto, T.; Edelbaum, D.; Morita, A.; Bergstresser, P.R.; Takashima, A. Identification of a novel, dendritic cell-associated molecule, Dectin-1, by subtractive cDNA cloning. J. Biol. Chem. 2000, 275, 20157–20167. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Reyes, C.N.; Becker, C.A.; Katsumoto, T.R.; Ma, J.; Wolf, A.J.; Bose, N.; Chan, A.S.H.; Magee, A.S.; Danielson, M.E. Activation of the innate immune receptor Dectin-1 upon formation of a “phagocytic synapse.”. Nature 2011, 472, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Hernanz-Falcón, P.; Joffre, O.; Williams, D.L.; Reis e Sousa, C. Internalization of Dectin-1 terminates induction of inflammatory responses. Eur. J. Immunol. 2009, 39, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Rosas, M.; Liddiard, K.; Kimberg, M.; Faro-Trindade, I.; McDonald, J.U.; Williams, D.L.; Brown, G.D.; Taylor, P.R. The induction of inflammation by Dectin-1 in vivo is dependent on myeloid cell programming and the progression of phagocytosis. J. Immunol. 2008, 181, 3549–3557. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M. Interaction of cells with immune complexes: Adherence, release of constituents, and tissue injury. J. Exp. Med. 1971, 134, 114s–135s. [Google Scholar] [PubMed]
- Rizzetto, L.; De Filippo, C.; Rivero, D.; Riccadonna, S.; Beltrame, L.; Cavalieri, D. Systems biology of host-mycobiota interactions: Dissecting Dectin-1 and Dectin-2 signalling in immune cells with DC-ATLAS. Immunobiology 2013, 218, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Drummond, R.A.; Brown, G.D. Signalling C-type lectins in antimicrobial immunity. PLoS Pathog. 2013, 9, e1003417. [Google Scholar] [CrossRef] [PubMed]
- Strasser, D.; Neumann, K.; Bergmann, H.; Marakalala, M.J.; Guler, R.; Rojowska, A.; Hopfner, K.-P.; Brombacher, F.; Urlaub, H.; Baier, G.; et al. Syk kinase-coupled C-type lectin receptors engage protein kinase C-σ to elicit Card9 adaptor-mediated innate immunity. Immunity 2012, 36, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Shimada, T.; Wolf, A.J.; Hsu, Y.-M.S.; Becker, C.A.; Lin, X.; Underhill, D.M. Differential Use of CARD9 by Dectin-1 in Macrophages and Dendritic Cells. J. Immunol. 2009, 182, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Wevers, B.A.; Kaptein, T.M.; Zijlstra-Willems, E.M.; Theelen, B.; Boekhout, T.; Geijtenbeek, T.B.H.; Gringhuis, S.I. Fungal engagement of the C-type lectin mincle suppresses Dectin-1-induced antifungal immunity. Cell Host Microbe 2014, 15, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Del Fresno, C.; Soulat, D.; Roth, S.; Blazek, K.; Udalova, I.; Sancho, D.; Ruland, J.; Ardavín, C. Interferon-β production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 2013, 38, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.-M.; Tang, B.; Zhu, L.-L.; Liu, Y.-H.; Zhao, X.-Q.; Gorjestani, S.; Hsu, Y.-M.S.; Yang, L.; Guan, J.-H.; Xu, G.-T.; et al. CARD9 mediates Dectin-1—Induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J. Exp. Med. 2014, 211, 2307–2321. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Theelen, B.; Van der Vlist, M.; Boekhout, T.; Geijtenbeek, T.B.H. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012, 13, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 2009, 5, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.; LeibundGut-Landmann, S.; Lochner, M.; Lahl, K.; Sparwasser, T.; Eberl, G.; Reis e Sousa, C. DC activated via Dectin-1 convert Treg into IL-17 producers. Eur. J. Immunol. 2008, 38, 3274–3281. [Google Scholar] [CrossRef] [PubMed]
- Vautier, S.; Drummond, R.A.; Redelinghuys, P.; Murray, G.I.; MacCallum, D.M.; Brown, G.D. Dectin-1 is not required for controlling Candida albicans colonization of the gastrointestinal tract. Infect. Immun. 2012, 80, 4216–4222. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Gow, N.A.R.; Joosten, L.A.B.; Verschueren, I.; Van der Meer, J.W.M.; Kullberg, B.J. Variable recognition of Candida albicans strains by TLR4 and lectin recognition receptors. Med. Mycol. 2010, 48, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, L.; Buschow, S.I.; Beltrame, L.; Figdor, C.G.; Schierer, S.; Schuler, G.; Cavalieri, D. The modular nature of dendritic cell responses to commensal and pathogenic fungi. PLoS ONE 2012, 7, e42430. [Google Scholar] [CrossRef] [PubMed]
- Ifrim, D.C.; Bain, J.M.; Reid, D.M.; Oosting, M.; Verschueren, I.; Gow, N.A.R.; Van Krieken, J.H.; Brown, G.D.; Kullberg, B.-J.; Joosten, L.A.B.; et al. Role of Dectin-2 for Host Defense against Systemic Infection with Candida glabrata. Infect. Immun. 2014, 82, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, B.; Willment, J.A.; Brown, G.D. The Dectin-2 family of C-type lectin-like receptors: An update. Int. Immunol. 2013, 25, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Yang, X.; Yudate, T.; Chung, J.-S.; Wu, J.; Luby-Phelps, K.; Kimberly, R.P.; Underhill, D.; Cruz, P.D.; Ariizumi, K. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J. Biol. Chem. 2006, 281, 38854–38866. [Google Scholar] [CrossRef] [PubMed]
- Ariizumi, K.; Shen, G.L.; Shikano, S.; Ritter, R.; Zukas, P.; Edelbaum, D.; Morita, A.; Takashima, A. Cloning of a second dendritic cell-associated C-type lectin (Dectin-2) and its alternatively spliced isoforms. J. Biol. Chem. 2000, 275, 11957–11963. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Reid, D.M.; Heinsbroek, S.E.M.; Brown, G.D.; Gordon, S.; Wong, S.Y.C. Dectin-2 is predominantly myeloid restricted and exhibits unique activation-dependent expression on maturing inflammatory monocytes elicited in vivo. Eur. J. Immunol. 2005, 35, 2163–2174. [Google Scholar] [CrossRef] [PubMed]
- Seeds, R.E.; Gordon, S.; Miller, J.L. Characterisation of myeloid receptor expression and interferon alpha/beta production in murine plasmacytoid dendritic cells by flow cytomtery. J. Immunol. Methods 2009, 350, 106–117. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.U.; Rosas, M.; Brown, G.D.; Jones, S.A.; Taylor, P.R. Differential dependencies of monocytes and neutrophils on Dectin-1, Dectin-2 and complement for the recognition of fungal particles in inflammation. PLoS ONE 2012, 7, e45781. [Google Scholar] [CrossRef] [PubMed]
- Gavino, A.C.P.; Chung, J.-S.; Sato, K.; Ariizumi, K.; Cruz, P.D. Identification and expression profiling of a human C-type lectin, structurally homologous to mouse Dectin-2. Exp. Dermatol. 2005, 14, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, N.; Tashiro, K.; Inaba, K.; Lutz, M.B.; Miyachi, Y. Molecular cloning of human Dectin-2. J. Invest. Dermatol. 2004, 122, 1522–1524. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Gojestani, S.; Wu, W.; Hsu, Y.-M.S.; Zhu, J.; Ariizumi, K.; Lin, X. CARD9 mediates Dectin-2-induced IkappaBalpha kinase ubiquitination leading to activation of NF-kappaB in response to stimulation by the hyphal form of Candida albicans. J. Biol. Chem. 2010, 285, 25969–25977. [Google Scholar] [CrossRef] [PubMed]
- Gorjestani, S.; Yu, M.; Tang, B.; Zhang, D.; Wang, D.; Lin, X. Phospholipase Cγ2 (PLCγ2) is key component in Dectin-2 signaling pathway, mediating anti-fungal innate immune responses. J. Biol. Chem. 2011, 286, 43651–43659. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Wevers, B.A.; Kaptein, T.M.; Van Capel, T.M.M.; Theelen, B.; Boekhout, T.; De Jong, E.C.; Geijtenbeek, T.B.H. Selective C-Rel activation via Malt1 controls anti-fungal T(H)-17 immunity by Dectin-1 and Dectin-2. PLoS Pathog. 2011, 7, e1001259. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xu, X.; Tian, X.; Shao, H.; Wu, X.; Wang, Q.; Su, X.; Shi, Y. Activation of NF-κB and respiratory burst following Aspergillus fumigatus stimulation of macrophages. Immunobiology 2014, 219, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Ritter, M.; Gross, O.; Kays, S.; Ruland, J.; Nimmerjahn, F.; Saijo, S.; Tschopp, J.; Layland, L.E.; Prazeres da Costa, C. Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc. Natl. Acad. Sci. USA. 2010, 107, 20459–20464. [Google Scholar] [CrossRef] [PubMed]
- Bugarcic, A.; Hitchens, K.; Beckhouse, A.G.; Wells, C.A.; Ashman, R.B.; Blanchard, H. Human and mouse macrophage-inducible C-type lectin (Mincle) bind Candida albicans. Glycobiology 2008, 18, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Cambi, A.; Gijzen, K.; De Vries, L.J.M.; Torensma, R.; Joosten, B.; Adema, G.J.; Netea, M.G.; Kullberg, B.-J.; Romani, L.; Figdor, C.G. The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur. J. Immunol. 2003, 33, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Brown, G.D.; Herre, J.; Williams, D.L.; Willment, J.A.; Gordon, S. The role of SIGNR1 and the beta-glucan receptor (Dectin-1) in the nonopsonic recognition of yeast by specific macrophages. J. Immunol. 2004, 172, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Marijnissen, R.J.; Kullberg, B.J.; Koenen, H.J.P.M.; Cheng, S.-C.; Joosten, I.; Van den Berg, W.B.; Williams, D.L.; Van der Meer, J.W.M.; Joosten, L.A.B.; et al. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 2009, 5, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.A.; Salvage-Jones, J.A.; Li, X.; Hitchens, K.; Butcher, S.; Murray, R.Z.; Beckhouse, A.G.; Lo, Y.-L.-S.; Manzanero, S.; Cobbold, C.; et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J. Immunol. 2008, 180, 7404–7413. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.; Reis e Sousa, C. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity 2011, 34, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Arce, I.; Martínez-Muñoz, L.; Roda-Navarro, P.; Fernández-Ruiz, E. The human C-type lectin CLECSF8 is a novel monocyte/macrophage endocytic receptor. Eur. J. Immunol. 2004, 34, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.M.; Brown, G.D. The Dectin-2 family of C-type lectins in immunity and homeostasis. Cytokine 2009, 48, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Kingeter, L.M.; Lin, X. C-type lectin receptor-induced NF-κB activation in innate immune and inflammatory responses. Cell. Mol. Immunol. 2012, 9, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Balch, S.G.; McKnight, A.J.; Seldin, M.F.; Gordon, S. Cloning of a novel C-type lectin expressed by murine macrophages. J. Biol. Chem. 1998, 273, 18656–18664. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.-L.; Zhao, X.-Q.; Jiang, C.; You, Y.; Chen, X.-P.; Jiang, Y.-Y.; Jia, X.-M.; Lin, X. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 2013, 39, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Dennehy, K.M.; Willment, J.A.; Williams, D.L.; Brown, G.D. Reciprocal regulation of IL-23 and IL-12 following co-activation of Dectin-1 and TLR signaling pathways. Eur. J. Immunol. 2009, 39, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Runglall, M.; Murciano, C.; Shen, C.; Nayar, D.; Thavaraj, S.; Kohli, A.; Islam, A.; Mora-Montes, H.; Challacombe, S.J.; et al. A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe 2010, 8, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Gow, N.A.R.; Van de Veerdonk, F.L.; Brown, A.J.P.; Netea, M.G. Candida albicans morphogenesis and host defence: Discriminating invasion from colonization. Nat. Rev. Microbiol. 2011, 10, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Igyártó, B.Z.; Haley, K.; Ortner, D.; Bobr, A.; Gerami-Nejad, M.; Edelson, B.T.; Zurawski, S.M.; Malissen, B.; Zurawski, G.; Berman, J.; et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 2011, 35, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Troutman, T.D.; Edukulla, R.; Pasare, C. Priming microenvironments dictate cytokine requirements for T helper 17 cell lineage commitment. Immunity 2011, 35, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Igyártó, B.Z.; Gerami-Nejad, M.; Kumamoto, Y.; Mohammed, J.; Jarrett, E.; Drummond, R.A.; Zurawski, S.M.; Zurawski, G.; Berman, J.; et al. Candida albicans morphology and dendritic cell subsets determine T helper cell differentiation. Immunity 2015, 42, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, E.; Vita, F.; Medic, N.; Soranzo, M.R.; Zabucchi, G.; Borelli, V. Mast cells kill Candida albicans in the extracellular environment but spare ingested fungi from death. Inflammation 2014, 37, 2174–2189. [Google Scholar] [CrossRef] [PubMed]
- St John, A.L.; Abraham, S.N. Innate immunity and its regulation by mast cells. J. Immunol. 2013, 190, 4458–4463. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Patlán, A.; Campillo-Navarro, M.; Rodríguez-Cortés, O.; Muñoz-Cruz, S.; Wong-Baeza, I.; Estrada-Parra, S.; Estrada-García, I.; Serafín-López, J.; Chacón-Salinas, R. Recognition of Candida albicans by Dectin-1 induces mast cell activation. Immunobiology 2015, 220, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Joly, S.; Ma, N.; Sadler, J.J.; Soll, D.R.; Cassel, S.L.; Sutterwala, F.S. Cutting Edge: Candida albicans Hyphae Formation Triggers Activation of the Nlrp3 Inflammasome. J. Immunol. 2009, 183, 3578–3581. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Santos, N.; Huppler, A.R.; Peterson, A.C.; Khader, S.A.; McKenna, K.C.; Gaffen, S.L. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol. 2013, 6, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Naglik, J.R. Mucosal immunity and Candida albicans infection. Clin. Dev. Immunol. 2011, 2011, 346307. [Google Scholar] [CrossRef] [PubMed]
- Glocker, E.-O.; Hennigs, A.; Nabavi, M.; Schäffer, A.A.; Woellner, C.; Salzer, U.; Pfeifer, D.; Veelken, H.; Warnatz, K.; Tahami, F.; et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009, 361, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Bishu, S.; Hernández-Santos, N.; Simpson-Abelson, M.R.; Huppler, A.R.; Conti, H.R.; Ghilardi, N.; Mamo, A.J.; Gaffen, S.L. The Adaptor CARD9 Is Required for Adaptive but Not Innate Immunity to Oral Mucosal Candida albicans Infections. Infect. Immun. 2014, 82, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Iliev, I.D.; Funari, V.A.; Taylor, K.D.; Nguyen, Q.; Reyes, C.N.; Strom, S.P.; Brown, J.; Becker, C.A.; Fleshner, P.R.; Dubinsky, M.; et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 2012, 336, 1314–1317. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kedar, S.; Baram, L.; Elad, H.; Brazowski, E.; Guzner-Gur, H.; Dotan, I. Human intestinal epithelial cells respond to β-glucans via Dectin-1 and Syk. Eur. J. Immunol. 2014, 44, 3729–3740. [Google Scholar] [CrossRef] [PubMed]
- Lowell, C.A. Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: Signaling cross talk. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Mócsai, A.; Ruland, J.; Tybulewicz, V.L.J. The SYK tyrosine kinase: A crucial player in diverse biological functions. Nat. Rev. Immunol. 2010, 10, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Vautier, S.; Drummond, R.A.; Chen, K.; Murray, G.I.; Kadosh, D.; Brown, A.J.P.; Gow, N.A.R.; MacCallum, D.M.; Kolls, J.K.; Brown, G.D. Candida albicans colonization and dissemination from the murine gastrointestinal tract: The influence of morphology and Th17 immunity. Cell. Microbiol. 2015, 17, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.Y.; Köhler, J.R.; Coggshall, K.T.; Van Rooijen, N.; Pier, G.B. Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog. 2008, 4, e35. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Tsoni, S.V.; Willment, J.A.; Dennehy, K.M.; Rosas, M.; Findon, H.; Haynes, K.; Steele, C.; Botto, M.; Gordon, S.; et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 2007, 8, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, J. Vaginal candidosis: Epidemiological and etiological factors. Int. J. Gynaecol. Obstet. Off. Organ Int. Fed. Gynaecol. Obstet. 2000, 71 (Suppl. 1), S21–S27. [Google Scholar] [CrossRef]
- Plantinga, T.S.; Van der Velden, W.J.F.M.; Ferwerda, B.; Van Spriel, A.B.; Adema, G.; Feuth, T.; Donnelly, J.P.; Brown, G.D.; Kullberg, B.-J.; Blijlevens, N.M.A.; et al. Early stop polymorphism in human DECTIN-1 is associated with increased candida colonization in hematopoietic stem cell transplant recipients. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2009, 49, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Usluogullari, B.; Gumus, I.; Gunduz, E.; Kaygusuz, I.; Simavli, S.; Acar, M.; Oznur, M.; Gunduz, M.; Kafali, H. The role of Human Dectin-1 Y238X Gene Polymorphism in recurrent vulvovaginal candidiasis infections. Mol. Biol. Rep. 2014, 41, 6763–6768. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Giovannini, G.; De Luca, A.; D’Angelo, C.; Casagrande, A.; Iannitti, R.G.; Ricci, G.; Cunha, C.; Romani, L. Dectin-1 isoforms contribute to distinct Th1/Th17 cell activation in mucosal candidiasis. Cell. Mol. Immunol. 2012, 9, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Heinsbroek, S.E.M.; Taylor, P.R.; Rosas, M.; Willment, J.A.; Williams, D.L.; Gordon, S.; Brown, G.D. Expression of functionally different Dectin-1 isoforms by murine macrophages. J. Immunol. 2006, 176, 5513–5518. [Google Scholar] [CrossRef] [PubMed]
- Rogers, N.C.; Slack, E.C.; Edwards, A.D.; Nolte, M.A.; Schulz, O.; Schweighoffer, E.; Williams, D.L.; Gordon, S.; Tybulewicz, V.L.; Brown, G.D.; et al. Syk-Dependent Cytokine Induction by Dectin-1 Reveals a Novel Pattern Recognition Pathway for C Type Lectins. Immunity 2005, 22, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.; Agrawal, S.; Banerjee, K.; Letterio, J.; Denning, T.L.; Oswald-Richter, K.; Kasprowicz, D.J.; Kellar, K.; Pare, J.; Van Dyke, T.; et al. Yeast zymosan, a stimulus for TLR2 and Dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J. Clin. Invest. 2006, 116, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaf, C.A.A.; Netea, M.G.; Verschueren, I.; Van der Meer, J.W.M.; Kullberg, B.J. Differential cytokine production and Toll-like receptor signaling pathways by Candida albicans blastoconidia and hyphae. Infect. Immun. 2005, 73, 7458–7464. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-C.; Van de Veerdonk, F.L.; Lenardon, M.; Stoffels, M.; Plantinga, T.; Smeekens, S.; Rizzetto, L.; Mukaremera, L.; Preechasuth, K.; Cavalieri, D.; et al. The Dectin-1/inflammasome pathway is responsible for the induction of protective T-helper 17 responses that discriminate between yeasts and hyphae of Candida albicans. J. Leukoc. Biol. 2011, 90, 357–366. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizzetto, L.; Weil, T.; Cavalieri, D. Systems Level Dissection of Candida Recognition by Dectins: A Matter of Fungal Morphology and Site of Infection. Pathogens 2015, 4, 639-661. https://doi.org/10.3390/pathogens4030639
Rizzetto L, Weil T, Cavalieri D. Systems Level Dissection of Candida Recognition by Dectins: A Matter of Fungal Morphology and Site of Infection. Pathogens. 2015; 4(3):639-661. https://doi.org/10.3390/pathogens4030639
Chicago/Turabian StyleRizzetto, Lisa, Tobias Weil, and Duccio Cavalieri. 2015. "Systems Level Dissection of Candida Recognition by Dectins: A Matter of Fungal Morphology and Site of Infection" Pathogens 4, no. 3: 639-661. https://doi.org/10.3390/pathogens4030639
APA StyleRizzetto, L., Weil, T., & Cavalieri, D. (2015). Systems Level Dissection of Candida Recognition by Dectins: A Matter of Fungal Morphology and Site of Infection. Pathogens, 4(3), 639-661. https://doi.org/10.3390/pathogens4030639