


Applying CRISPR Technologies for the Treatment of Human Herpesvirus Infections: A Scoping Review

Abstract
1. Introduction
2. Methods
2.1. Search Strategy, Eligibility Criteria, and Selection Process
2.2. Data Charting Process and Synthesis of Results
3. Results
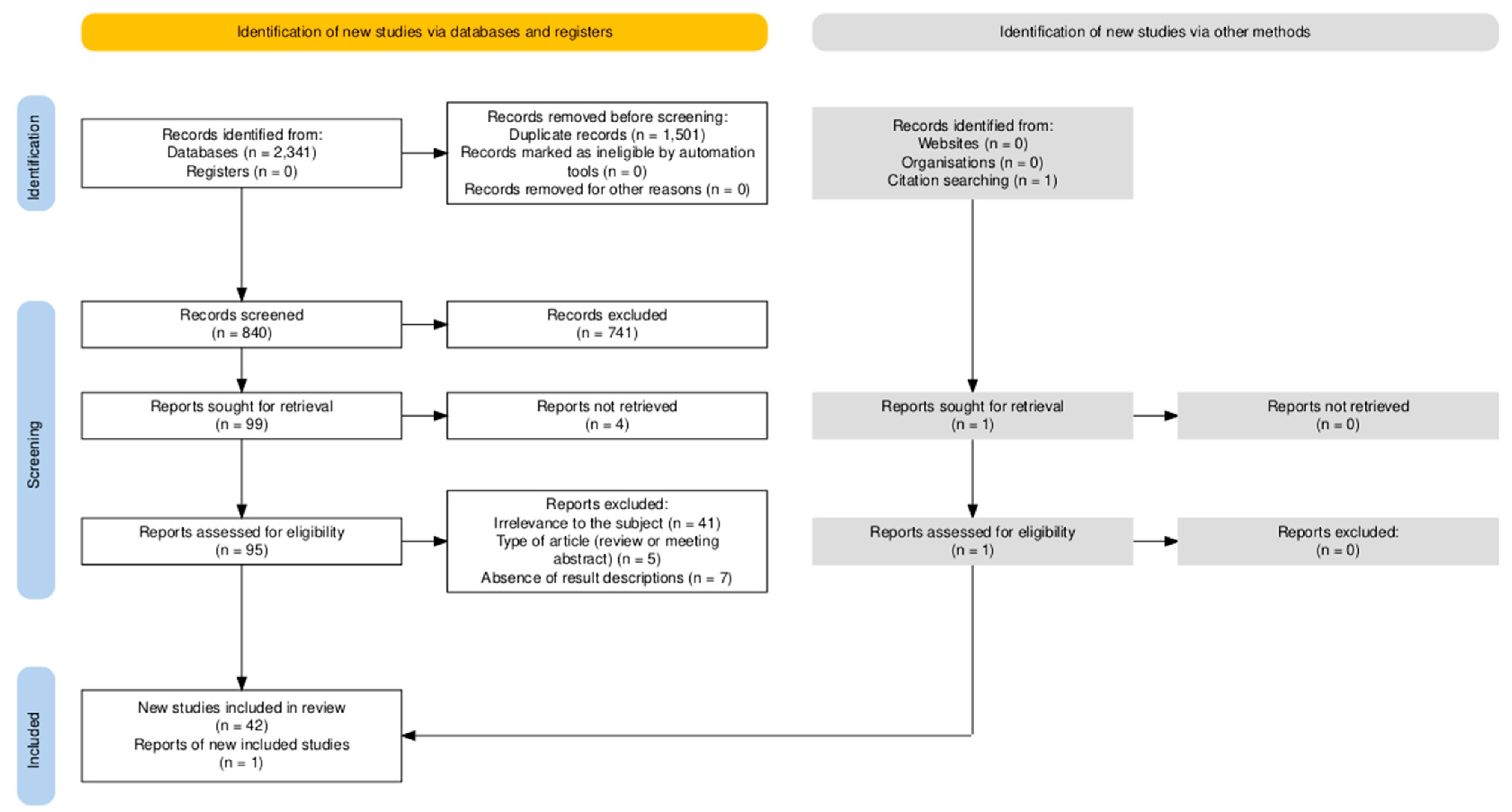
3.1. Screening Results
3.2. Overview of CRISPR Strategies Used to Target Different Human Herpesviruses
3.2.1. CRISPR Systems Targeting Herpes Simplex Virus Type 1
The Application of CRISPR Systems to Inhibit Lytic HSV-1 Replication
The Application of CRISPR Systems to Edit Quiescent HSV-1 Genomes and Disrupt Viral Reactivation
The Application of CRISPR Systems for the Prevention or Treatment of Herpes Simplex Keratitis
3.2.2. CRISPR Systems Targeting the Lytic Reproduction Cycle and Reactivation of Varicella-Zoster Virus
3.2.3. CRISPR Systems Targeting Epstein-Barr Virus
The Application of CRISPR Systems to Edit EBV Latency-Associated Genes
The Application of CRISPR Systems to Induce the Reactivation of Latent EBV Infection
3.2.4. CRISPR Systems Targeting Lytic Human Cytomegalovirus Replication and Reactivation
3.2.5. CRISPR Systems Targeting the Integrated Latent Human Herpesvirus 6A Genome
3.2.6. CRISPR Systems Targeting Kaposi’s Sarcoma-Associated Herpesvirus
The Application of CRISPR Systems to Inhibit Lytic KSHV Replication
The Application of CRISPR Systems Targeting KSHV Genes Important for Latency Maintenance
The Application of CRISPR Systems to Induce the Reactivation of Latent KSHV Infection
3.2.7. Applying CRISPR Systems for the Enhanced Efficiency or Development of Cellular Immunotherapy

{kind=link}
{kind=link}
| Virus | Mechanism | Target | CRISPR System | Vector | Test Subject | Reference |
|---|---|---|---|---|---|---|
| HSV-1 | Inhibiting lytic viral replication | Immediate early (IE) viral genes | ||||
| Singleplex | ||||||
| ICP0 | CRISPR-Cas9 | Plasmid transfection | In vitro: Vero L7 cells | P. C. Roehm et al. [42] | ||
| Lentivirus | In vitro: Human oligodendroglioma TC620 cells | |||||
| Lentivirus | In vitro: Vero cells | D. S. Karpov et al. (2022) [41] | ||||
| CRISPR-Cas9, CRISPR-Cas9 | Lentivirus | In vitro: Vero cells | Y. Chen et al. [52] | |||
| ICP4 | CRISPR-Cas9, CRISPR-Cas9 | Lentivirus | In vitro: Vero cells | Y. Chen et al. [52] | ||
| CRISPR-Cas9 | AAV-1 | In vitro: Murine primary trigeminal ganglion neuronal cells | ||||
| Lentivirus | In vitro: Human foreskin fibroblasts | H. S. Oh et al. [56] | ||||
| ALICECas9+gRNA | AAVrh10 | In vivo: Herpes simplex keratitis mouse model | Y. D. Wang et al. [59] | |||
| ICP27 | CRISPR-Cas9 | Lentivirus | In vitro: Human foreskin fibroblasts | H. S. Oh et al. [56] | ||
| CRISPR-Cas9 | Lentivirus | In vitro: Vero cells | F. R. van Diemen et al. [45] | |||
| Multiplex | ||||||
| ICP0 (2 sites), ICP27 (2 sites) | CRISPR-Cas9 | Plasmid transfection | In vitro: Vero cells | N. Amrani et al. [50] | ||
| AAV2 | In vitro: Vero cells | N. Amrani et al. [50] | ||||
| A. Bellizzi et al. [51] | ||||||
| ICP0-ICP27 | CRISPR-Cas9 | Lentivirus | In vitro: Human oligodendroglioma TC620 cells | P. C. Roehm et al. [42] | ||
| CRISPR-Cas9 | - AAV8-Y733 - AAV9 | In vivo: HSV-1 latently infected rabbit keratitis model | N. Amrani et al. [50] | |||
| ICP0-ICP4, IPC4-ICP27 | CRISPR-Cas9 | Lentivirus | In vitro: Human oligodendroglioma TC620 cells | P. C. Roehm et al. [42] | ||
| Early (E) viral genes | ||||||
| Singleplex | ||||||
| UL5, UL8, UL9, UL42, or UL52 | CRISPR-Cas9 | Lentivirus | In vitro: Vero cells | F. R. van Diemen et al. [45] | ||
| UL19 | CRISPR-Cas9 | Plasmid transfection | In vitro: Vero cells | D. S. Karpov et al. (2022) [41] | ||
| UL29 | CRISPR-Cas9 | Lentivirus | In vitro: Human foreskin fibroblasts | H. S. Oh et al. [56] | ||
| CRISPR-Cas9 | Engineered extracellular vesicles | In vitro: - Vero cells - Hela cells - Neuro HT22 cells In vivo: Mouse model | Y. Wan et al. [58] | |||
| UL30 | CRISPR-Cas9 | Lentivirus | In vitro: Vero cells | F. R. van Diemen et al. [45] | ||
| CRISPR-Cas9 | Lentivirus | In vitro: Human foreskin fibroblasts | H. S. Oh et al. [56] | |||
| CRISPR-Cas9, CRISPR-CasX | Plasmid transfection | In vitro: Vero cells | D. S. Karpov et al. (2019 and 2022) [41,54] | |||
| UL39 | CRISPR-Cas9 | Plasmid transfection | In vitro: Vero cells | J. Vasques Raposo et al. [57] | ||
| In vivo: BALB/c mouse model | R. M. P. de Sousa [53] | |||||
| Multiplex | ||||||
| UL8-UL29 | CRISPR-Cas9 | Lentivirus | In vitro: - Vero cells - MRC5 human lung fibroblasts | F. R. van Diemen et al. [45] | ||
| CRISPR-Cas9 | Plasmid transfection | In vitro: Vero cells | D. S. Karpov et al. (2019 and 2022) [41,54] | |||
| HELP (HSV-1-erasing lentiviral particle) | In vitro: - HEK293T cells - HaCaT cells - Murine primary corneal stromal cells In vivo: Herpes simplex keratitis mouse model Ex vivo: Human cornea | D. Yin et al. [49] | ||||
| Clinical trial in humans | A. Wei et al. [47] | |||||
| UL29-UL52 | CRISPR-Cas9 | Lentivirus | In vitro: - Vero cells - MRC5 human lung fibroblasts | F. R. van Diemen et al. [45] | ||
| CRISPR-Cas9 | Plasmid transfection | In vitro: Vero cells | D. S. Karpov et al. (2019 and 2022) [41,54] | |||
| UL8-UL52 | CRISPR-Cas9 | Lentivirus | In vitro: Vero cells | F. R. van Diemen et al. [45] | ||
| UL19-UL30 | CRISPR-Cas9 | Plasmid transfection | In vitro: Vero cells | D. S. Karpov et al. (2019 and 2022) [41,54] | ||
| Late (L) viral genes | ||||||
| gD | CRISPR-Cas9 | Plasmid transfection | In vitro: HEK293-AD cells | N. Khodadad et al. [55] | ||
| UL15, UL27, UL36, or UL37 | CRISPR-Cas9 | Lentivirus | In vitro: Vero cells | F. R. van Diemen et al. [45] | ||
| Nonessential viral genes/intergenic regions | ||||||
| US3 or US8 | CRISPR-Cas9 | Lentivirus | In vitro: Vero cells | F. R. van Diemen et al. [45] | ||
| Intergenic regions between UL26-27 and UL37-38 | CRISPR-Cas9 | Lentivirus | In vitro: Human foreskin fibroblasts | H. S. Oh et al. [56] | ||
| Host genes | ||||||
| NECTIN-1 | CRISPR-Cas9 | Lentivirus | In vitro: Human corneal epithelial cells | Y. Li et al. [62] | ||
| DUX4 | CRISPR-Cas9 | Lentivirus | In vitro: - HEK 293T cells - HAP1 cells | E. Neugebauer et al. [61] | ||
| Multiplex strategy (IE, E, L, and/or nonessential viral genes) | ||||||
| ICP4, ICP27, VP16, and/or gD | CRISPR-Cas9 | Plasmid transfection Lentivirus | In vitro: BHK-21 cells In vivo: Mouse models | M. Ying et al. [60] | ||
| UL29, UL52, and US8 | ALICECas9+gRNA | Plasmid transfection | In vitro: HEK293 T cells In vivo: Mouse models | Y. D. Wang et al. [59] | ||
| Targeting latent viral genomes for the inhibition of viral reactivation | Singleplex | |||||
| UL8 or UL52 | CRISPR-Cas9 | Lentivirus | In vitro: MRC5 human lung fibroblasts | F. R. van Diemen et al. [45] | ||
| UL29 | CRISPR-Cas9 | Lentivirus | In vitro: MRC5 human lung fibroblasts | F. R. van Diemen et al. [45] | ||
| CRISPR-Cas9 | Lentivirus | In vitro: Human foreskin fibroblasts | H. S. Oh et al. [56] | |||
| UL30 | CRISPR-Cas9 | Lentivirus | In vitro: Human foreskin fibroblasts | H. S. Oh et al. [56] | ||
| Multiplex | ||||||
| ICP0 (2 sites) or ICP27 (2 sites) | CRISPR-Cas9 | AAV2 | In vitro: Human induced pluripotent stem cell-derived cerebral organoids | A. Bellizzi et al. [51] | ||
| UL29-UL30 | CRISPR-Cas9 | Lentivirus | In vitro: Human foreskin fibroblasts | H. S. Oh et al. [56] | ||
| VZV | Inhibiting lytic viral replication and targeting latent viral genomes for the inhibition of viral reactivation | ORF62/71 | CRISPR-Cas9 | AAV2 | In vitro: - Retinal-pigmented epithelial (ARPE-19) cells - Human embryonic stem cell (hESC)-derived neuron cultures | B. Wu et al. [48] |
| EBV | Targeting viral genes important for latency maintenance | Singleplex | ||||
| EBNA1 or OriP | CRISPR-Cas9 | Plasmid transfection | In vitro: - NPC C666-1 cells - HEK293M81 cells | K. S. Yuen et al. [63] | ||
| Lentivirus | In vitro: Burkitt’s lymphoma Akata-Bx1 cells | F. R. van Diemen et al. [45] | ||||
| W repeats | CRISPR-Cas9 | Plasmid transfection | In vitro: - NPC C666-1 cells - HEK293M81 cells | K. S. Yuen et al. [63] | ||
| LMP1 | CRISPR-Cas9 | Plasmid transfection | In vitro: Human NPC CNE-2 cells | H. Huo and G. Hu [64] | ||
| PBAE-plasmid polyplex nanoparticles | In vitro: - Nasopharyngeal carcinoma C666-1 cells In vivo: - Mouse C666-1 xenograft tumor model | C. Yuan et al. [65] | ||||
| In vivo: KM mouse model | J. Ding et al. [39] | |||||
| LMP2 | CRISPR-Cas9 | Plasmid transfection | In vitro: Human NPC CNE-2 cells | H. Huo and G. Hu [64] | ||
| Multiplex | ||||||
| EBNA1, EBNA-LP, EBNA-LP/PstI/125bp Repeats, LMP1, and EBNA3C | CRISPR-Cas9 | Plasmid transfection | In vitro: Raji cells | J. Wang and S.R. Quake [46] | ||
| EBNA1 (2 sites) and EBNA1-OriP | CRISPR-Cas9 | Lentivirus | In vitro: Burkitt’s lymphoma Akata-Bx1 cells | F. R. van Diemen et al. [45] | ||
| Purposefully inducing viral reactivation | Promotor of ZAT | CRISPRa | Lentivirus Plasmid transfection | In vitro: - Akata (EBV+)/P3HR1Burkitt lymphoma cells - EBV+ gastric cancer SNU-719 cells - EBV+ nasopharyngeal carcinoma HK-1 cells | F.G. Sugiokto and R. Li [66] | |
| Enhancing the efficiency of cellular immunotherapy | EBV-VSTs | |||||
| NR3C1 | CRISPR-Cas9 | Ribonucleoprotein electroporation | In vitro: Peripheral blood mononuclear cells | K. Koukoulias et al. [77] | ||
| CCR5 | CRISPR-Cas9 | - Ribonucleoprotein nucleofection - AAV6 | In vitro: Peripheral blood mononuclear cells | D. Palianina et al. [78] | ||
| EBV-LMP2A-CTLs | ||||||
| PD-1 | CRISPR-Cas9 | Plasmid transfection | In vitro: Human peripheral blood lymphocytes In vivo: Mouse xenograft model | S. Su et al. [43] | ||
| Developing cellular immunotherapy | CAR-T cells | |||||
| TRAC | CRISPR-Cas9 | Electroporation | In vitro: Peripheral blood mononuclear cells In vivo: Mouse xenograft models | T. Braun et al. [79] | ||
| HCMV | Inhibiting lytic viral replication and/or targeting latent viral genomes for the inhibition of viral reactivation | Immediate early (IE) viral genes | ||||
| Singleplex | ||||||
| UL122/123 | CRISPR-Cas9 | Lentivirus | In vitro: MRC5 primary fibroblasts | J. Gergen et al. [67] | ||
| IE (exon 1) | CRISPR-Cas9 | Lentivirus | In vitro: - Human foreskin fibroblasts - THP-1 cells | J. Xiao et al. [68] | ||
| Multiplex | ||||||
| UL122/123 | CRISPR-Cas9 | Lentivirus | In vitro: - MRC5 primary fibroblasts - U-251 MG astrocytoma cells | J. Gergen et al. [67] | ||
| Early (E) viral genes | ||||||
| UL44, UL54, UL57, UL70, UL84, or UL105 | CRISPR-Cas9 | Lentivirus | In vitro: MRC5 human lung fibroblasts | F. R. van Diemen et al. [45] | ||
| Late (L) viral genes | ||||||
| UL86 | CRISPR-Cas9 | Lentivirus | In vitro: MRC5 human lung fibroblasts | F. R. van Diemen et al. [45] | ||
| UL23, UL26, or UL35 | CRISPR-Cas9 | Gene drive viruses | In vitro: Human foreskin fibroblasts | M. Walter et al. (2020, 2021) [69,70] | ||
| Nonessential viral genes | ||||||
| US6, US7, or US11 | CRISPR-Cas9 | Lentivirus | In vitro: MRC5 human lung fibroblasts | F. R. van Diemen et al. [45] | ||
| Enhancing the efficiency of cellular immunotherapy | HCMV-VSTs | |||||
| NR3C1 | CRISPR-Cas9 | Ribonucleo-protein electroporation | In vitro: Peripheral blood mononuclear cells | T. Kaeuferle et al. [76] | ||
| K. Koukoulias et al. [77] | ||||||
| In vitro: - Peripheral blood mononuclear cells In vivo: - Mouse model | R. Basar et al. [75] | |||||
| HHV-6A | Targeting the integrated latent virus | Noncoding region of DRs between the DR1 and the DR6/7 locus | CRISPR-Cas9 | - Lentivirus - Plasmid transfection | In vitro: - HEK293T cells - Smooth muscle cells | G. Aimola et al. [37] |
| KSHV | Inhibiting lytic viral replication | ORF54 | CRISPR-Cas9 | Lentivirus | In vitro: Human renal cell carcinoma SLK cells | C. O. Haddad et al. [72] |
| ORF57 | CRISPR-Cas9 | Puro vector | In vitro: BCBL-1 cells (body-cavity-based lymphoma cell line) iSLK/Bac16 cells | A. BeltCappellino et al. [71] | ||
| CRISPRi | Lentivirus | In vitro: iSLK-219 cells | K. Brackett et al. [38] | |||
| ORF59 | CRISPRi | Lentivirus | In vitro: iSLK-219 cells | K. Brackett et al. [38] | ||
| Targeting viral genes important for latency maintenance | ORF73 | CRISPR-Cas9 | AAV5 | In vitro: - Vero219 cells - L1T2 human endothelial cells - BC3 human pleural effusion B lymphoblasts | F. Y. Tso et al. [44] | |
| - Lentivirus - Liposome | In vitro: - KSHV-transformed rat embryonic metanephric mesenchymal precursor cells (KMM cells) - BCBL-1 cells | E. Ju et al. [73] | ||||
| Lentivirus | In vitro: Human renal cell carcinoma SLK cells | C. O. Haddad et al. [72] | ||||
| LANA promoter LTc and/or LTi | CRISPRi | Lentivirus | In vitro: - iSLK-219 cells - BCBL-1 cells | K. Brackett et al. [38] | ||
| Purposefully inducing viral reactivation | ORF50 or ORF57 | CRISPRa | Plasmid transfection | In vitro: HEK293 cells | E. Elbasani et al. [40] | |
| miR-K12-1 to -9 and miR-K12-11 | CRISPR-Cas9 | Lentivirus | In vitro: - BCBL-1 cells - BCP-1 cells | Z. P. Liang et al. [74] | ||
4. Discussion
4.1. Five Anti-Herpesviral Mechanisms of CRISPR Technologies
4.2. Four CRISPR Systems Are Used as an Anti-Herpesviral Strategy
4.3. Delivery of CRISPR Systems
4.4. Treatment Window
4.5. Singleplex Versus Multiplex Strategy
4.6. Barriers to Clinical Implementation
5. Limitations of the Study
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| ALICE | Autonomous, intelligent, virus-inducible immune-like |
| ARPE-19 | Adult retinal pigment epithelial cells clone 19 |
| BAC | Bacterial artificial chromosome |
| BALB/c | Bagg albino laboratory-bred substrain c |
| BCBL | Body cavity-based lymphoma |
| BHK | Baby hamster kidney |
| Cas | CRISPR-associated protein |
| CAR | Chimeric antigen receptor |
| CD | Cluster of differentiation |
| CLEAR | Coordinated lifecycle elimination against viral replication |
| CNE-2 | Cantonese nasopharyngeal epithelial cells |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CRISPRa | CRISPR activation |
| CRISPRi | CRISPR interference |
| CTLs | Cytotoxic T cells |
| dCas9 | Deactivated Cas9 |
| DR | Direct repeat region |
| dsDNA | Double-stranded DNA |
| DUX4 | Double homeobox 4 |
| E | Early |
| EBNA | EBV nuclear antigen |
| EBV | Epstein-Barr virus |
| EBVaGC | EBV-associated gastric carcinoma cells |
| EVs | Extracellular vesicles |
| gD | glycoprotein D |
| GFP | Green fluorescent protein |
| GMP | Good manufacturing practice |
| gp | Glycoprotein |
| GR | Glucocorticoid receptor |
| gRNA | guide RNA |
| HaCaT cells | Spontaneously immortalized human keratinocyte cells |
| HAP1 | Haploid human cell line 1 |
| HCMV | Human cytomegalovirus |
| HDR | Homology-directed repair |
| HEK293 | Human embryonic kidney cells |
| HEK293T | Human embryonic kidney cells with SV40 large T antigen |
| HELP | HSV-1-erasing lentiviral particles |
| hESC | Human embryonic stem cell |
| HHV | Human herpesvirus |
| HSCT | Hematopoietic stem cell transplant |
| HSK | Herpes simplex keratitis |
| HSV | Herpes simplex virus |
| HT22 | Hippocampal terminal cells |
| iciHHV-6 | Inherited chromosomally integrated human herpesvirus 6 |
| ICP | Infected cell protein |
| IE | Immediate early |
| IgG | Immunoglobulin G |
| INDELs | Insertions or deletions |
| (i)SLK | (Inducible) cell line from a Kaposi’s sarcoma lesion |
| KO | Knock-out |
| KRAB | Krüppel-associated box |
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| L | Late |
| LANA | Latency-associated nuclear antigen |
| LAT | Latency-associated transcript |
| LMP | Latent membrane protein |
| LP | Leader protein |
| LT | Latent transcript |
| MeSH | Medical subject heading |
| miRNA | microRNA |
| MOI | Multiplicity of infection |
| MRC5 | Medical Research Council strain 5 |
| mRNA | Messenger RNA |
| NECTIN-1 | Nectin cell adhesion molecule 1 |
| NHEJ | Non-homologous end joining |
| NPC | Nasopharyngeal carcinoma |
| NPs | Nanoparticles |
| NR3C1 | Nuclear receptor subfamily 3 group C member 1 |
| ORF | Open reading frame |
| OriP | Latent origin of replication |
| OSF | Open Science Framework |
| PBAE | Poly(β-amino ester) |
| PBMCs | Peripheral blood mononuclear cells |
| PD-1 | Programmed cell death protein 1 |
| PEL | Primary effusion lymphoma |
| PML | Promyelocytic leukemia |
| PRISMA-ScR | Preferred Reporting Items for Systematic Reviews and Meta-Analyses extension for Scoping Reviews |
| RVG | Rabies virus glycoprotein |
| ScFv | Single-chain variable fragment |
| SMC | Smooth muscle cells |
| SNU-719 | Seoul National University cell line 719 |
| STING | Stimulator of IFN genes |
| TC620 | Human oligodendroglioma Tübingen cells |
| THP-1 | Human monocytic leukemia cell line |
| TRAC | T cell receptor-α constant |
| UL | Unique long region |
| US | Unique short region |
| VP16 | Viral protein 16 |
| VSTs | Virus-specific T cells |
| VZV | Varicella-zoster virus |
| WT | Wild-type |
| ZTA | Z trans-activator |
References
- Evans, C.M.; Kudesia, G.; McKendrick, M. Management of herpesvirus infections. Int. J. Antimicrob. Agents 2013, 42, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Agut, H.; Bonnafous, P.; Gautheret-Dejean, A. Human Herpesviruses 6A, 6B, and 7. Microbiol. Spectr. 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Freer, G.; Pistello, M. Varicella-zoster virus infection: Natural history, clinical manifestations, immunity and current and future vaccination strategies. New Microbiol. 2018, 41, 95–105. [Google Scholar]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Krueger, G.R.; Koch, B.; Leyssens, N.; Berneman, Z.; Rojo, J.; Horwitz, C.; Sloots, T.; Margalith, M.; Conradie, J.; Imai, S. Comparison of seroprevalences of human herpesvirus-6 and-7 in healthy blood donors from nine countries. Vox Sang. 1998, 75, 193–197. [Google Scholar] [CrossRef]
- Minhas, V.; Wood, C. Epidemiology and transmission of Kaposi’s sarcoma-associated herpesvirus. Viruses 2014, 6, 4178–4194. [Google Scholar] [CrossRef] [PubMed]
- Smatti, M.K.; Al-Sadeq, D.W.; Ali, N.H.; Pintus, G.; Abou-Saleh, H.; Nasrallah, G.K. Epstein–Barr virus epidemiology, serology, and genetic variability of LMP-1 oncogene among healthy population: An update. Front. Oncol. 2018, 8, 211. [Google Scholar] [CrossRef]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef]
- Zhu, S.; Viejo-Borbolla, A. Pathogenesis and virulence of herpes simplex virus. Virulence 2021, 12, 2670–2702. [Google Scholar] [CrossRef]
- Pantry, S.N.; Medveczky, P.G. Latency, Integration, and Reactivation of Human Herpesvirus-6. Viruses 2017, 9, 194. [Google Scholar] [CrossRef]
- Stoeger, T.; Adler, H. “Novel” Triggers of Herpesvirus Reactivation and Their Potential Health Relevance. Front. Microbiol. 2018, 9, 3207. [Google Scholar] [CrossRef] [PubMed]
- Agut, H.; Bonnafous, P.; Gautheret-Dejean, A. Update on infections with human herpesviruses 6A, 6B, and 7. Med. Mal. Infect. 2017, 47, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Drago, F.; Aragone, M.G.; Lugani, C.; Rebora, A. Cytomegalovirus infection in normal and immunocompromised humans. A review. Dermatol. 2000, 200, 189–195. [Google Scholar] [CrossRef]
- Fatahzadeh, M.; Schwartz, R.A. Human herpes simplex virus infections: Epidemiology, pathogenesis, symptomatology, diagnosis, and management. J. Am. Acad. Dermatol. 2007, 57, 737–763; quiz 764-736. [Google Scholar] [CrossRef]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef]
- Iftode, N.; Rădulescu, M.A.; Aramă, Ș.S.; Aramă, V. Update on Kaposi sarcoma-associated herpesvirus (KSHV or HHV8)-review. Rom. J. Intern. Med. 2020, 58, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.E.; Gershon, A.A. Clinical Features of Varicella-Zoster Virus Infection. Viruses 2018, 10, 609. [Google Scholar] [CrossRef]
- Naughton, P.; Healy, M.; Enright, F.; Lucey, B. Infectious Mononucleosis: Diagnosis and clinical interpretation. Br. J. Biomed. Sci. 2021, 78, 107–116. [Google Scholar] [CrossRef]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Ward, K. The natural history and laboratory diagnosis of human herpesviruses-6 and -7 infections in the immunocompetent. J. Clin. Virol. 2005, 32, 183–193. [Google Scholar] [CrossRef]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Cole, S. Herpes Simplex Virus: Epidemiology, Diagnosis, and Treatment. Nurs. Clin. N. Am. 2020, 55, 337–345. [Google Scholar] [CrossRef]
- Cunha, B.A. Cytomegalovirus pneumonia: Community-acquired pneumonia in immunocompetent hosts. Infect. Dis. Clin. N. Am. 2010, 24, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Beckham, J.D.; Piquet, A.L.; Tyler, K.L.; Chauhan, L.; Pastula, D.M. Herpesvirus-Associated Encephalitis: An Update. Curr. Trop. Med. Rep. 2022, 9, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Nakase, H.; Herfarth, H. Cytomegalovirus Colitis, Cytomegalovirus Hepatitis and Systemic Cytomegalovirus Infection: Common Features and Differences. Inflamm. Intest. Dis. 2016, 1, 15–23. [Google Scholar] [CrossRef]
- Katano, H. Pathological Features of Kaposi’s Sarcoma-Associated Herpesvirus Infection. Adv. Exp. Med. Biol. 2018, 1045, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Robertson, E.S. Epstein–Barr Virus History and Pathogenesis. Viruses 2023, 15, 714. [Google Scholar] [CrossRef]
- Sadowski, L.A.; Upadhyay, R.; Greeley, Z.W.; Margulies, B.J. Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and-2. Viruses 2021, 13, 1228. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yao, Y.; Zhang, Y.; Fan, G. CRISPR system: Discovery, development and off-target detection. Cell Signal 2020, 70, 109577. [Google Scholar] [CrossRef]
- Horodecka, K.; Düchler, M. CRISPR/Cas9: Principle, Applications, and Delivery through Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 6072. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. CRISPR/Cas9-Based Antiviral Strategy: Current Status and the Potential Challenge. Molecules 2019, 24, 1349. [Google Scholar] [CrossRef]
- Liu, J.J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 2019, 566, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Bendixen, L.; Jensen, T.I.; Bak, R.O. CRISPR-Cas-mediated transcriptional modulation: The therapeutic promises of CRISPRa and CRISPRi. Mol. Ther. 2023, 31, 1920–1937. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef]
- Haddaway, N.R.; Page, M.J.; Pritchard, C.C.; McGuinness, L.A. PRISMA2020: An R package and Shiny app for producing PRISMA 2020-compliant flow diagrams, with interactivity for optimised digital transparency and Open Synthesis. Campbell Syst. Rev. 2022, 18, e1230. [Google Scholar] [CrossRef]
- Aimola, G.; Wight, D.J.; Flamand, L.; Kaufer, B.B. Excision of Integrated Human Herpesvirus 6A Genomes Using CRISPR/Cas9 Technology. Microbiol. Spectr. 2023, 11, e0076423. [Google Scholar] [CrossRef]
- Brackett, K.; Mungale, A.; Lopez-Isidro, M.; Proctor, D.A.; Najarro, G.; Arias, C. CRISPR Interference Efficiently Silences Latent and Lytic Viral Genes in Kaposi’s Sarcoma-Associated Herpesvirus-Infected Cells. Viruses 2021, 13, 783. [Google Scholar] [CrossRef]
- Ding, J.; Zhang, H.; Dai, T.; Gao, X.; Yin, Z.; Wang, Q.; Long, M.; Tan, S. TPGS-b-PBAE Copolymer-Based Polyplex Nanoparticles for Gene Delivery and Transfection In Vivo and In Vitro. Pharmaceutics 2024, 16, 213. [Google Scholar] [CrossRef]
- Elbasani, E.; Falasco, F.; Gramolelli, S.; Nurminen, V.; Günther, T.; Weltner, J.; Balboa, D.; Grundhoff, A.; Otonkoski, T.; Ojala, P.M. Kaposi’s Sarcoma-Associated Herpesvirus Reactivation by Targeting of a dCas9-Based Transcription Activator to the ORF50 Promoter. Viruses 2020, 12, 952. [Google Scholar] [CrossRef]
- Karpov, D.S.; Demidova, N.A.; Kulagin, K.A.; Shuvalova, A.I.; Kovalev, M.A.; Simonov, R.A.; Karpov, V.L.; Snezhkina, A.V.; Kudryavtseva, A.V.; Klimova, R.R.; et al. Complete and Prolonged Inhibition of Herpes Simplex Virus Type 1 Infection In Vitro by CRISPR/Cas9 and CRISPR/CasX Systems. Int. J. Mol. Sci. 2022, 23, 14847. [Google Scholar] [CrossRef] [PubMed]
- Roehm, P.C.; Shekarabi, M.; Wollebo, H.S.; Bellizzi, A.; He, L.F.; Salkind, J.; Khalili, K. Inhibition of HSV-1 Replication by Gene Editing Strategy. Sci. Rep. 2016, 6, 23146. [Google Scholar] [CrossRef]
- Su, S.; Zou, Z.; Chen, F.; Ding, N.; Du, J.; Shao, J.; Li, L.; Fu, Y.; Hu, B.; Yang, Y.; et al. CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology 2017, 6, e1249558. [Google Scholar] [CrossRef] [PubMed]
- Tso, F.Y.; West, J.T.; Wood, C. Reduction of Kaposi’s Sarcoma-Associated Herpesvirus Latency Using CRISPR-Cas9 To Edit the Latency-Associated Nuclear Antigen Gene. J. Virol. 2019, 93, 7. [Google Scholar] [CrossRef]
- van Diemen, F.R.; Kruse, E.M.; Hooykaas, M.J.; Bruggeling, C.E.; Schürch, A.C.; van Ham, P.M.; Imhof, S.M.; Nijhuis, M.; Wiertz, E.J.; Lebbink, R.J. CRISPR/Cas9-Mediated Genome Editing of Herpesviruses Limits Productive and Latent Infections. PLoS Pathog. 2016, 12, e1005701. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Quake, S.R. RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc. Natl. Acad. Sci. USA 2014, 111, 13157–13162. [Google Scholar] [CrossRef]
- Wei, A.; Yin, D.; Zhai, Z.; Ling, S.; Le, H.; Tian, L.; Xu, J.; Paludan, S.R.; Cai, Y.; Hong, J. In vivo CRISPR gene editing in patients with herpetic stromal keratitis. Mol. Ther. 2023, 31, 3163–3175. [Google Scholar] [CrossRef]
- Wu, B.; Yee, M.B.; Goldstein, R.S.; Kinchington, P.R. Antiviral Targeting of Varicella Zoster Virus Replication and Neuronal Reactivation Using CRISPR/Cas9 Cleavage of the Duplicated Open Reading Frames 62/71. Virusesl 2022, 14, 378. [Google Scholar] [CrossRef]
- Yin, D.; Ling, S.; Wang, D.; Dai, Y.; Jiang, H.; Zhou, X.; Paludan, S.R.; Hong, J.; Cai, Y. Targeting herpes simplex virus with CRISPR-Cas9 cures herpetic stromal keratitis in mice. Nat. Biotechnol. 2021, 39, 567–577. [Google Scholar] [CrossRef]
- Amrani, N.; Luk, K.; Singh, P.; Shipley, M.; Isik, M.; Donadoni, M.; Bellizzi, A.; Khalili, K.; Sariyer, I.K.; Neumann, D.; et al. CRISPR-Cas9-mediated genome editing delivered by a single AAV9 vector inhibits HSV-1 reactivation in a latent rabbit keratitis model. Mol. Ther. Methods Clin. Dev. 2024, 32, 101303. [Google Scholar] [CrossRef]
- Bellizzi, A.; Çakır, S.; Donadoni, M.; Sariyer, R.; Liao, S.; Liu, H.; Ruan, G.X.; Gordon, J.; Khalili, K.; Sariyer, I.K. Suppression of HSV-1 infection and viral reactivation by CRISPR-Cas9 gene editing in 2D and 3D culture models. Mol. Ther. Nucleic Acids 2024, 35, 102282. [Google Scholar] [CrossRef]
- Chen, Y.; Zhi, S.; Liang, P.; Zheng, Q.; Liu, M.; Zhao, Q.; Ren, J.; Cui, J.; Huang, J.; Liu, Y.; et al. Single AAV-Mediated CRISPR-SaCas9 Inhibits HSV-1 Replication by Editing ICP4 in Trigeminal Ganglion Neurons. Mol. Ther.-Methods Clin. Dev. 2020, 18, 33–43. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, R.M.P.; Garcia, L.S.; Lemos, F.S.; de Campos, V.S.; Machado Ferreira, E.; de Almeida, N.A.A.; Maron-Gutierrez, T.; de Souza, E.M.; de Paula, V.S. CRISPR/Cas9 Eye Drop HSV-1 Treatment Reduces Brain Viral Load: A Novel Application to Prevent Neuronal Damage. Pathogens 2024, 13, 1087. [Google Scholar] [CrossRef]
- Karpov, D.S.; Karpov, V.L.; Klimova, R.R.; Demidova, N.A.; Kushch, A.A. [A Plasmid-Expressed CRISPR/Cas9 System Suppresses Replication of HSV Type I in a Vero Cell Culture]. Mol. Biol. 2019, 53, 91–100. [Google Scholar] [CrossRef]
- Khodadad, N.; Fani, M.; Jamehdor, S.; Nahidsamiei, R.; Makvandi, M.; Kaboli, S.; Teimoori, A.; Thekkiniath, J. A knockdown of the herpes simplex virus type-1 gene in all-in-one CRISPR vectors. Folia Histochem. Cytobiol. 2020, 58, 174–181. [Google Scholar] [CrossRef]
- Oh, H.S.; Neuhausser, W.M.; Eggan, P.; Angelova, M.; Kirchner, R.; Eggan, K.C.; Knipe, D.M. Herpesviral lytic gene functions render the viral genome susceptible to novel editing by CRISPR/Cas9. Elife 2019, 8, e51662. [Google Scholar] [CrossRef]
- Raposo, J.V.; Barros, L.R.C.; Torres, L.R.; Pinto, R.B.d.S.; Lopes, A.d.O.; de Souza, E.M.; Bonamino, M.H.; de Paula, V.S. CRISPR/Cas-9 vector system: Targets UL-39 and inhibits Simplexvirus humanalpha1 (HSV-1) replication in vitro. Cell Mol. Biol. 2023, 69, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Li, L.; Chen, R.; Han, J.; Lei, Q.; Chen, Z.; Tang, X.; Wu, W.; Liu, S.; Yao, X. Engineered extracellular vesicles efficiently deliver CRISPR-Cas9 ribonucleoprotein (RNP) to inhibit herpes simplex virus1 infection in vitro and in vivo. Acta Pharm. Sin. B 2024, 14, 1362–1379. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Tan, C.W.; Qiao, L.; Ni Chia, W.; Zhang, H.; Huang, Q.; Deng, Z.; Wang, Z.; Wang, X.; et al. Engineering antiviral immune-like systems for autonomous virus detection and inhibition in mice. Nat. Commun. 2022, 13, 7629. [Google Scholar] [CrossRef]
- Ying, M.; Wang, H.; Liu, T.; Han, Z.; Lin, K.; Shi, Q.; Zheng, N.; Ye, T.; Gong, H.; Xu, F. CLEAR Strategy Inhibited HSV Proliferation Using Viral Vectors Delivered CRISPR-Cas9. Pathogens 2023, 12, 814. [Google Scholar] [CrossRef]
- Neugebauer, E.; Walter, S.; Tan, J.; Drayman, N.; Franke, V.; van Gent, M.; Pennisi, S.; Veratti, P.; Stein, K.S.; Welker, I.; et al. Herpesviruses mimic zygotic genome activation to promote viral replication. Nat. Commun. 2025, 16, 710. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, Y.; Li, G.; Huang, S.; Xu, J.; Ding, Q.; Hong, J. Targeting NECTIN-1 Based on CRISPR/Cas9 System Attenuated the Herpes Simplex Virus Infection in Human Corneal Epithelial Cells In Vitro. Transl. Vis. Sci. Technol. 2022, 11, 8. [Google Scholar] [CrossRef]
- Yuen, K.S.; Wang, Z.M.; Wong, N.H.M.; Zhang, Z.Q.; Cheng, T.F.; Lui, W.Y.; Chan, C.P.; Jin, D.Y. Suppression of Epstein-Barr virus DNA load in latently infected nasopharyngeal carcinoma cells by CRISPR/Cas9. Virus Res. 2018, 244, 296–303. [Google Scholar] [CrossRef]
- Huo, H.; Hu, G. CRISPR/Cas9-mediated LMP1 knockout inhibits Epstein-Barr virus infection and nasopharyngeal carcinoma cell growth. Infect. Agents Cancer 2019, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Chang, S.; Zhang, C.; Dong, D.; Ding, J.; Mahdavian, A.R.; Hu, Z.; Sun, L.; Tan, S. Post cross-linked ROS-responsive poly(β-amino ester)-plasmid polyplex NPs for gene therapy of EBV-associated nasopharyngeal carcinoma. J. Mater. Chem. B 2024, 12, 3129–3143. [Google Scholar] [CrossRef] [PubMed]
- Sugiokto, F.G.; Li, R. Targeted eradication of EBV-positive cancer cells by CRISPR/dCas9-mediated EBV reactivation in combination with ganciclovir. mBio 2024, 15, e0079524. [Google Scholar] [CrossRef]
- Gergen, J.; Coulon, F.; Creneguy, A.; Elain-Duret, N.; Gutierrez, A.; Pinkenburg, O.; Verhoeyen, E.; Anegon, I.; Nguyen, T.H.; Halary, F.A.; et al. Multiplex CRISPR/Cas9 system impairs HCMV replication by excising an essential viral gene. PLoS ONE 2018, 13, e0192602. [Google Scholar] [CrossRef]
- Xiao, J.; Deng, J.; Zhang, Q.; Ma, P.; Lv, L.; Zhang, Y.; Li, C.; Zhang, Y. Targeting human cytomegalovirus IE genes by CRISPR/Cas9 nuclease effectively inhibits viral replication and reactivation. Arch. Virol. 2020, 165, 1827–1835. [Google Scholar] [CrossRef]
- Walter, M.; Perrone, R.; Verdin, E. Targeting Conserved Sequences Circumvents the Evolution of Resistance in a Viral Gene Drive against Human Cytomegalovirus. J. Virol. 2021, 95, e0080221. [Google Scholar] [CrossRef]
- Walter, M.; Verdin, E. Viral gene drive in herpesviruses. Nat. Commun. 2020, 11, 4884. [Google Scholar] [CrossRef]
- BeltCappellino, A.; Majerciak, V.; Lobanov, A.; Lack, J.; Cam, M.; Zheng, Z.M. CRISPR/Cas9-Mediated Knockout and In Situ Inversion of the ORF57 Gene from All Copies of the Kaposi’s Sarcoma-Associated Herpesvirus Genome in BCBL-1 Cells. J. Virol. 2019, 93, 21. [Google Scholar] [CrossRef] [PubMed]
- Haddad, C.O.; Kalt, I.; Shovman, Y.; Xia, L.; Schlesinger, Y.; Sarid, R.; Parnas, O. Targeting the Kaposi’s sarcoma-associated herpesvirus genome with the CRISPR-Cas9 platform in latently infected cells. Virol. J. 2021, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Ju, E.; Li, T.; da Silva, S.R.; Markazi, A.; Gao, S. Reversible switching of primary cells between normal and malignant state by oncogenic virus KSHV and CRISPR/Cas9-mediated targeting of a major viral latent protein. J. Med. Virol. 2021, 93, 5065–5075. [Google Scholar] [CrossRef]
- Liang, Z.; Qin, Z.; Riker, A.I.; Xi, Y. CRISPR/Cas9 ablating viral microRNA promotes lytic reactivation of Kaposi’s sarcoma-associated herpesvirus. Biochem. Biophys. Res. Commun. 2020, 533, 1400–1405. [Google Scholar] [CrossRef]
- Basar, R.; Daher, M.; Uprety, N.; Gokdemir, E.; Alsuliman, A.; Ensley, E.; Ozcan, G.; Mendt, M.; Sanabria, M.H.; Kerbauy, L.N.; et al. Large-scale GMP-compliant CRISPR-Cas9-mediated deletion of the glucocorticoid receptor in multivirus-specific T cells. Blood Adv. 2020, 4, 3357–3367. [Google Scholar] [CrossRef]
- Kaeuferle, T.; Deisenberger, L.; Jablonowski, L.; Stief, T.A.; Blaeschke, F.; Willier, S.; Feuchtinger, T. CRISPR-Cas9-Mediated Glucocorticoid Resistance in Virus-Specific T Cells for Adoptive T Cell Therapy Posttransplantation. Mol. Ther. 2020, 28, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Koukoulias, K.; Papayanni, P.G.; Georgakopoulou, A.; Alvanou, M.; Laidou, S.; Kouimtzidis, A.; Pantazi, C.; Gkoliou, G.; Vyzantiadis, T.A.; Spyridonidis, A.; et al. “Cerberus” T Cells: A Glucocorticoid-Resistant, Multi-Pathogen Specific T Cell Product to Fight Infections in Severely Immunocompromised Patients. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Palianina, D.; Di Roberto, R.B.; Castellanos-Rueda, R.; Schlatter, F.; Reddy, S.T.; Khanna, N. A method for polyclonal antigen-specific T cell-targeted genome editing (TarGET) for adoptive cell transfer applications. Mol. Ther. Methods Clin. Dev. 2023, 30, 147–160. [Google Scholar] [CrossRef]
- Braun, T.; Pruene, A.; Darguzyte, M.; Stein, A.F.V.; Nguyen, P.-H.; Wagner, D.L.; Kath, J.; Roig-Merino, A.; Heuser, M.; Riehm, L.L.; et al. Non-viral TRAC-knocked-in CD19(KI)CAR-T and gp350(KI)CAR-T cells tested against Burkitt lymphomas with type 1 or 2 EBV infection: In vivo cellular dynamics and potency. Front. Immunol. 2023, 14, 1086433. [Google Scholar] [CrossRef] [PubMed]
- Baddeley, H.J.E.; Isalan, M. The Application of CRISPR/Cas Systems for Antiviral Therapy. Front. Genome Ed. 2021, 3, 745559. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, Y.; Yang, J.; Li, W.; Zhang, M.; Wang, Q.; Zhang, L.; Wei, G.; Tian, Y.; Zhao, K.; et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature 2022, 609, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qiu, S.; Zhang, X.; Chen, W. Optimized DNA electroporation for primary human T cell engineering. BMC Biotechnol. 2018, 18, 4. [Google Scholar] [CrossRef]
- Bu, W.; Li, Y. In Vivo Gene Delivery into Mouse Mammary Epithelial Cells Through Mammary Intraductal Injection. J. Vis. Exp. JoVE 2023, 10, 192. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2020, 29, 464–488. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, F.; Gao, G. CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell 2020, 181, 136–150. [Google Scholar] [CrossRef]
- Alexander, E.; Leong, K.W. Discovery of nanobodies: A comprehensive review of their applications and potential over the past five years. J. Nanobiotechnol. 2024, 22, 661. [Google Scholar] [CrossRef]
- Di Ianni, E.; Obuchi, W.; Breyne, K.; Breakefield, X.O. Extracellular vesicles for the delivery of gene therapy. Nat. Rev. Bioeng. 2025, 3, 360–373. [Google Scholar] [CrossRef]
- Eichhoff, A.M.; Börner, K.; Albrecht, B.; Schäfer, W.; Baum, N.; Haag, F.; Körbelin, J.; Trepel, M.; Braren, I.; Grimm, D.; et al. Nanobody-Enhanced Targeting of AAV Gene Therapy Vectors. Mol. Ther. Methods Clin. Dev. 2019, 15, 211–220. [Google Scholar] [CrossRef]
- Frank, A.M.; Buchholz, C.J. Surface-Engineered Lentiviral Vectors for Selective Gene Transfer into Subtypes of Lymphocytes. Mol. Therapy Methods Clin. Dev. 2019, 12, 19–31. [Google Scholar] [CrossRef]
- Li, X.; Stuckert, P.; Bosch, I.; Marks, J.D.; Marasco, W. Single-chain antibody-mediated gene delivery into ErbB2-positive human breast cancer cells. Cancer Gene Ther. 2001, 8, 555–565. [Google Scholar] [CrossRef]
- Cohen, J.I. Herpesvirus latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef] [PubMed]

| Virus Type | Search | Number of Search Results |
|---|---|---|
| PubMed | ||
| CRISPR* AND (herpes* OR HHV) | 422 | |
| ((“Clustered Regularly Interspaced Short Palindromic Repeats”[MeSH]) OR (“CRISPR-Cas Systems”[MeSH])) AND “Herpesviridae”[MeSH] | 202 | |
| Herpes simplex virus type 1 and type 2/Human herpesvirus 1 and 2 | CRISPR* AND HSV | 115 |
| ((“Clustered Regularly Interspaced Short Palindromic Repeats”[MeSH]) OR (“CRISPR-Cas Systems”[MeSH])) AND ((“Simplexvirus”[MeSH]) OR (“Herpesvirus 1, Human”[MeSH]) OR (“Herpesvirus 2, Human”[MeSH])) | 61 | |
| Varicella-zoster virus/Human herpesvirus 3 | CRISPR* AND (varicella-zoster virus OR VZV) | 5 |
| ((“Clustered Regularly Interspaced Short Palindromic Repeats”[MeSH]) OR (“CRISPR-Cas Systems”[MeSH])) AND “Herpesvirus 3, Human”[MeSH] | 2 | |
| Epstein-Barr virus/Human herpesvirus 4 | CRISPR* AND (Epstein-Barr virus OR EBV) | 136 |
| ((“Clustered Regularly Interspaced Short Palindromic Repeats”[MeSH]) OR (“CRISPR-Cas Systems”[MeSH])) AND “Herpesvirus 4, Human”[MeSH] | 43 | |
| Human cytomegalovirus/Human herpesvirus 5 | CRISPR* AND (cytomegalovirus OR CMV) | 148 |
| ((“Clustered Regularly Interspaced Short Palindromic Repeats”[MeSH]) OR (“CRISPR-Cas Systems”[MeSH])) AND “Cytomegalovirus”[MeSH] | 27 | |
| Human herpesvirus 6 | ((“Clustered Regularly Interspaced Short Palindromic Repeats”[MeSH]) OR (“CRISPR-Cas Systems”[MeSH])) AND “Herpesvirus 6, Human”[MeSH] | 0 |
| Human herpesvirus 7 | ((“Clustered Regularly Interspaced Short Palindromic Repeats”[MeSH]) OR (“CRISPR-Cas Systems”[MeSH])) AND “Herpesvirus 7, Human”[MeSH] | 0 |
| Kaposi’s sarcoma-associated herpesvirus/Human herpesvirus 8 | CRISPR* AND (Kaposi’s sarcoma-associated herpesvirus OR KSHV) | 54 |
| ((“Clustered Regularly Interspaced Short Palindromic Repeats”[MeSH]) OR (“CRISPR-Cas Systems”[MeSH])) AND “Herpesvirus 8, Human”[MeSH] | 19 | |
| Web of Science | ||
| CRISPR* (All Fields) and herpes* (All Fields) | 316 | |
| CRISPR* (All Fields) and HHV* (All Fields) | 53 | |
| Herpes simplex virus type 1 and type 2/Human herpesvirus 1 and 2 | CRISPR* (All Fields) and HSV* (All Fields) | 174 |
| Varicella-zoster virus/Human herpesvirus 3 | CRISPR* (All Fields) and varicella-zoster virus (All Fields) | 6 |
| CRISPR* (All Fields) and VZV (All Fields) | 5 | |
| Epstein-Barr virus/Human herpesvirus 4 | CRISPR* (All Fields) and Epstein-Barr virus (All Fields) | 166 |
| CRISPR* (All Fields) and EBV (All Fields) | 98 | |
| Human cytomegalovirus/Human herpesvirus 5 | CRISPR* (All Fields) and cytomegalovirus (All Fields) | 103 |
| CRISPR* (All Fields) and CMV (All Fields) | 98 | |
| Human herpesvirus 6 | Included in the general search | 0 |
| Human herpesvirus 7 | Included in the general search | 0 |
| Kaposi’s sarcoma-associated herpesvirus/Human herpesvirus 8 | CRISPR* (All Fields) and Kaposi’s sarcoma-associated herpesvirus (All Fields) | 44 |
| CRISPR* (All Fields) and KSHV (All Fields) | 44 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanssens, C.; Van Cleemput, J. Applying CRISPR Technologies for the Treatment of Human Herpesvirus Infections: A Scoping Review. Pathogens 2025, 14, 654. https://doi.org/10.3390/pathogens14070654
Hanssens C, Van Cleemput J. Applying CRISPR Technologies for the Treatment of Human Herpesvirus Infections: A Scoping Review. Pathogens. 2025; 14(7):654. https://doi.org/10.3390/pathogens14070654
Chicago/Turabian StyleHanssens, Chloë, and Jolien Van Cleemput. 2025. "Applying CRISPR Technologies for the Treatment of Human Herpesvirus Infections: A Scoping Review" Pathogens 14, no. 7: 654. https://doi.org/10.3390/pathogens14070654
APA StyleHanssens, C., & Van Cleemput, J. (2025). Applying CRISPR Technologies for the Treatment of Human Herpesvirus Infections: A Scoping Review. Pathogens, 14(7), 654. https://doi.org/10.3390/pathogens14070654