
Human Cytomegalovirus Immune Evasion of Natural Killer Cells: A Virus for All Seasons?

 , , , , , and
, , , , , and
Abstract
1. Human Cytomegalovirus (HCMV)
2. Natural Killer Cell Activation—Innate Immunity
3. Natural Killer Cell Activation—Adaptive Immunity
4. Importance of NK Cells in Controlling HCMV Infection
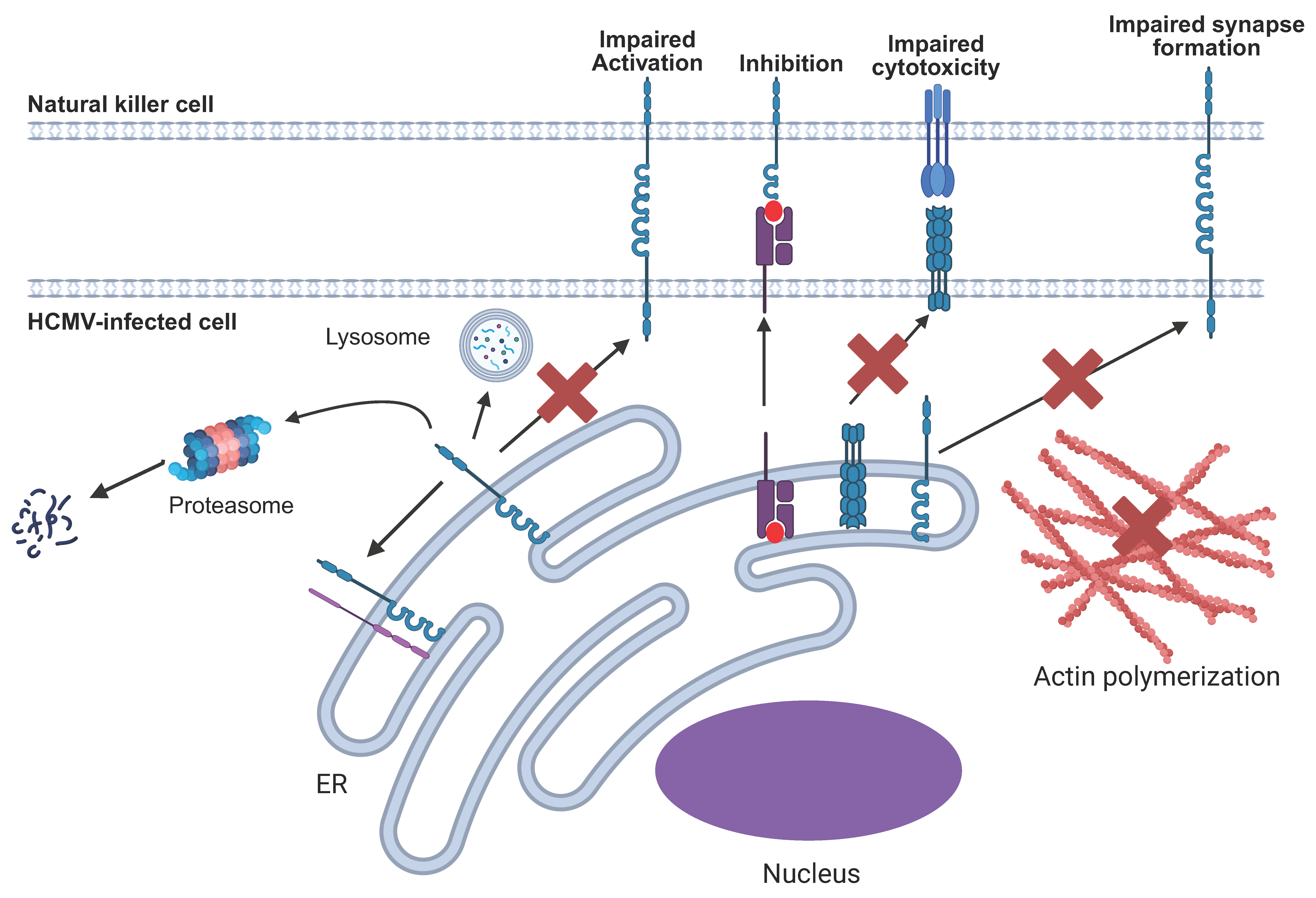
5. Broad Mechanisms of NK Immune Evasins
6. Specific HCMV NK Immune Evasin Mechanisms
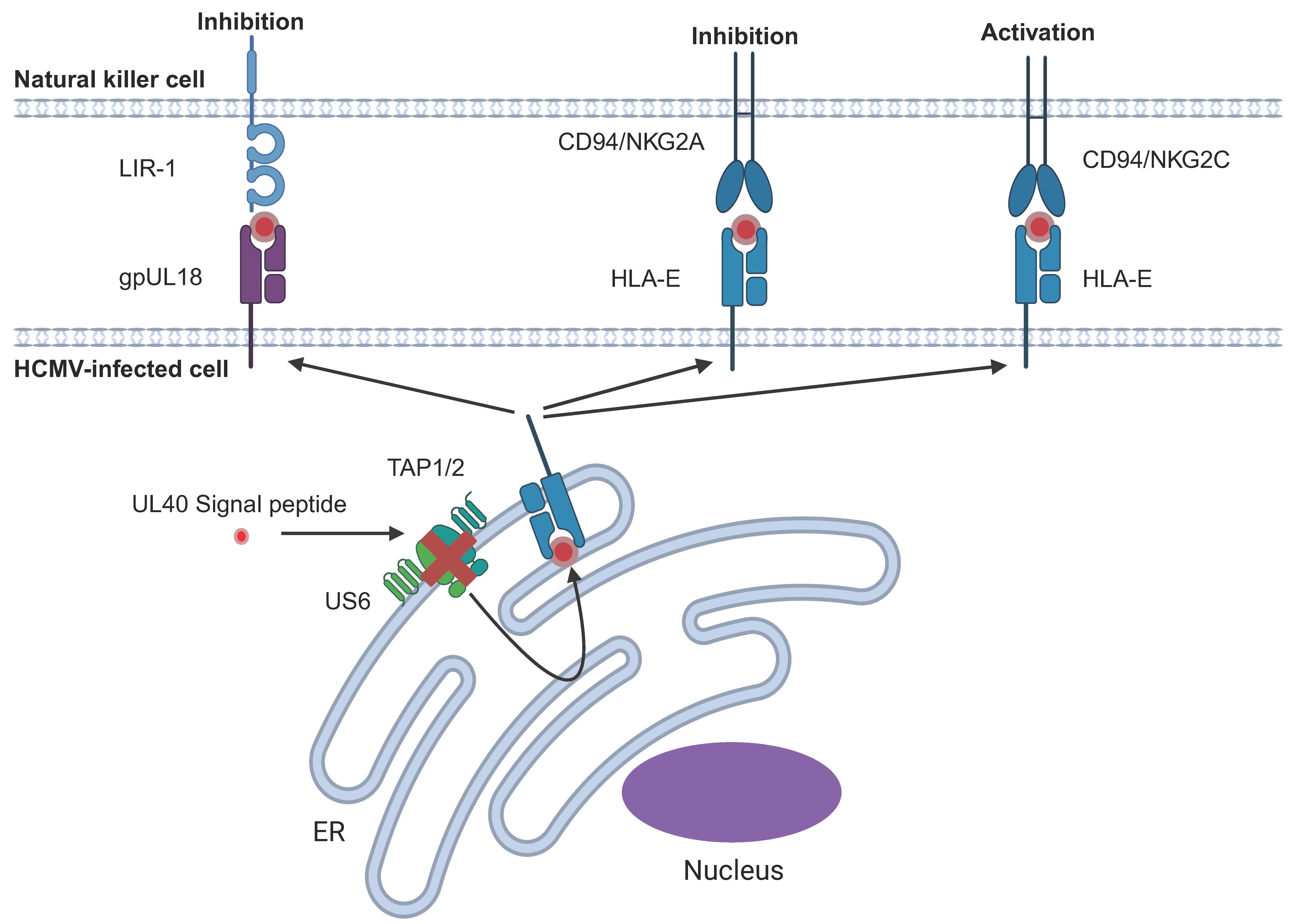
6.1. HCMV-Encoded HLA-I Homologues
6.2. Upregulation of Endogenous HLA-E Cell Surface Expression
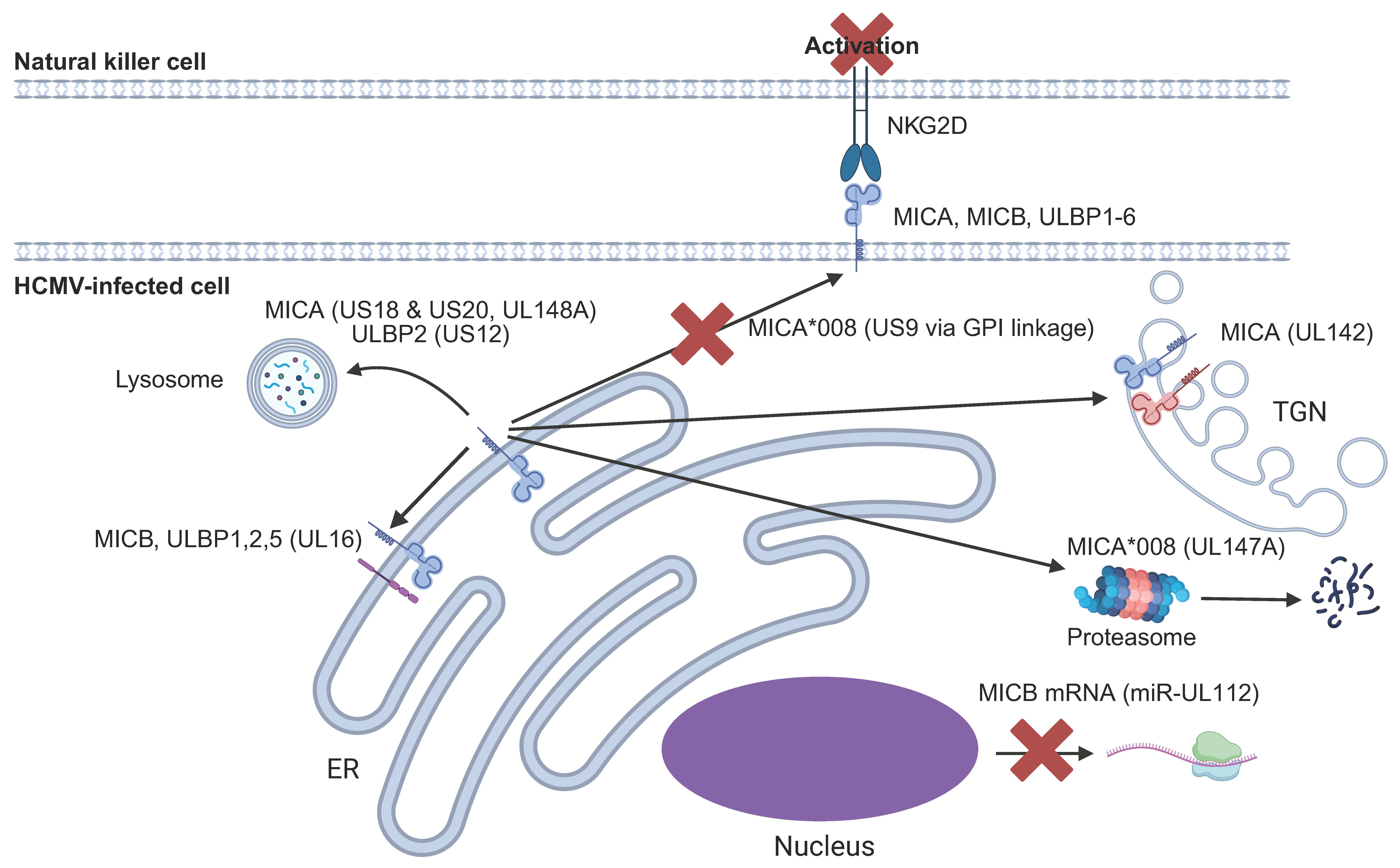
6.3. NKG2D Ligands (NKG2DL)
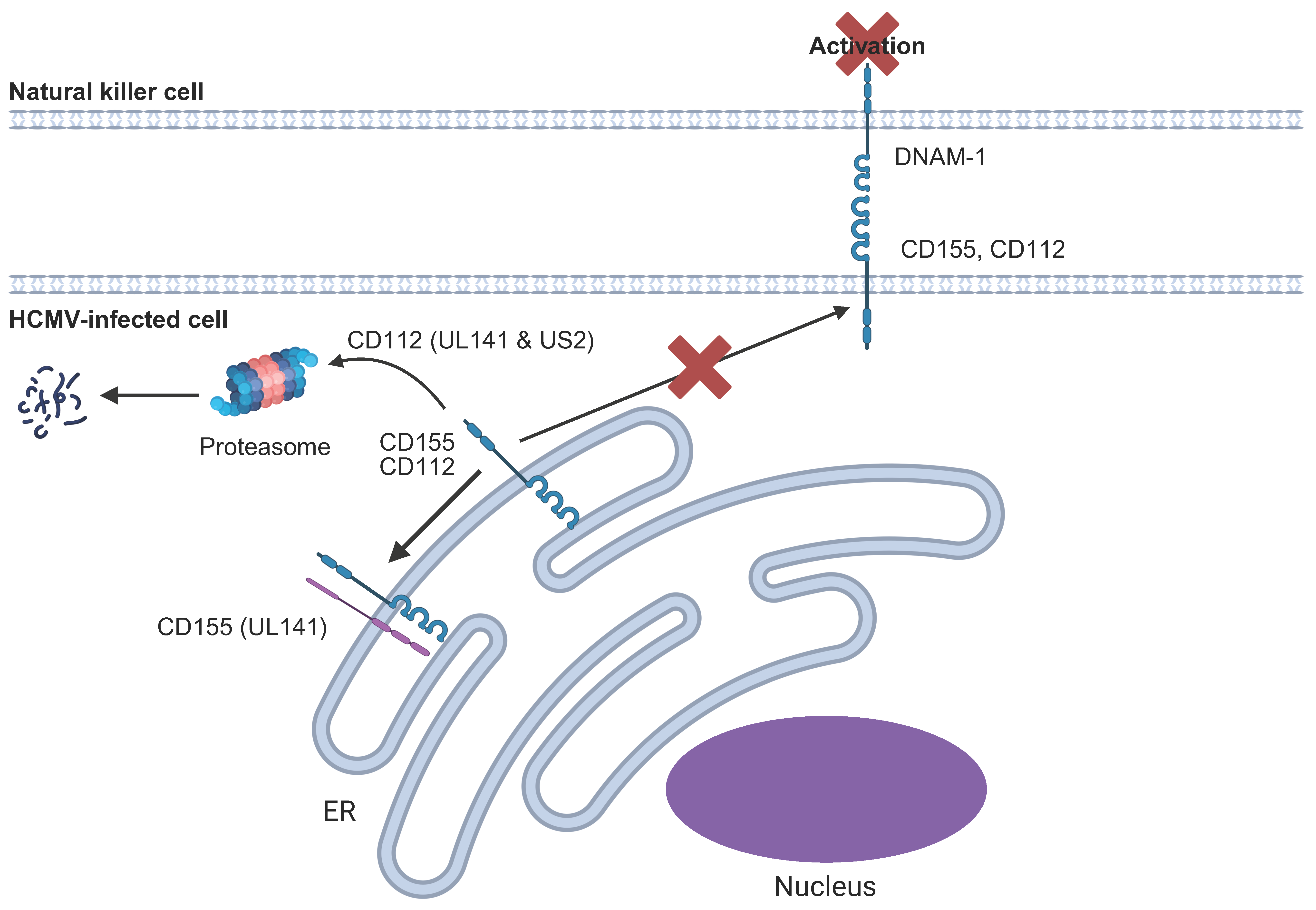
6.4. DNAM1/CD226 Ligands
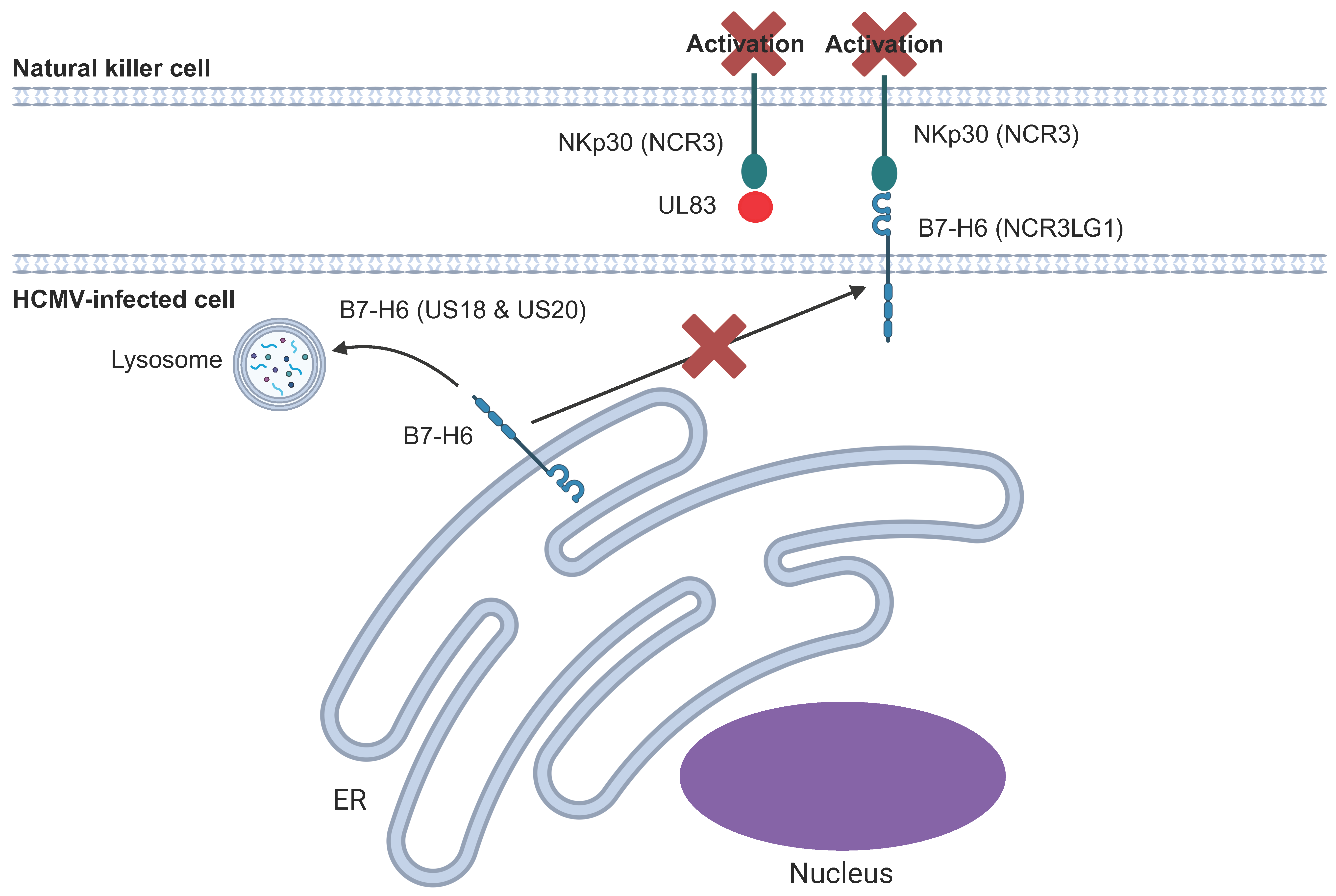
6.5. NKp30 and Its Ligands
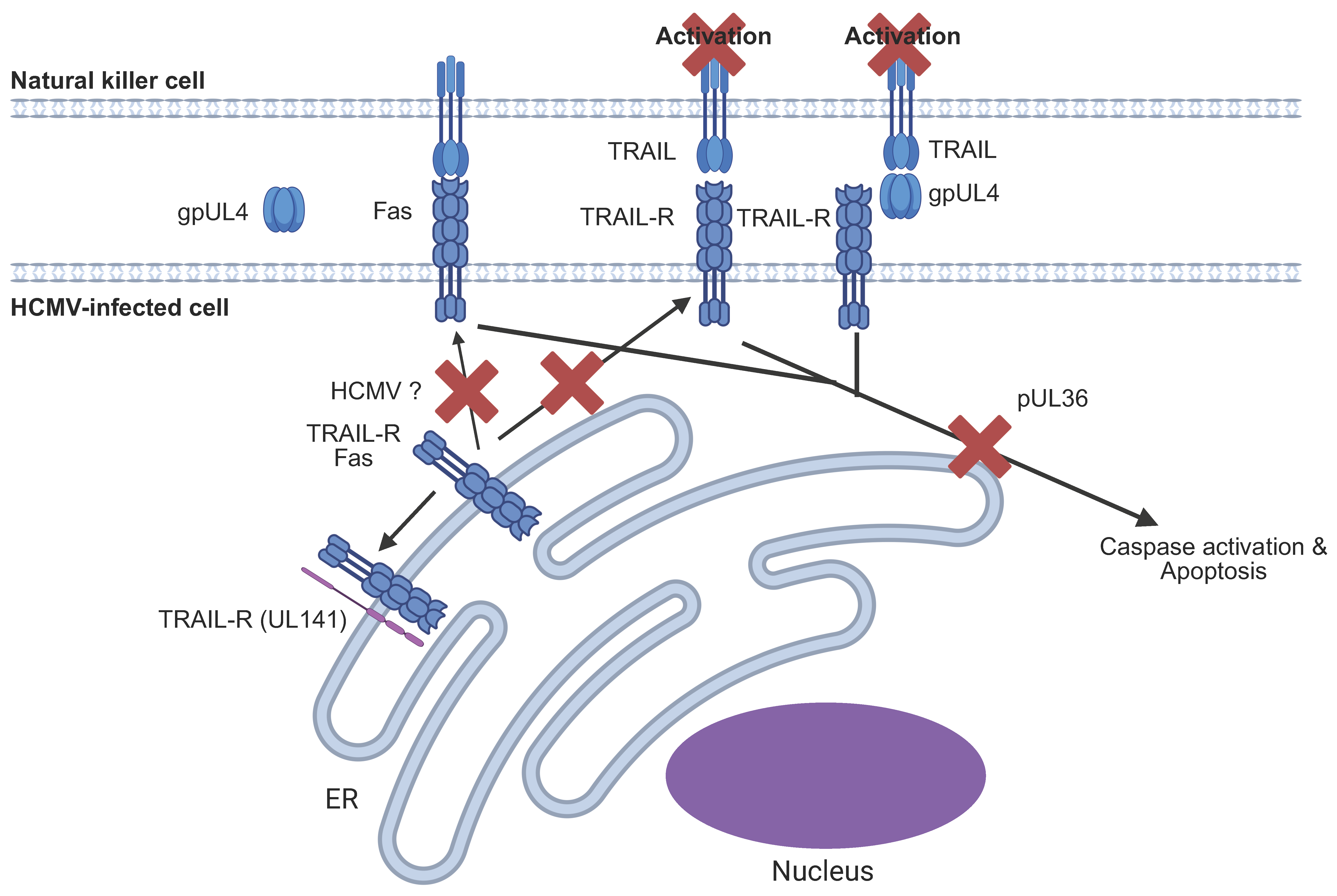
6.6. HCMV Targeting of the Extrinsic Apoptotic Pathway
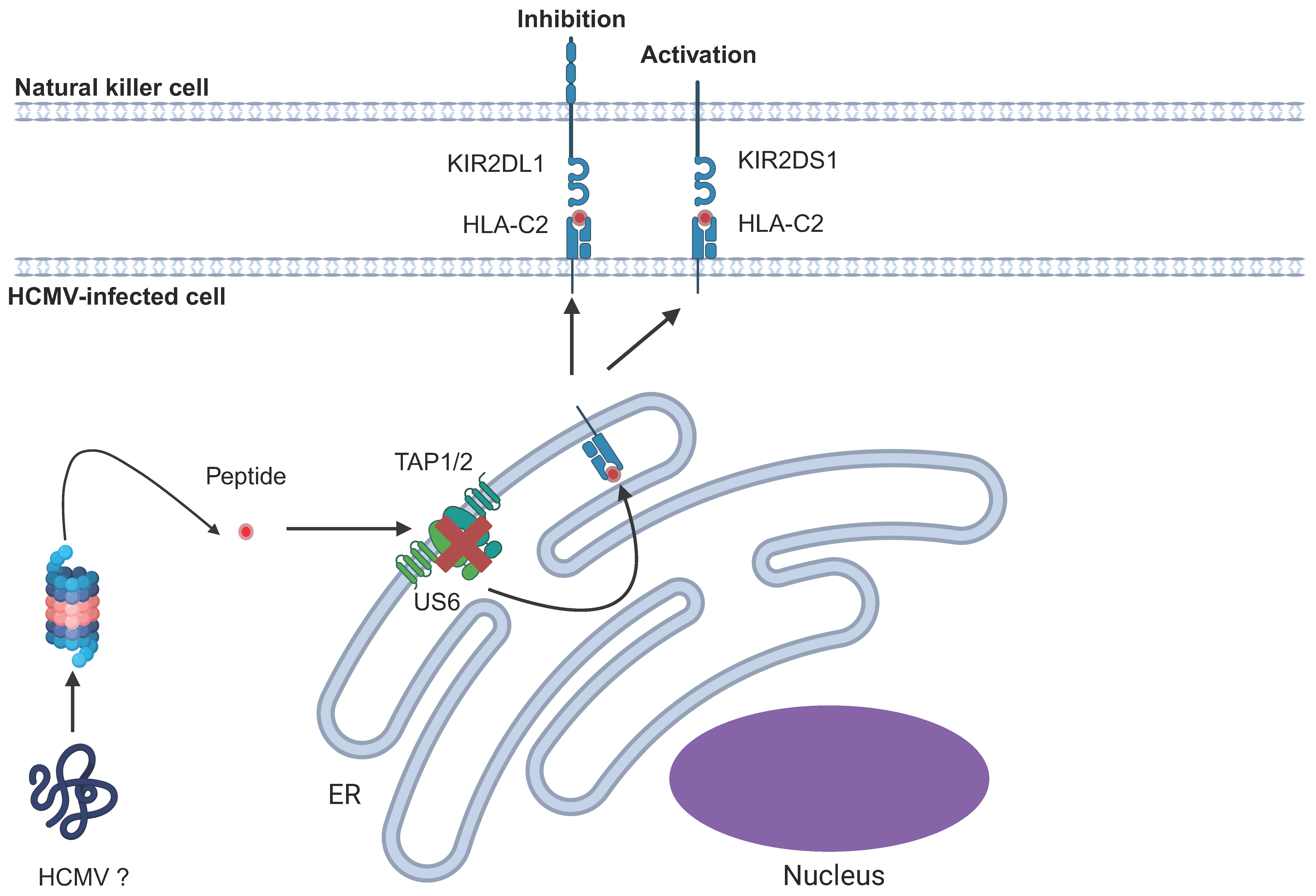
6.7. Killer Immunoglobulin-Like Receptors (KIRs)
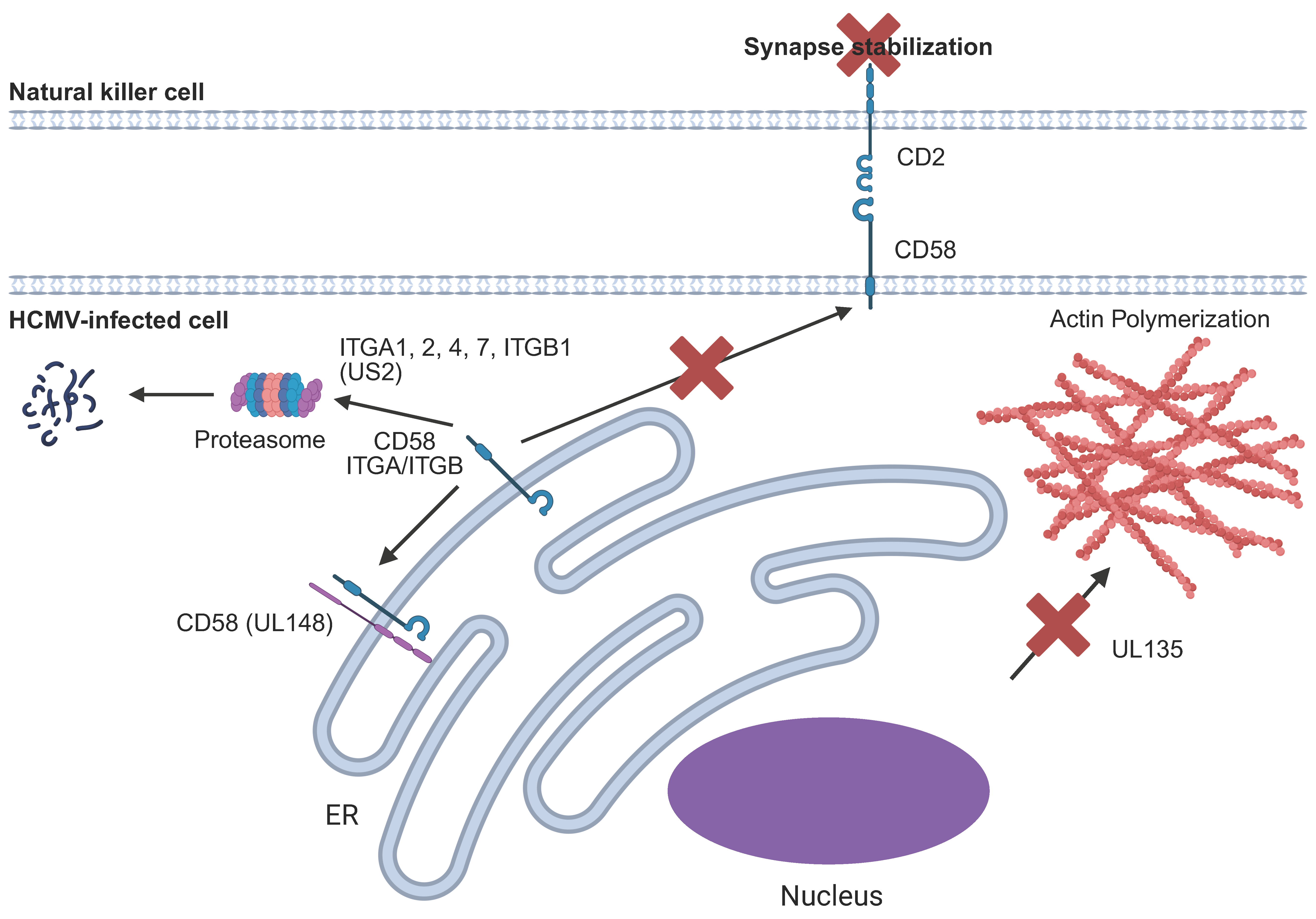
6.8. Disruption of the NK Immune Synapse
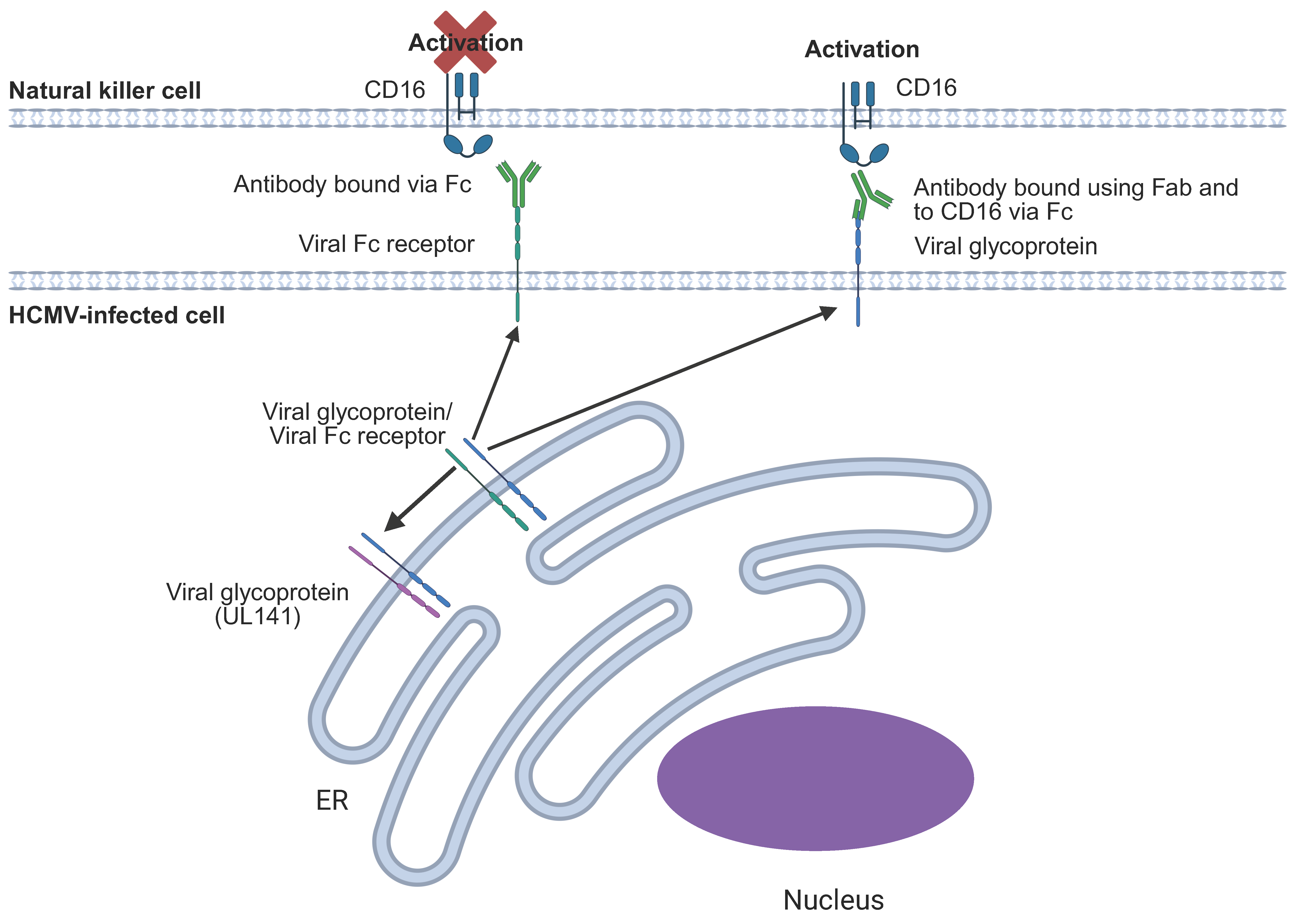
6.9. Evasion of ADCC
7. Overcoming HCMV-Mediated Immune Evasion by ADCC
8. Future Perspectives
8.1. Why Does HCMV Encode So Many NK Immune Evasins?
8.2. Have We Mapped All HCMV-Encoded Immune Evasins and the Full Range of Effects?
8.3. What Is the Role of Natural Sequence Variation in Both the Host and the Virus?
8.4. Does Targeting NK Activation Using Non-Neutralising Antibodies Represent a Therapeutic Approach to Overcome HCMV-Mediation NK Cell Evasion?
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADCC | Antibody-dependent cellular cytotoxicity |
| CD | Cluster of differentiation |
| Cen | Centromeric |
| cCMV | Congenital cytomegalovirus infection |
| DNAM1 | DNAX accessory protein-1 |
| FcγR | Fc gamma receptor |
| G | Glycoprotein |
| Gp | Glycoprotein |
| GPI | Glycosylphosphatidylinositol |
| HCMV | Human cytomegalovirus |
| HLA | Human leukocyte antigen |
| IFN | Interferon |
| IL | Interleukin |
| ITAM | immunoreceptor tyrosine-based activation motif |
| ITG | Integrin |
| ITIM | Immunoreceptor tyrosine-based inhibitory motif |
| KIRs | Killer immunoglobulin-like receptors |
| KIR2DL | KIR 2 domain long |
| KIR2DS | KIR 2 domain short |
| KIR3DL | KIR 3 domain long |
| KIR3DS | KIR 3 domain short |
| LILRB1 | Leukocyte immunoglobulin-like receptor subfamily B member 1 |
| LIR-1 | Leukocyte immunoglobulin-like receptor 1 |
| MIC | MHC Class I polypeptide-related sequence |
| miR | microRNA |
| NK | Natural Killer |
| NKG2A | Natural killer group 2 member A |
| NKG2C | Natural killer group 2 member C |
| NKG2D | Natural killer group 2 member D |
| NKG2DL | NKG2D ligand |
| NKp30 | Natural killer p30-related protein |
| NKRs | Natural killer cell receptors |
| ORFs | Open reading frames |
| Pp65 | 65 kDa phosphoprotein |
| RL | Terminal repeat long |
| TAP | Transporter associated with antigen processing |
| Tel | Telomeric |
| TNF | Tumor necrosis factor |
| TRAIL | TNF-related apoptosis-inducing ligand |
| TRAIL-R | TRAIL receptor |
| UL | Unique long |
| ULBP | UL16-binding protein |
| US | Unique short |
References
- Griffiths, P.D.; McLean, A.; Emery, V.C. Encouraging prospects for immunisation against primary cytomegalovirus infection. Vaccine 2001, 19, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Musonda, K.G.; Nyonda, M.; Filteau, S.; Kasonka, L.; Monze, M.; Gompels, U.A. Increased Cytomegalovirus Secretion and Risks of Infant Infection by Breastfeeding Duration From Maternal Human Immunodeficiency Virus Positive Compared to Negative Mothers in Sub-Saharan Africa. J. Pediatr. Infect. Dis. Soc. 2016, 5, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef]
- Stowell, J.D.; Mask, K.; Amin, M.; Clark, R.; Levis, D.; Hendley, W.; Lanzieri, T.M.; Dollard, S.C.; Cannon, M.J. Cross-sectional study of cytomegalovirus shedding and immunological markers among seropositive children and their mothers. BMC Infect. Dis. 2014, 14, 568. [Google Scholar] [CrossRef] [PubMed]
- Staras, S.A.; Flanders, W.D.; Dollard, S.C.; Pass, R.F.; McGowan, J.E., Jr.; Cannon, M.J. Cytomegalovirus seroprevalence and childhood sources of infection: A population-based study among pre-adolescents in the United States. J. Clin. Virol. 2008, 43, 266–271. [Google Scholar] [CrossRef]
- Staras, S.A.; Flanders, W.D.; Dollard, S.C.; Pass, R.F.; McGowan, J.E., Jr.; Cannon, M.J. Influence of sexual activity on cytomegalovirus seroprevalence in the United States, 1988–1994. Sex. Transm. Dis. 2008, 35, 472–479. [Google Scholar] [CrossRef]
- Wills, M.R.; Poole, E.; Lau, B.; Krishna, B.; Sinclair, J.H. The immunology of human cytomegalovirus latency: Could latent infection be cleared by novel immunotherapeutic strategies? Cell. Mol. Immunol. 2015, 12, 128–138. [Google Scholar] [CrossRef]
- Spector, S.A.; Hirata, K.K.; Newman, T.R. Identification of multiple cytomegalovirus strains in homosexual men with acquired immunodeficiency syndrome. J. Infect. Dis. 1984, 150, 953–956. [Google Scholar] [CrossRef]
- Chandler, S.H.; Handsfield, H.H.; McDougall, J.K. Isolation of multiple strains of cytomegalovirus from women attending a clinic for sexually transmitted disease. J. Infect. Dis. 1987, 155, 655–660. [Google Scholar] [CrossRef]
- Kanesaki, T.; Baba, K.; Tanaka, K.; Ishibashi, M.; Yabuuchi, H. Characterization of cytomegalovirus isolates recovered during repeated infection in renal transplant recipients. J. Med. Virol. 1989, 28, 140–143. [Google Scholar] [CrossRef]
- Hansen, S.G.; Powers, C.J.; Richards, R.; Ventura, A.B.; Ford, J.C.; Siess, D.; Axthelm, M.K.; Nelson, J.A.; Jarvis, M.A.; Picker, L.J.; et al. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 2010, 328, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Cudini, J.; Roy, S.; Houldcroft, C.J.; Bryant, J.M.; Depledge, D.P.; Tutill, H.; Veys, P.; Williams, R.; Worth, A.J.J.; Tamuri, A.U.; et al. Human cytomegalovirus haplotype reconstruction reveals high diversity due to superinfection and evidence of within-host recombination. Proc. Natl. Acad. Sci. USA 2019, 116, 5693–5698. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef]
- Ssentongo, P.; Hehnly, C.; Birungi, P.; Roach, M.A.; Spady, J.; Fronterre, C.; Wang, M.; Murray-Kolb, L.E.; Al-Shaar, L.; Chinchilli, V.M.; et al. Congenital Cytomegalovirus Infection Burden and Epidemiologic Risk Factors in Countries With Universal Screening: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e2120736. [Google Scholar] [CrossRef]
- Krause, P.R.; Bialek, S.R.; Boppana, S.B.; Griffiths, P.D.; Laughlin, C.A.; Ljungman, P.; Mocarski, E.S.; Pass, R.F.; Read, J.S.; Schleiss, M.R.; et al. Priorities for CMV vaccine development. Vaccine 2013, 32, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Pinninti, S.G.; Britt, W.J.; Boppana, S.B. Auditory and Vestibular Involvement in Congenital Cytomegalovirus Infection. Pathogens 2024, 13, 1019. [Google Scholar] [CrossRef]
- Hill, R.B., Jr.; Rowlands, D.T., Jr.; Rifkind, D. Infectious Pulmonary Disease in Patients Receiving Immunosuppressive Therapy for Organ Transplantation. N. Engl. J. Med. 1964, 271, 1021–1027. [Google Scholar] [CrossRef]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef]
- Herbein, G. The Human Cytomegalovirus, from Oncomodulation to Oncogenesis. Viruses 2018, 10, 408. [Google Scholar] [CrossRef]
- Herbein, G. Tumors and Cytomegalovirus: An Intimate Interplay. Viruses 2022, 14, 812. [Google Scholar] [CrossRef]
- Johnsen, J.I.; Baryawno, N.; Soderberg-Naucler, C. Is human cytomegalovirus a target in cancer therapy? Oncotarget 2011, 2, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef]
- Brito, A.F.; Baele, G.; Nahata, K.D.; Grubaugh, N.D.; Pinney, J.W. Intrahost speciations and host switches played an important role in the evolution of herpesviruses. Virus Evol. 2021, 7, veab025. [Google Scholar] [CrossRef]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [PubMed]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef]
- Wilkinson, G.W.; Tomasec, P.; Stanton, R.J.; Armstrong, M.; Prod’homme, V.; Aicheler, R.; McSharry, B.P.; Rickards, C.R.; Cochrane, D.; Llewellyn-Lacey, S.; et al. Modulation of natural killer cells by human cytomegalovirus. J. Clin. Virol. 2008, 41, 206–212. [Google Scholar] [CrossRef]
- Mancini, M.; Vidal, S.M. Mechanisms of Natural Killer Cell Evasion Through Viral Adaptation. Annu. Rev. Immunol. 2020, 38, 511–539. [Google Scholar] [CrossRef]
- Dell’Oste, V.; Biolatti, M.; Galitska, G.; Griffante, G.; Gugliesi, F.; Pasquero, S.; Zingoni, A.; Cerboni, C.; De Andrea, M. Tuning the Orchestra: HCMV vs. Innate Immunity. Front. Microbiol. 2020, 11, 661. [Google Scholar] [CrossRef]
- Berry, R.; Watson, G.M.; Jonjic, S.; Degli-Esposti, M.A.; Rossjohn, J. Modulation of innate and adaptive immunity by cytomegaloviruses. Nat. Rev. Immunol. 2020, 20, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Paolini, R.; Bernardini, G.; Molfetta, R.; Santoni, A. NK cells and interferons. Cytokine Growth Factor. Rev. 2015, 26, 113–120. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Cooper, M.A.; Nuovo, G.J.; Cella, M.; Facchetti, F.; Colonna, M.; Caligiuri, M.A. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: A potential new link between adaptive and innate immunity. Blood 2003, 101, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Fitz, L.; Ryan, M.; Hewick, R.M.; Clark, S.C.; Chan, S.; Loudon, R.; Sherman, F.; Perussia, B.; Trinchieri, G. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J. Exp. Med. 1989, 170, 827–845. [Google Scholar] [CrossRef] [PubMed]
- Carson, W.E.; Giri, J.G.; Lindemann, M.J.; Linett, M.L.; Ahdieh, M.; Paxton, R.; Anderson, D.; Eisenmann, J.; Grabstein, K.; Caligiuri, M.A. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J. Exp. Med. 1994, 180, 1395–1403. [Google Scholar] [CrossRef]
- Wu, Y.; Tian, Z.; Wei, H. Developmental and Functional Control of Natural Killer Cells by Cytokines. Front. Immunol. 2017, 8, 930. [Google Scholar] [CrossRef]
- Parrish-Novak, J.; Dillon, S.R.; Nelson, A.; Hammond, A.; Sprecher, C.; Gross, J.A.; Johnston, J.; Madden, K.; Xu, W.; West, J.; et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature 2000, 408, 57–63. [Google Scholar] [CrossRef]
- Ziblat, A.; Domaica, C.I.; Spallanzani, R.G.; Iraolagoitia, X.L.; Rossi, L.E.; Avila, D.E.; Torres, N.I.; Fuertes, M.B.; Zwirner, N.W. IL-27 stimulates human NK-cell effector functions and primes NK cells for IL-18 responsiveness. Eur. J. Immunol. 2015, 45, 192–202. [Google Scholar] [CrossRef]
- Pende, D.; Falco, M.; Vitale, M.; Cantoni, C.; Vitale, C.; Munari, E.; Bertaina, A.; Moretta, F.; Del Zotto, G.; Pietra, G.; et al. Killer Ig-Like Receptors (KIRs): Their Role in NK Cell Modulation and Developments Leading to Their Clinical Exploitation. Front. Immunol. 2019, 10, 1179. [Google Scholar] [CrossRef]
- Moretta, A.; Vitale, M.; Bottino, C.; Orengo, A.M.; Morelli, L.; Augugliaro, R.; Barbaresi, M.; Ciccone, E.; Moretta, L. P58 molecules as putative receptors for major histocompatibility complex (MHC) class I molecules in human natural killer (NK) cells. Anti-p58 antibodies reconstitute lysis of MHC class I-protected cells in NK clones displaying different specificities. J. Exp. Med. 1993, 178, 597–604. [Google Scholar] [CrossRef]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, A.; Garcia-Beltran, W.F.; Norman, P.J.; Watson, G.M.; Holzemer, A.; Chapel, A.; Richert, L.; Pommerening-Roser, A.; Korner, C.; Ozawa, M.; et al. A subset of HLA-DP molecules serve as ligands for the natural cytotoxicity receptor NKp44. Nat. Immunol. 2019, 20, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Gaggero, S.; Bruschi, M.; Petretto, A.; Parodi, M.; Del Zotto, G.; Lavarello, C.; Prato, C.; Santucci, L.; Barbuto, A.; Bottino, C.; et al. Nidogen-1 is a novel extracellular ligand for the NKp44 activating receptor. Oncoimmunology 2018, 7, e1470730. [Google Scholar] [CrossRef]
- Ito, K.; Shiraishi, R.; Higai, K. Globo-A Binds to the Recombinant Natural Cytotoxicity Receptor NKp44. Biol. Pharm. Bull. 2018, 41, 1480–1484. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, A.; Kundu, K.; Peleg, R.; Yossef, R.; Kaplanov, I.; Ghosh, S.; Khrapunsky, Y.; Gershoni-Yahalom, O.; Rabinski, T.; Cerwenka, A.; et al. NKp44-Derived Peptide Binds Proliferating Cell Nuclear Antigen and Mediates Tumor Cell Death. Front. Immunol. 2018, 9, 1114. [Google Scholar] [CrossRef]
- Baychelier, F.; Sennepin, A.; Ermonval, M.; Dorgham, K.; Debre, P.; Vieillard, V. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood 2013, 122, 2935–2942. [Google Scholar] [CrossRef]
- Rosental, B.; Brusilovsky, M.; Hadad, U.; Oz, D.; Appel, M.Y.; Afergan, F.; Yossef, R.; Rosenberg, L.A.; Aharoni, A.; Cerwenka, A.; et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J. Immunol. 2011, 187, 5693–5702. [Google Scholar] [CrossRef]
- Horowitz, A.; Strauss-Albee, D.M.; Leipold, M.; Kubo, J.; Nemat-Gorgani, N.; Dogan, O.C.; Dekker, C.L.; Mackey, S.; Maecker, H.; Swan, G.E.; et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci. Transl. Med. 2013, 5, 208ra145. [Google Scholar] [CrossRef]
- Merino, A.M.; Kim, H.; Miller, J.S.; Cichocki, F. Unraveling exhaustion in adaptive and conventional NK cells. J. Leukoc. Biol. 2020, 108, 1361–1368. [Google Scholar] [CrossRef]
- Sohn, H.; Cooper, M.A. Metabolic regulation of NK cell function: Implications for immunotherapy. Immunometabolism 2023, 5, e00020. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Forrest, C.; Chase, T.J.G.; Cuff, A.O.; Maroulis, D.; Motallebzadeh, R.; Gander, A.; Davidson, B.; Griffiths, P.; Male, V.; Reeves, M. Control of human cytomegalovirus replication by liver resident natural killer cells. Nat. Commun. 2023, 14, 1409. [Google Scholar] [CrossRef]
- Amand, M.; Iserentant, G.; Poli, A.; Sleiman, M.; Fievez, V.; Sanchez, I.P.; Sauvageot, N.; Michel, T.; Aouali, N.; Janji, B.; et al. Human CD56(dim)CD16(dim) Cells As an Individualized Natural Killer Cell Subset. Front. Immunol. 2017, 8, 699. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Gabrielli, S.; Ortolani, C.; Del Zotto, G.; Luchetti, F.; Canonico, B.; Buccella, F.; Artico, M.; Papa, S.; Zamai, L. The Memories of NK Cells: Innate-Adaptive Immune Intrinsic Crosstalk. J. Immunol. Res. 2016, 2016, 1376595. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef]
- Sanseviero, E. NK Cell-Fc Receptors Advance Tumor Immunotherapy. J. Clin. Med. 2019, 8, 1667. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef]
- Topham, N.J.; Hewitt, E.W. Natural killer cell cytotoxicity: How do they pull the trigger? Immunology 2009, 128, 7–15. [Google Scholar] [CrossRef]
- Tomescu, C.; Kroll, K.; Colon, K.; Papasavvas, E.; Frank, I.; Tebas, P.; Mounzer, K.; Reeves, R.K.; Montaner, L.J. Identification of the predominant human NK cell effector subset mediating ADCC against HIV-infected targets coated with BNAbs or plasma from PLWH. Eur. J. Immunol. 2021, 51, 2051–2061. [Google Scholar] [CrossRef]
- Haroun-Izquierdo, A.; Vincenti, M.; Netskar, H.; van Ooijen, H.; Zhang, B.; Bendzick, L.; Kanaya, M.; Momayyezi, P.; Li, S.; Wiiger, M.T.; et al. Adaptive single-KIR(+)NKG2C(+) NK cells expanded from select superdonors show potent missing-self reactivity and efficiently control HLA-mismatched acute myeloid leukemia. J. Immunother. Cancer 2022, 10, e005577. [Google Scholar] [CrossRef]
- Kristensen, A.B.; Wragg, K.M.; Vanderven, H.A.; Lee, W.S.; Silvers, J.; Kent, H.E.; Grant, M.D.; Kelleher, A.D.; Juno, J.A.; Kent, S.J.; et al. Phenotypic and functional characteristics of highly differentiated CD57+NKG2C+ NK cells in HIV-1-infected individuals. Clin. Exp. Immunol. 2022, 210, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Cabrera, C.; Erkizia, I.; Bofill, M.; Clotet, B.; Ruiz, L.; Lopez-Botet, M. Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV-1-positive patients. J. Infect. Dis. 2006, 194, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Jing, S.; Sheng, G.; Jia, F. The basic biology of NK cells and its application in tumor immunotherapy. Front. Immunol. 2024, 15, 1420205. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.B.; Yu, J.H.; Caligiuri, M.A. Natural killer cell-based immunotherapy for cancer. J. Immunol. 2025, vkaf036. [Google Scholar] [CrossRef]
- Vietzen, H.; Pollak, K.; Honsig, C.; Jaksch, P.; Puchhammer-Stöckl, E. NKG2C Deletion Is a Risk Factor for Human Cytomegalovirus Viremia and Disease After Lung Transplantation. J. Infect. Dis. 2018, 217, 802–806. [Google Scholar] [CrossRef]
- Foley, B.; Cooley, S.; Verneris, M.R.; Pitt, M.; Curtsinger, J.; Luo, X.; Lopez-Vergès, S.; Lanier, L.L.; Weisdorf, D.; Miller, J.S. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 2012, 119, 2665–2674. [Google Scholar] [CrossRef]
- Giordano, C.; Carlomagno, S.; Falco, M.; Cantoni, C.; Vitale, M.; Caruana, I.; Dirks, J.; Serio, A.; Muccio, L.; Bartalucci, G.; et al. CD94-driven in vitro expansion of highly functional adaptive NKG2C(+) NKG2A(-) CD57(+) NK cells from CMV(+) healthy donors. Front. Immunol. 2025, 16, 1481745. [Google Scholar] [CrossRef]
- Guma, M.; Angulo, A.; Vilches, C.; Gomez-Lozano, N.; Malats, N.; Lopez-Botet, M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004, 104, 3664–3671. [Google Scholar] [CrossRef]
- Basílio-Queirós, D.; Venturini, L.; Luther-Wolf, S.; Dammann, E.; Ganser, A.; Stadler, M.; Falk, C.S.; Weissinger, E.M. Adaptive NK cells undergo a dynamic modulation in response to human cytomegalovirus and recruit T cells in in vitro migration assays. Bone Marrow Transpl. 2022, 57, 712–720. [Google Scholar] [CrossRef]
- Medjouel Khlifi, H.; Guia, S.; Vivier, E.; Narni-Mancinelli, E. Role of the ITAM-Bearing Receptors Expressed by Natural Killer Cells in Cancer. Front. Immunol. 2022, 13, 898745. [Google Scholar] [CrossRef]
- Luetke-Eversloh, M.; Hammer, Q.; Durek, P.; Nordström, K.; Gasparoni, G.; Pink, M.; Hamann, A.; Walter, J.; Chang, H.-D.; Dong, J.; et al. Human Cytomegalovirus Drives Epigenetic Imprinting of the IFNG Locus in NKG2Chi Natural Killer Cells. PLOS Pathog. 2014, 10, e1004441. [Google Scholar] [CrossRef] [PubMed]
- Schlums, H.; Cichocki, F.; Tesi, B.; Theorell, J.; Beziat, V.; Holmes, T.D.; Han, H.; Chiang, S.C.; Foley, B.; Mattsson, K.; et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 2015, 42, 443–456. [Google Scholar] [CrossRef]
- Zhang, T.; Scott, J.M.; Hwang, I.; Kim, S. Cutting edge: Antibody-dependent memory-like NK cells distinguished by FcRγ deficiency. J. Immunol. 2013, 190, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Zhang, T.; Scott, J.M.; Kim, A.R.; Lee, T.; Kakarla, T.; Kim, A.; Sunwoo, J.B.; Kim, S. Identification of human NK cells that are deficient for signaling adaptor FcRγ and specialized for antibody-dependent immune functions. Int. Immunol. 2012, 24, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Budt, M.; Saez, A.; Brckalo, T.; Hengel, H.; Angulo, A.; Lopez-Botet, M. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 2006, 107, 3624–3631. [Google Scholar] [CrossRef]
- Hammer, Q.; Rückert, T.; Borst, E.M.; Dunst, J.; Haubner, A.; Durek, P.; Heinrich, F.; Gasparoni, G.; Babic, M.; Tomic, A.; et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat. Immunol. 2018, 19, 453–463. [Google Scholar] [CrossRef]
- Mace, E.M.; Orange, J.S. Genetic Causes of Human NK Cell Deficiency and Their Effect on NK Cell Subsets. Front. Immunol. 2016, 7, 545. [Google Scholar] [CrossRef]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef]
- Foley, B.; Cooley, S.; Verneris, M.R.; Curtsinger, J.; Luo, X.; Waller, E.K.; Anasetti, C.; Weisdorf, D.; Miller, J.S. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J. Immunol. 2012, 189, 5082–5088. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Miller, J.S.; Stuart, R.K.; Cooley, S.; Salhotra, A.; Curtsinger, J.; Westervelt, P.; DiPersio, J.F.; Hillman, T.M.; Silver, N.; et al. A Phase 1 Trial of CNDO-109-Activated Natural Killer Cells in Patients with High-Risk Acute Myeloid Leukemia. Biol. Blood Marrow Transplant. 2018, 24, 1581–1589. [Google Scholar] [CrossRef]
- Kottaridis, P.D.; North, J.; Tsirogianni, M.; Marden, C.; Samuel, E.R.; Jide-Banwo, S.; Grace, S.; Lowdell, M.W. Two-Stage Priming of Allogeneic Natural Killer Cells for the Treatment of Patients with Acute Myeloid Leukemia: A Phase I Trial. PLoS ONE 2015, 10, e0123416. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; Baars, P.A.; Dantin, C.; van den Burg, M.; van Lier, R.A.W.; Roosnek, E. Human NK cells can control CMV infection in the absence of T cells. Blood 2008, 112, 914–915. [Google Scholar] [CrossRef] [PubMed]
- Redondo-Pachon, D.; Crespo, M.; Yelamos, J.; Muntasell, A.; Perez-Saez, M.J.; Perez-Fernandez, S.; Vila, J.; Vilches, C.; Pascual, J.; Lopez-Botet, M. Adaptive NKG2C+ NK Cell Response and the Risk of Cytomegalovirus Infection in Kidney Transplant Recipients. J. Immunol. 2017, 198, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Ataya, M.; Redondo-Pachon, D.; Llinas-Mallol, L.; Yelamos, J.; Heredia, G.; Perez-Saez, M.J.; Vila, J.; Costa-Garcia, M.; Raich-Regue, D.; Vilches, C.; et al. Pretransplant adaptive NKG2C+ NK cells protect against cytomegalovirus infection in kidney transplant recipients. Am. J. Transplant. 2020, 20, 663–676. [Google Scholar] [CrossRef]
- Harpur, C.M.; Stankovic, S.; Kanagarajah, A.; Widjaja, J.M.L.; Levvey, B.J.; Cristiano, Y.; Snell, G.I.; Brooks, A.G.; Westall, G.P.; Sullivan, L.C. Enrichment of Cytomegalovirus-induced NKG2C+ Natural Killer Cells in the Lung Allograft. Transplantation 2019, 103, 1689–1699. [Google Scholar] [CrossRef]
- Yu, X.X.; Shang, Q.N.; Liu, X.F.; He, M.; Pei, X.Y.; Mo, X.D.; Lv, M.; Han, T.T.; Huo, M.R.; Zhao, X.S.; et al. Donor NKG2C homozygosity contributes to CMV clearance after haploidentical transplantation. JCI Insight 2022, 7, e149120. [Google Scholar] [CrossRef]
- Davis, Z.B.; Cooley, S.A.; Cichocki, F.; Felices, M.; Wangen, R.; Luo, X.; DeFor, T.E.; Bryceson, Y.T.; Diamond, D.J.; Brunstein, C.; et al. Adaptive Natural Killer Cell and Killer Cell Immunoglobulin–Like Receptor–Expressing T Cell Responses are Induced by Cytomegalovirus and Are Associated with Protection against Cytomegalovirus Reactivation after Allogeneic Donor Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2015, 21, 1653–1662. [Google Scholar] [CrossRef]
- Kheav, V.D.; Busson, M.; Scieux, C.; Peffault de Latour, R.; Maki, G.; Haas, P.; Mazeron, M.C.; Carmagnat, M.; Masson, E.; Xhaard, A.; et al. Favorable impact of natural killer cell reconstitution on chronic graft-versus-host disease and cytomegalovirus reactivation after allogeneic hematopoietic stem cell transplantation. Haematologica 2014, 99, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Noyola, D.E.; Fortuny, C.; Muntasell, A.; Noguera-Julian, A.; Munoz-Almagro, C.; Alarcon, A.; Juncosa, T.; Moraru, M.; Vilches, C.; Lopez-Botet, M. Influence of congenital human cytomegalovirus infection and the NKG2C genotype on NK-cell subset distribution in children. Eur. J. Immunol. 2012, 42, 3256–3266. [Google Scholar] [CrossRef] [PubMed]
- Noyola, D.E.; Alarcón, A.; Noguera-Julian, A.; Muntasell, A.; Muñoz-Almagro, C.; García, J.; Mur, A.; Fortuny, C.; López-Botet, M. Dynamics of the NK-cell subset redistribution induced by cytomegalovirus infection in preterm infants. Hum. Immunol. 2015, 76, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Vaaben, A.V.; Levan, J.; Nguyen, C.B.T.; Callaway, P.C.; Prahl, M.; Warrier, L.; Nankya, F.; Musinguzi, K.; Kakuru, A.; Muhindo, M.K.; et al. In Utero Activation of Natural Killer Cells in Congenital Cytomegalovirus Infection. J. Infect. Dis. 2022, 226, 566–575. [Google Scholar] [CrossRef]
- Fowler, K.B.; Stagno, S.; Pass, R.F. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA J. Am. Med. Assoc. 2003, 289, 1008–1011. [Google Scholar] [CrossRef]
- Simonazzi, G.; Curti, A.; Cervi, F.; Gabrielli, L.; Contoli, M.; Capretti, M.G.; Rizzo, N.; Guerra, B.; Farina, A.; Lazzarotto, T. Perinatal Outcomes of Non-Primary Maternal Cytomegalovirus Infection: A 15-Year Experience. Fetal Diagn. Ther. 2018, 43, 138–142. [Google Scholar] [CrossRef]
- Pighi, C.; Rotili, A.; De Luca, M.; Chiurchiù, S.; Calò Carducci, F.I.; Rossetti, C.; Cifaldi, L.; Bei, R.; Caforio, L.; Bernardi, S.; et al. Characterization of Natural Killer Cell Profile in a Cohort of Infected Pregnant Women and Their Babies and Its Relation to CMV Transmission. Viruses 2024, 16, 780. [Google Scholar] [CrossRef]
- Cosman, D.; Fanger, N.; Borges, L.; Kubin, M.; Chin, W.; Peterson, L.; Hsu, M.L. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 1997, 7, 273–282. [Google Scholar] [CrossRef]
- Prod’homme, V.; Griffin, C.; Aicheler, R.J.; Wang, E.C.; McSharry, B.P.; Rickards, C.R.; Stanton, R.J.; Borysiewicz, L.K.; Lopez-Botet, M.; Wilkinson, G.W.; et al. The human cytomegalovirus MHC class I homolog UL18 inhibits LIR-1+ but activates LIR-1- NK cells. J. Immunol. 2007, 178, 4473–4481. [Google Scholar] [CrossRef]
- Beck, S.; Barrell, B.G. Human cytomegalovirus encodes a glycoprotein homologous to MHC class-I antigens. Nature 1988, 331, 269–272. [Google Scholar] [CrossRef]
- Browne, H.; Smith, G.; Beck, S.; Minson, T. A complex between the MHC class I homologue encoded by human cytomegalovirus and beta 2 microglobulin. Nature 1990, 347, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.; Wang, E.C.Y.; McSharry, B.P.; Rickards, C.; Browne, H.; Wilkinson, G.W.G.; Tomasec, P. Characterization of a highly glycosylated form of the human cytomegalovirus HLA class I homologue gpUL18. J. Gen. Virol. 2005, 86, 2999–3008. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Choi, S.E.; Yun, I.H.; Kim, J.Y.; Ahn, C.; Kim, S.J.; Ha, J.; Hwang, E.S.; Cha, C.Y.; Miyagawa, S.; et al. Human cytomegalovirus UL18 alleviated human NK-mediated swine endothelial cell lysis. Biochem. Biophys. Res. Commun. 2004, 315, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.E.; Thomas, L.M.; Bjorkman, P.J. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat. Immunol. 2003, 4, 913–919. [Google Scholar] [CrossRef]
- Yang, Z.; Bjorkman, P.J. Structure of UL18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 10095–10100. [Google Scholar] [CrossRef]
- Vales-Gomez, M.; Shiroishi, M.; Maenaka, K.; Reyburn, H.T. Genetic variability of the major histocompatibility complex class I homologue encoded by human cytomegalovirus leads to differential binding to the inhibitory receptor ILT2. J. Virol. 2005, 79, 2251–2260. [Google Scholar] [CrossRef]
- Kuroki, K.; Tsuchiya, N.; Shiroishi, M.; Rasubala, L.; Yamashita, Y.; Matsuta, K.; Fukazawa, T.; Kusaoi, M.; Murakami, Y.; Takiguchi, M.; et al. Extensive polymorphisms of LILRB1 (ILT2, LIR1) and their association with HLA-DRB1 shared epitope negative rheumatoid arthritis. Hum. Mol. Genet. 2005, 14, 2469–2480. [Google Scholar] [CrossRef]
- Cerboni, C.; Achour, A.; Warnmark, A.; Mousavi-Jazi, M.; Sandalova, T.; Hsu, M.L.; Cosman, D.; Karre, K.; Carbone, E. Spontaneous mutations in the human CMV HLA class I homologue UL18 affect its binding to the inhibitory receptor LIR-1/ILT2/CD85j. Eur. J. Immunol. 2006, 36, 732–741. [Google Scholar] [CrossRef]
- Yu, K.; Davidson, C.L.; Wojtowicz, A.; Lisboa, L.; Wang, T.; Airo, A.M.; Villard, J.; Buratto, J.; Sandalova, T.; Achour, A.; et al. LILRB1 polymorphisms influence posttransplant HCMV susceptibility and ligand interactions. J. Clin. Investig. 2018, 128, 1523–1537. [Google Scholar] [CrossRef]
- Leong, C.C.; Chapman, T.L.; Bjorkman, P.J.; Formankova, D.; Mocarski, E.S.; Phillips, J.H.; Lanier, L.L. Modulation of natural killer cell cytotoxicity in human cytomegalovirus infection: The role of endogenous class I major histocompatibility complex and a viral class I homolog. J. Exp. Med. 1998, 187, 1681–1687. [Google Scholar] [CrossRef]
- Tomasec, P.; Braud, V.M.; Rickards, C.; Powell, M.B.; McSharry, B.P.; Gadola, S.; Cerundolo, V.; Borysiewicz, L.K.; McMichael, A.J.; Wilkinson, G.W. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 2000, 287, 1031. [Google Scholar] [CrossRef]
- Ulbrecht, M.; Martinozzi, S.; Grzeschik, M.; Hengel, H.; Ellwart, J.W.; Pla, M.; Weiss, E.H. Cutting edge: The human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J. Immunol. 2000, 164, 5019–5022. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.C.Y.; McSharry, B.; Retiere, C.; Tomasec, P.; Williams, S.; Borysiewicz, L.K.; Braud, V.M.; Wilkinson, G.W.G. UL40-mediated NK evasion during productive infection with human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2002, 99, 7570–7575. [Google Scholar] [CrossRef]
- Beziat, V.; Liu, L.L.; Malmberg, J.A.; Ivarsson, M.A.; Sohlberg, E.; Bjorklund, A.T.; Retiere, C.; Sverremark-Ekstrom, E.; Traherne, J.; Ljungman, P.; et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 2013, 121, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Asenjo, J.; Moraru, M.; Al-Akioui-Sanz, K.; Altadill, M.; Muntasell, A.; Lopez-Botet, M.; Vilches, C. Diversity of NKG2C genotypes in a European population: Conserved and recombinant haplotypes in the coding, promoter, and 3′-untranslated regions. HLA 2022, 100, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Strong, R.K.; Holmes, M.A.; Li, P.; Braun, L.; Lee, N.; Geraghty, D.E. HLA-E allelic variants. Correlating differential expression, peptide affinities, crystal structures, and thermal stabilities. J. Biol. Chem. 2003, 278, 5082–5090. [Google Scholar] [CrossRef]
- Heatley, S.L.; Pietra, G.; Lin, J.; Widjaja, J.M.L.; Harpur, C.M.; Lester, S.; Rossjohn, J.; Szer, J.; Schwarer, A.; Bradstock, K.; et al. Polymorphism in human cytomegalovirus UL40 impacts on recognition of human leukocyte antigen-E (HLA-E) by natural killer cells. J. Biol. Chem. 2013, 288, 8679–8690. [Google Scholar] [CrossRef]
- Moraru, M.; Cisneros, E.; Gomez-Lozano, N.; de Pablo, R.; Portero, F.; Canizares, M.; Vaquero, M.; Roustan, G.; Millan, I.; Lopez-Botet, M.; et al. Host genetic factors in susceptibility to herpes simplex type 1 virus infection: Contribution of polymorphic genes at the interface of innate and adaptive immunity. J. Immunol. 2012, 188, 4412–4420. [Google Scholar] [CrossRef]
- Muntasell, A.; Pupuleku, A.; Cisneros, E.; Vera, A.; Moraru, M.; Vilches, C.; Lopez-Botet, M. Relationship of NKG2C Copy Number with the Distribution of Distinct Cytomegalovirus-Induced Adaptive NK Cell Subsets. J. Immunol. 2016, 196, 3818–3827. [Google Scholar] [CrossRef]
- Hikami, K.; Tsuchiya, N.; Yabe, T.; Tokunaga, K. Variations of human killer cell lectin-like receptors: Common occurrence of NKG2-C deletion in the general population. Genes. Immun. 2003, 4, 160–167. [Google Scholar] [CrossRef]
- Miyashita, R.; Tsuchiya, N.; Hikami, K.; Kuroki, K.; Fukazawa, T.; Bijl, M.; Kallenberg, C.G.; Hashimoto, H.; Yabe, T.; Tokunaga, K. Molecular genetic analyses of human NKG2C (KLRC2) gene deletion. Int. Immunol. 2004, 16, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Moraru, M.; Canizares, M.; Muntasell, A.; de Pablo, R.; Lopez-Botet, M.; Vilches, C. Assessment of copy-number variation in the NKG2C receptor gene in a single-tube and characterization of a reference cell panel, using standard polymerase chain reaction. Tissue Antigens 2012, 80, 184–187. [Google Scholar] [CrossRef]
- Muntasell, A.; Vilches, C.; Angulo, A.; Lopez-Botet, M. Adaptive reconfiguration of the human NK-cell compartment in response to cytomegalovirus: A different perspective of the host-pathogen interaction. Eur. J. Immunol. 2013, 43, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.R.; Arunachalam, J.; Bhardwaj, A.; Saifullah, A.; Lakhchaura, R.; Soni, M.; Bhagawati, G.; Chakrabarti, S. Impact of adaptive natural killer cells, KLRC2 genotype and cytomegalovirus reactivation on late mortality in patients with severe COVID-19 lung disease. Clin. Transl. Immunol. 2022, 11, e1359. [Google Scholar] [CrossRef]
- Raulet, D.H.; Marcus, A.; Coscoy, L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol. Rev. 2017, 280, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.A.; Aicheler, R.; Stanton, R.J.; Wang, E.C.; Han, S.; Seirafian, S.; Davies, J.; McSharry, B.P.; Weekes, M.P.; Antrobus, P.R.; et al. Two novel human cytomegalovirus NK cell evasion functions target MICA for lysosomal degradation. PLoS Pathog. 2014, 10, e1004058. [Google Scholar] [CrossRef]
- Pignoloni, B.; Fionda, C.; Dell’Oste, V.; Luganini, A.; Cippitelli, M.; Zingoni, A.; Landolfo, S.; Gribaudo, G.; Santoni, A.; Cerboni, C. Distinct Roles for Human Cytomegalovirus Immediate Early Proteins IE1 and IE2 in the Transcriptional Regulation of MICA and PVR/CD155 Expression. J. Immunol. 2016, 197, 4066–4078. [Google Scholar] [CrossRef]
- Cosman, D.; Mullberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Kubin, M.; Cassiano, L.; Chalupny, J.; Chin, W.; Cosman, D.; Fanslow, W.; Mullberg, J.; Rousseau, A.M.; Ulrich, D.; Armitage, R. ULBP1, 2, 3: Novel MHC class I-related molecules that bind to human cytomegalovirus glycoprotein UL16, activate NK cells. Eur. J. Immunol. 2001, 31, 1428–1437. [Google Scholar] [CrossRef]
- Rolle, A.; Mousavi-Jazi, M.; Eriksson, M.; Odeberg, J.; Soderberg-Naucler, C.; Cosman, D.; Karre, K.; Cerboni, C. Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: Up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein. J. Immunol. 2003, 171, 902–908. [Google Scholar] [CrossRef]
- Welte, S.A.; Sinzger, C.; Lutz, S.Z.; Singh-Jasuja, H.; Sampaio, K.L.; Eknigk, U.; Rammensee, H.G.; Steinle, A. Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur. J. Immunol. 2003, 33, 194–203. [Google Scholar] [CrossRef]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; et al. Host immune system gene targeting by a viral miRNA. Science 2007, 317, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Petersdorf, E.W.; Shuler, K.B.; Longton, G.M.; Spies, T.; Hansen, J.A. Population study of allelic diversity in the human MHC class I-related MIC-A gene. Immunogenetics 1999, 49, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lazaro, A.M.; Lavingia, B.; Stastny, P. Typing for all known MICA alleles by group-specific PCR and SSOP. Hum. Immunol. 2001, 62, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lazaro, A.M.; Zou, Y.; Lavingia, B.; Moraes, E.M.; Moraes, R.J.; Stastny, P. MICA polymorphism in South American Indians. Immunogenetics 2002, 53, 900–906. [Google Scholar] [CrossRef]
- Ashiru, O.; Lopez-Cobo, S.; Fernandez-Messina, L.; Pontes-Quero, S.; Pandolfi, R.; Reyburn, H.T.; Vales-Gomez, M. A GPI anchor explains the unique biological features of the common NKG2D-ligand allele MICA*008. Biochem. J. 2013, 454, 295–302. [Google Scholar] [CrossRef]
- Wills, M.R.; Ashiru, O.; Reeves, M.B.; Okecha, G.; Trowsdale, J.; Tomasec, P.; Wilkinson, G.W.; Sinclair, J.; Sissons, J.G. Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J. Immunol. 2005, 175, 7457–7465. [Google Scholar] [CrossRef]
- Ashiru, O.; Bennett, N.J.; Boyle, L.H.; Thomas, M.; Trowsdale, J.; Wills, M.R. NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J. Virol. 2009, 83, 12345–12354. [Google Scholar] [CrossRef]
- Bennett, N.J.; Ashiru, O.; Morgan, F.J.; Pang, Y.; Okecha, G.; Eagle, R.A.; Trowsdale, J.; Sissons, J.G.; Wills, M.R. Intracellular sequestration of the NKG2D ligand ULBP3 by human cytomegalovirus. J. Immunol. 2010, 185, 1093–1102. [Google Scholar] [CrossRef]
- Chalupny, N.J.; Rein-Weston, A.; Dosch, S.; Cosman, D. Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem. Biophys. Res. Commun. 2006, 346, 175–181. [Google Scholar] [CrossRef]
- Fielding, C.A.; Weekes, M.P.; Nobre, L.V.; Ruckova, E.; Wilkie, G.S.; Paulo, J.A.; Chang, C.; Suarez, N.M.; Davies, J.A.; Antrobus, R.; et al. Control of immune ligands by members of a cytomegalovirus gene expansion suppresses natural killer cell activation. Elife 2017, 6, e22206. [Google Scholar] [CrossRef]
- Dassa, L.; Seidel, E.; Oiknine-Djian, E.; Yamin, R.; Wolf, D.G.; Le-Trilling, V.T.K.; Mandelboim, O. The Human Cytomegalovirus Protein UL148A Downregulates the NK Cell-Activating Ligand MICA To Avoid NK Cell Attack. J. Virol. 2018, 92, e00162-18. [Google Scholar] [CrossRef]
- Seidel, E.; Le, V.T.K.; Bar-On, Y.; Tsukerman, P.; Enk, J.; Yamin, R.; Stein, N.; Schmiedel, D.; Oiknine Djian, E.; Weisblum, Y.; et al. Dynamic Co-evolution of Host and Pathogen: HCMV Downregulates the Prevalent Allele MICA *008 to Escape Elimination by NK Cells. Cell Rep. 2015, 10, 968–982. [Google Scholar] [CrossRef] [PubMed]
- Seidel, E.; Dassa, L.; Kahlon, S.; Tirosh, B.; Halenius, A.; Seidel Malkinson, T.; Mandelboim, O. A slowly cleaved viral signal peptide acts as a protein-integral immune evasion domain. Nat. Commun. 2021, 12, 2061. [Google Scholar] [CrossRef] [PubMed]
- Kol, I.; Rishiq, A.; Cohen, M.; Kahlon, S.; Pick, O.; Dassa, L.; Stein, N.; Bar-On, Y.; Wolf, D.G.; Seidel, E.; et al. CLPTM1L is a GPI-anchoring pathway component targeted by HCMV. J. Cell Biol. 2023, 222, e202207104. [Google Scholar] [CrossRef]
- Seidel, E.; Dassa, L.; Schuler, C.; Oiknine-Djian, E.; Wolf, D.G.; Le-Trilling, V.T.K.; Mandelboim, O. The human cytomegalovirus protein UL147A downregulates the most prevalent MICA allele: MICA*008, to evade NK cell-mediated killing. PLoS Pathog. 2021, 17, e1008807. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Banas, H.; Casas-Aviles, I.; Duran, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef]
- Shibuya, A.; Campbell, D.; Hannum, C.; Yssel, H.; Franz-Bacon, K.; McClanahan, T.; Kitamura, T.; Nicholl, J.; Sutherland, G.R.; Lanier, L.L.; et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 1996, 4, 573–581. [Google Scholar] [CrossRef]
- Tomasec, P.; Wang, E.C.; Davison, A.J.; Vojtesek, B.; Armstrong, M.; Griffin, C.; McSharry, B.P.; Morris, R.J.; Llewellyn-Lacey, S.; Rickards, C.; et al. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat. Immunol. 2005, 6, 181–188. [Google Scholar] [CrossRef]
- Prod’homme, V.; Sugrue, D.M.; Stanton, R.J.; Nomoto, A.; Davies, J.; Rickards, C.R.; Cochrane, D.; Moore, M.; Wilkinson, G.W.G.; Tomasec, P. Human cytomegalovirus UL141 promotes efficient downregulation of the natural killer cell activating ligand CD112. J. Gen. Virol. 2010, 91, 2034–2039. [Google Scholar] [CrossRef]
- Hsu, J.L.; van den Boomen, D.J.; Tomasec, P.; Weekes, M.P.; Antrobus, R.; Stanton, R.J.; Ruckova, E.; Sugrue, D.; Wilkie, G.S.; Davison, A.J.; et al. Plasma membrane profiling defines an expanded class of cell surface proteins selectively targeted for degradation by HCMV US2 in cooperation with UL141. PLoS Pathog. 2015, 11, e1004811. [Google Scholar] [CrossRef]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front. Immunol. 2019, 10, 909. [Google Scholar] [CrossRef] [PubMed]
- Pogge von Strandmann, E.; Simhadri, V.R.; von Tresckow, B.; Sasse, S.; Reiners, K.S.; Hansen, H.P.; Rothe, A.; Boll, B.; Simhadri, V.L.; Borchmann, P.; et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 2007, 27, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, H.; Geng, J.; Zheng, X.; Wei, H.; Sun, R.; Tian, Z. Tumor-released Galectin-3, a soluble inhibitory ligand of human NKp30, plays an important role in tumor escape from NK cell attack. J. Biol. Chem. 2014, 289, 33311–33319. [Google Scholar] [CrossRef]
- Arnon, T.I.; Achdout, H.; Levi, O.; Markel, G.; Saleh, N.; Katz, G.; Gazit, R.; Gonen-Gross, T.; Hanna, J.; Nahari, E.; et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat. Immunol. 2005, 6, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Penner, I.; Buscher, N.; Dejung, M.; Freiwald, A.; Butter, F.; Plachter, B. Subviral Dense Bodies of Human Cytomegalovirus Induce an Antiviral Type I Interferon Response. Cells 2022, 11, 4028. [Google Scholar] [CrossRef]
- Charpak-Amikam, Y.; Kubsch, T.; Seidel, E.; Oiknine-Djian, E.; Cavaletto, N.; Yamin, R.; Schmiedel, D.; Wolf, D.; Gribaudo, G.; Messerle, M.; et al. Human cytomegalovirus escapes immune recognition by NK cells through the downregulation of B7-H6 by the viral genes US18 and US20. Sci. Rep. 2017, 7, 8661. [Google Scholar] [CrossRef]
- Hofle, J.; Trenkner, T.; Kleist, N.; Schwane, V.; Vollmers, S.; Barcelona, B.; Niehrs, A.; Fittje, P.; Huynh-Tran, V.H.; Sauter, J.; et al. Engagement of TRAIL triggers degranulation and IFNgamma production in human natural killer cells. EMBO Rep. 2022, 23, e54133. [Google Scholar] [CrossRef]
- Smith, W.; Tomasec, P.; Aicheler, R.; Loewendorf, A.; Nemcovicova, I.; Wang, E.C.; Stanton, R.J.; Macauley, M.; Norris, P.; Willen, L.; et al. Human cytomegalovirus glycoprotein UL141 targets the TRAIL death receptors to thwart host innate antiviral defenses. Cell Host Microbe 2013, 13, 324–335. [Google Scholar] [CrossRef]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA 2001, 98, 7829–7834. [Google Scholar] [CrossRef]
- Vlachava, V.M.; Seirafian, S.; Fielding, C.A.; Kollnberger, S.; Aicheler, R.J.; Hughes, J.; Baker, A.; Weekes, M.P.; Forbes, S.; Wilkinson, G.W.G.; et al. HCMV-secreted glycoprotein gpUL4 inhibits TRAIL-mediated apoptosis and NK cell activation. Proc. Natl. Acad. Sci. USA 2023, 120, e2309077120. [Google Scholar] [CrossRef] [PubMed]
- Nemcovicova, I.; Benedict, C.A.; Zajonc, D.M. Structure of human cytomegalovirus UL141 binding to TRAIL-R2 reveals novel, non-canonical death receptor interactions. PLoS Pathog. 2013, 9, e1003224. [Google Scholar] [CrossRef]
- Seirafian, S.; Prod’homme, V.; Sugrue, D.; Davies, J.; Fielding, C.; Tomasec, P.; Wilkinson, G.W.G. Human cytomegalovirus suppresses Fas expression and function. J. Gen. Virol. 2014, 95, 933–939. [Google Scholar] [CrossRef]
- Arnoult, D.; Bartle, L.M.; Skaletskaya, A.; Poncet, D.; Zamzami, N.; Park, P.U.; Sharpe, J.; Youle, R.J.; Goldmacher, V.S. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. USA 2004, 101, 7988–7993. [Google Scholar] [CrossRef]
- Patrone, M.; Percivalle, E.; Secchi, M.; Fiorina, L.; Pedrali-Noy, G.; Zoppe, M.; Baldanti, F.; Hahn, G.; Koszinowski, U.H.; Milanesi, G.; et al. The human cytomegalovirus UL45 gene product is a late, virion-associated protein and influences virus growth at low multiplicities of infection. J. Gen. Virol. 2003, 84, 3359–3370. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.H.; Yang, Y.P.; Lin, J.C.; Hsu, C.H.; Jhang, H.C.; Yang, Y.T.; Lee, C.H.; Ho, L.L.; Hsu, W.M.; Ku, H.H.; et al. The immediate early 2 protein of human cytomegalovirus (HCMV) mediates the apoptotic control in HCMV retinitis through up-regulation of the cellular FLICE-inhibitory protein expression. J. Immunol. 2006, 177, 6199–6206. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Long, E.O. Understanding how combinations of HLA and KIR genes influence disease. J. Exp. Med. 2005, 201, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Parham, P. MHC class I molecules and KIRs in human history, health and survival. Nat. Rev. Immunol. 2005, 5, 201–214. [Google Scholar] [CrossRef]
- Di Bona, D.; Scafidi, V.; Plaia, A.; Colomba, C.; Nuzzo, D.; Occhino, C.; Tuttolomondo, A.; Giammanco, G.; De Grazia, S.; Montalto, G.; et al. HLA and killer cell immunoglobulin-like receptors influence the natural course of CMV infection. J. Infect. Dis. 2014, 210, 1083–1089. [Google Scholar] [CrossRef]
- Stern, M.; Hadaya, K.; Honger, G.; Martin, P.Y.; Steiger, J.; Hess, C.; Villard, J. Telomeric rather than centromeric activating KIR genes protect from cytomegalovirus infection after kidney transplantation. Am. J. Transplant. 2011, 11, 1302–1307. [Google Scholar] [CrossRef]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef]
- Ponte, M.; Cantoni, C.; Biassoni, R.; Tradori-Cappai, A.; Bentivoglio, G.; Vitale, C.; Bertone, S.; Moretta, A.; Moretta, L.; Mingari, M.C. Inhibitory receptors sensing HLA-G1 molecules in pregnancy: Decidua-associated natural killer cells express LIR-1 and CD94/NKG2A and acquire p49, an HLA-G1-specific receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 5674–5679. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Gao, X.; Lee, J.H.; Nelson, G.W.; Detels, R.; Goedert, J.J.; Buchbinder, S.; Hoots, K.; Vlahov, D.; Trowsdale, J.; et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat. Genet. 2002, 31, 429–434. [Google Scholar] [CrossRef] [PubMed]
- de Rham, C.; Hadaya, K.; Bandelier, C.; Ferrari-Lacraz, S.; Villard, J. Expression of killer cell immunoglobulin-like receptors (KIRs) by natural killer cells during acute CMV infection after kidney transplantation. Transpl. Immunol. 2014, 31, 157–164. [Google Scholar] [CrossRef]
- Jones, D.C.; Peacock, S.; Hughes, D.; Traherne, J.A.; Allen, R.L.; Barnardo, M.C.; Friend, P.; Taylor, C.J.; Fuggle, S.; Trowsdale, J.; et al. Killer immunoglobulin-like receptor gene repertoire influences viral load of primary human cytomegalovirus infection in renal transplant patients. Genes. Immun. 2014, 15, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; He, B.; Liu, H.; Muhammad, I.; Cai, J.; Wang, F. Immunoglobulin-like receptor genotype-associated protection from cytomegalovirus infection after liver transplantation. Transpl. Immunol. 2025, 88, 102171. [Google Scholar] [CrossRef]
- Gao, F.; Shi, Z.; Shi, J.; Luo, Y.; Yu, J.; Fu, H.; Lai, X.; Liu, L.; Yuan, Z.; Zheng, Z.; et al. Donor aKIR genes influence the risk of EBV and CMV reactivation after anti-thymocyte globulin-based haploidentical hematopoietic stem cell transplantation. HLA 2024, 103, e15320. [Google Scholar] [CrossRef]
- Cook, M.; Briggs, D.; Craddock, C.; Mahendra, P.; Milligan, D.; Fegan, C.; Darbyshire, P.; Lawson, S.; Boxall, E.; Moss, P. Donor KIR genotype has a major influence on the rate of cytomegalovirus reactivation following T-cell replete stem cell transplantation. Blood 2006, 107, 1230–1232. [Google Scholar] [CrossRef]
- Deborska-Materkowska, D.; Perkowska-Ptasinska, A.; Sadowska-Jakubowicz, A.; Gozdowska, J.; Ciszek, M.; Pazik, J.; Ostaszewska, A.; Kosieradzki, M.; Nowak, J.; Durlik, M. Killer Immunoglobulin-Like Receptor 2DS2 (KIR2DS2), KIR2DL2-HLA-C1, and KIR2DL3 as Genetic Markers for Stratifying the Risk of Cytomegalovirus Infection in Kidney Transplant Recipients. Int. J. Mol. Sci. 2019, 20, 546. [Google Scholar] [CrossRef]
- van der Ploeg, K.; Chang, C.; Ivarsson, M.A.; Moffett, A.; Wills, M.R.; Trowsdale, J. Modulation of Human Leukocyte Antigen-C by Human Cytomegalovirus Stimulates KIR2DS1 Recognition by Natural Killer Cells. Front. Immunol. 2017, 8, 298. [Google Scholar] [CrossRef]
- Santoni, G.; Amantini, C.; Santoni, M.; Maggi, F.; Morelli, M.B.; Santoni, A. Mechanosensation and Mechanotransduction in Natural Killer Cells. Front. Immunol. 2021, 12, 688918. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.J.; Prod’homme, V.; Purbhoo, M.A.; Moore, M.; Aicheler, R.J.; Heinzmann, M.; Bailer, S.M.; Haas, J.; Antrobus, R.; Weekes, M.P.; et al. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe 2014, 16, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.C.Y.; Pjechova, M.; Nightingale, K.; Vlahava, V.M.; Patel, M.; Ruckova, E.; Forbes, S.K.; Nobre, L.; Antrobus, R.; Roberts, D.; et al. Suppression of costimulation by human cytomegalovirus promotes evasion of cellular immune defenses. Proc. Natl. Acad. Sci. USA 2018, 115, 4998–5003. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Hoffmann, K.; Sievert, A.; Reinhard, H.; Merce-Maldonado, E.; Le-Trilling, V.T.K.; Halenius, A.; Gütle, D.; Hengel, H. Human cytomegalovirus antagonizes activation of Fcγ receptors by distinct and synergizing modes of IgG manipulation. eLife 2021, 10, e63877. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.; Calo, S.; D’Aurizio, R.; Lilja, A.; Pacchiani, N.; Merola, M. Recombinant human cytomegalovirus (HCMV) RL13 binds human immunoglobulin G Fc. PLoS ONE 2012, 7, e50166. [Google Scholar] [CrossRef]
- Corrales-Aguilar, E.; Trilling, M.; Hunold, K.; Fiedler, M.; Le, V.T.; Reinhard, H.; Ehrhardt, K.; Merce-Maldonado, E.; Aliyev, E.; Zimmermann, A.; et al. Human cytomegalovirus Fcgamma binding proteins gp34 and gp68 antagonize Fcgamma receptors I, II and III. PLoS Pathog. 2014, 10, e1004131. [Google Scholar] [CrossRef]
- Bentley, K.; Statkute, E.; Murrell, I.; Fielding, C.; Antrobus, R.; Preston, H.; Kerr-Jones, L.; Cochrane, D.; Brizic, I.; Lehner, P.J.; et al. Whole Virion Proteomics of HCMV Reveals that gpUL141 is a Virion Component that Regulates Envelope Glycoprotein Composition to Protect Against Humoral Immunity. bioRxiv, 2024. [Google Scholar] [CrossRef]
- Davies, E.L.; Noor, M.; Lim, E.Y.; Houldcroft, C.J.; Okecha, G.; Atkinson, C.; Reeves, M.B.; Jackson, S.E.; Wills, M.R. HCMV carriage in the elderly diminishes anti-viral functionality of the adaptive immune response resulting in virus replication at peripheral sites. Front. Immunol. 2022, 13, 1083230. [Google Scholar] [CrossRef]
- Germer, M.; Herbener, P.; Schuttrumpf, J. Functional Properties of Human Cytomegalovirus Hyperimmunoglobulin and Standard Immunoglobulin Preparations. Ann. Transplant. 2016, 21, 558–564. [Google Scholar] [CrossRef]
- Patel, H.D.; Nikitin, P.; Gesner, T.; Lin, J.J.; Barkan, D.T.; Ciferri, C.; Carfi, A.; Akbarnejad Yazdi, T.; Skewes-Cox, P.; Wiedmann, B.; et al. in vitro Characterization of Human Cytomegalovirus-Targeting Therapeutic Monoclonal Antibodies LJP538 and LJP539. Antimicrob. Agents Chemother. 2016, 60, 4961–4971. [Google Scholar] [CrossRef]
- Freed, D.C.; Tang, Q.; Tang, A.; Li, F.; He, X.; Huang, Z.; Meng, W.; Xia, L.; Finnefrock, A.C.; Durr, E.; et al. Pentameric complex of viral glycoprotein H is the primary target for potent neutralization by a human cytomegalovirus vaccine. Proc. Natl. Acad. Sci. USA 2013, 110, E4997–E5005. [Google Scholar] [CrossRef]
- Macagno, A.; Bernasconi, N.L.; Vanzetta, F.; Dander, E.; Sarasini, A.; Revello, M.G.; Gerna, G.; Sallusto, F.; Lanzavecchia, A. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J. Virol. 2010, 84, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Monroe, J.; Linton, C.; Archer, J.; Beard, C.W.; Barnett, S.W.; Palladino, G.; Mason, P.W.; Carfi, A.; Lilja, A.E. Human cytomegalovirus gH/gL/UL128/UL130/UL131A complex elicits potently neutralizing antibodies in mice. Vaccine 2014, 32, 3796–3804. [Google Scholar] [CrossRef] [PubMed]
- Wussow, F.; Chiuppesi, F.; Martinez, J.; Campo, J.; Johnson, E.; Flechsig, C.; Newell, M.; Tran, E.; Ortiz, J.; La Rosa, C.; et al. Human Cytomegalovirus Vaccine Based on the Envelope gH/gL Pentamer Complex. PLoS Pathog. 2014, 10, e1004524. [Google Scholar] [CrossRef] [PubMed]
- Wussow, F.; Yue, Y.; Martinez, J.; Deere, J.D.; Longmate, J.; Herrmann, A.; Barry, P.A.; Diamond, D.J. A vaccine based on the rhesus cytomegalovirus UL128 complex induces broadly neutralizing antibodies in rhesus macaques. J. Virol. 2013, 87, 1322–1332. [Google Scholar] [CrossRef]
- Kabanova, A.; Perez, L.; Lilleri, D.; Marcandalli, J.; Agatic, G.; Becattini, S.; Preite, S.; Fuschillo, D.; Percivalle, E.; Sallusto, F.; et al. Antibody-driven design of a human cytomegalovirus gHgLpUL128L subunit vaccine that selectively elicits potent neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2014, 111, 17965–17970. [Google Scholar] [CrossRef]
- Barten, M.J.; Baldanti, F.; Staus, A.; Hüber, C.M.; Glynou, K.; Zuckermann, A. Effectiveness of Prophylactic Human Cytomegalovirus Hyperimmunoglobulin in Preventing Cytomegalovirus Infection following Transplantation: A Systematic Review and Meta-Analysis. Life 2022, 12, 361. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Stanton, A.; McCarrell, E.; Smith, C.; Osman, M.; Harber, M.; Davenport, A.; Jones, G.; Wheeler, D.C.; O’Beirne, J.; et al. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: A phase 2 randomised placebo-controlled trial. Lancet 2011, 377, 1256–1263. [Google Scholar] [CrossRef]
- Cui, X.; Meza, B.P.; Adler, S.P.; McVoy, M.A. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine 2008, 26, 5760–5766. [Google Scholar] [CrossRef]
- Fierro, C.; Brune, D.; Shaw, M.; Schwartz, H.; Knightly, C.; Lin, J.; Carfi, A.; Natenshon, A.; Kalidindi, S.; Reuter, C.; et al. Safety and Immunogenicity of a Messenger RNA-Based Cytomegalovirus Vaccine in Healthy Adults: Results From a Phase 1 Randomized Clinical Trial. J. Infect. Dis. 2024, 230, e668–e678. [Google Scholar] [CrossRef]
- Nelson, C.S.; Huffman, T.; Jenks, J.A.; Cisneros de la Rosa, E.; Xie, G.; Vandergrift, N.; Pass, R.F.; Pollara, J.; Permar, S.R. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc. Natl. Acad. Sci. USA 2018, 115, 6267–6272. [Google Scholar] [CrossRef]
- Dorfman, J.R.; Balla, S.R.; Pathirana, J.; Groome, M.J.; Madhi, S.A.; Moore, P.L. In Utero Human Cytomegalovirus Infection Is Associated With Increased Levels of Putatively Protective Maternal Antibodies in Nonprimary Infection: Evidence for Boosting but Not Protection. Clin. Infect. Dis. 2021, 73, e981–e987. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Chin, A.L.; Liu, J.; Jardetzky, T.S.; Mudd, J.O.; Orloff, S.L.; Streblow, D.; Mussi-Pinhata, M.M.; Yamamoto, A.Y.; Duarte, G.; et al. HCMV trimer- and pentamer-specific antibodies synergize for virus neutralization but do not correlate with congenital transmission. Proc. Natl. Acad. Sci. USA 2019, 116, 3728–3733. [Google Scholar] [CrossRef]
- Hughes, B.L.; Clifton, R.G.; Rouse, D.J.; Saade, G.R.; Dinsmoor, M.J.; Reddy, U.M.; Pass, R.; Allard, D.; Mallett, G.; Fette, L.M.; et al. A Trial of Hyperimmune Globulin to Prevent Congenital Cytomegalovirus Infection. N. Engl. J. Med. 2021, 385, 436–444. [Google Scholar] [CrossRef]
- Revello, M.G.; Lazzarotto, T.; Guerra, B.; Spinillo, A.; Ferrazzi, E.; Kustermann, A.; Guaschino, S.; Vergani, P.; Todros, T.; Frusca, T.; et al. A Randomized Trial of Hyperimmune Globulin to Prevent Congenital Cytomegalovirus. N. Engl. J. Med. 2014, 370, 1316–1326. [Google Scholar] [CrossRef]
- Maertens, J.; Logan, A.C.; Jang, J.; Long, G.; Tang, J.L.; Hwang, W.Y.K.; Koh, L.P.; Chemaly, R.; Gerbitz, A.; Winkler, J.; et al. Phase 2 Study of Anti-Human Cytomegalovirus Monoclonal Antibodies for Prophylaxis in Hematopoietic Cell Transplantation. Antimicrob. Agents Chemother. 2020, 64, 10.1128. [Google Scholar] [CrossRef] [PubMed]
- Ishida, J.H.; Patel, A.; Mehta, A.K.; Gatault, P.; McBride, J.M.; Burgess, T.; Derby, M.A.; Snydman, D.R.; Emu, B.; Feierbach, B.; et al. Phase 2 Randomized, Double-Blind, Placebo-Controlled Trial of RG7667, a Combination Monoclonal Antibody, for Prevention of Cytomegalovirus Infection in High-Risk Kidney Transplant Recipients. Antimicrob. Agents Chemother. 2017, 61, e01794-16. [Google Scholar] [CrossRef] [PubMed]
- Jabs, D.A.; Gilpin, A.M.K.; Min, Y.-I.; Erice, A.; Kempen, J.H.; Quinn, T.C.; Studies of Ocular Complications of AIDS Research Group. HIV and cytomegalovirus viral load and clinical outcomes in AIDS and cytomegalovirus retinitis patients: Monoclonal Antibody Cytomegalovirus Retinitis Trial. AIDS 2002, 16, 877–887. [Google Scholar] [CrossRef]
- Kagan, K.O.; Enders, M.; Schampera, M.S.; Baeumel, E.; Hoopmann, M.; Geipel, A.; Berg, C.; Goelz, R.; De Catte, L.; Wallwiener, D.; et al. Prevention of maternal-fetal transmission of cytomegalovirus after primary maternal infection in the first trimester by biweekly hyperimmunoglobulin administration. Ultrasound Obs. Gynecol. 2019, 53, 383–389. [Google Scholar] [CrossRef]
- Dargan, D.J.; Douglas, E.; Cunningham, C.; Jamieson, F.; Stanton, R.J.; Baluchova, K.; McSharry, B.P.; Tomasec, P.; Emery, V.C.; Percivalle, E.; et al. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J. Gen. Virol. 2010, 91, 1535–1546. [Google Scholar] [CrossRef]
- Ziemann, M.; Hennig, H. Prevention of Transfusion-Transmitted Cytomegalovirus Infections: Which is the Optimal Strategy? Transfus. Med. Hemother. 2014, 41, 40–44. [Google Scholar] [CrossRef]
- Jackson, J.W.; Sparer, T. There Is Always Another Way! Cytomegalovirus’ Multifaceted Dissemination Schemes. Viruses 2018, 10, 383. [Google Scholar] [CrossRef] [PubMed]
- Sattentau, Q. Avoiding the void: Cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008, 6, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Murrell, I.; Bedford, C.; Ladell, K.; Miners, K.L.; Price, D.A.; Tomasec, P.; Wilkinson, G.W.G.; Stanton, R.J. The pentameric complex drives immunologically covert cell–cell transmission of wild-type human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2017, 114, 6104–6109. [Google Scholar] [CrossRef]
- Hu, X.; Karthigeyan, K.P.; Herbek, S.; Valencia, S.M.; Jenks, J.A.; Webster, H.; Miller, I.G.; Connors, M.; Pollara, J.; Andy, C.; et al. Human Cytomegalovirus mRNA-1647 Vaccine Candidate Elicits Potent and Broad Neutralization and Higher Antibody-Dependent Cellular Cytotoxicity Responses Than the gB/MF59 Vaccine. J. Infect. Dis. 2024, 230, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Weekes, M.P.; Tomasec, P.; Huttlin, E.L.; Fielding, C.A.; Nusinow, D.; Stanton, R.J.; Wang, E.C.Y.; Aicheler, R.; Murrell, I.; Wilkinson, G.W.G.; et al. Quantitative temporal viromics: An approach to investigate host-pathogen interaction. Cell 2014, 157, 1460–1472. [Google Scholar] [CrossRef]
- Vlahava, V.M.; Murrell, I.; Zhuang, L.; Aicheler, R.J.; Lim, E.; Miners, K.L.; Ladell, K.; Suarez, N.M.; Price, D.A.; Davison, A.J.; et al. Monoclonal antibodies targeting nonstructural viral antigens can activate ADCC against human cytomegalovirus. J. Clin. Investig. 2021, 131, e139296. [Google Scholar] [CrossRef]
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010. [Google Scholar] [CrossRef]
- Semmes, E.C.; Miller, I.G.; Rodgers, N.; Phan, C.T.; Hurst, J.H.; Walsh, K.M.; Stanton, R.J.; Pollara, J.; Permar, S.R. ADCC-activating antibodies correlate with decreased risk of congenital human cytomegalovirus transmission. JCI Insight 2023, 8, e167768. [Google Scholar] [CrossRef]
- Jennewein, M.F.; Goldfarb, I.; Dolatshahi, S.; Cosgrove, C.; Noelette, F.J.; Krykbaeva, M.; Das, J.; Sarkar, A.; Gorman, M.J.; Fischinger, S.; et al. Fc Glycan-Mediated Regulation of Placental Antibody Transfer. Cell 2019, 178, 202–215.e214. [Google Scholar] [CrossRef]
- Martinez, D.R.; Fong, Y.; Li, S.H.; Yang, F.; Jennewein, M.F.; Weiner, J.A.; Harrell, E.A.; Mangold, J.F.; Goswami, R.; Seage, G.R., III; et al. Fc Characteristics Mediate Selective Placental Transfer of IgG in HIV-Infected Women. Cell 2019, 178, 190–201.e111. [Google Scholar] [CrossRef]
- Semmes, E.C.; Nettere, D.R.; Nelson, A.N.; Hurst, J.H.; Cain, D.W.; Burt, T.D.; Kurtzberg, J.; Reeves, R.K.; Coyne, C.B.; Fouda, G.G.; et al. In utero human cytomegalovirus infection expands NK-like FcγRIII+CD8+ T cells that mediate Fc antibody functions. J. Clin. Investig. 2025, 135, e181342. [Google Scholar] [CrossRef]
- Sinclair, J.; Reeves, M. The intimate relationship between human cytomegalovirus and the dendritic cell lineage. Front. Microbiol. 2014, 5, 389. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Sinclair, J. Sleepless latency of human cytomegalovirus. Med. Microbiol. Immunol. 2015, 204, 421–429. [Google Scholar] [CrossRef]
- Pereira, B.I.; De Maeyer, R.P.H.; Covre, L.P.; Nehar-Belaid, D.; Lanna, A.; Ward, S.; Marches, R.; Chambers, E.S.; Gomes, D.C.O.; Riddell, N.E.; et al. Sestrins induce natural killer function in senescent-like CD8(+) T cells. Nat. Immunol. 2020, 21, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Houldcroft, C.J.; Jackson, S.E.; Lim, E.Y.; Sedikides, G.X.; Davies, E.L.; Atkinson, C.; McIntosh, M.; Remmerswaal, E.B.M.; Okecha, G.; Bemelman, F.J.; et al. Assessing Anti-HCMV Cell Mediated Immune Responses in Transplant Recipients and Healthy Controls Using a Novel Functional Assay. Front. Cell Infect. Microbiol. 2020, 10, 275. [Google Scholar] [CrossRef] [PubMed]
- Rubina, A.; Patel, M.; Nightingale, K.; Potts, M.; Fielding, C.A.; Kollnberger, S.; Lau, B.; Ladell, K.; Miners, K.L.; Nichols, J.; et al. ADAM17 targeting by human cytomegalovirus remodels the cell surface proteome to simultaneously regulate multiple immune pathways. Proc. Natl. Acad. Sci. USA 2023, 120, e2303155120. [Google Scholar] [CrossRef]
- Venturini, C.; Breuer, J. Cytomegalovirus Genetic Diversity and Evolution: Insights into Genotypes and Their Role in Viral Pathogenesis. Pathogens 2025, 14, 50. [Google Scholar] [CrossRef]
- Sijmons, S.; Thys, K.; Mbong Ngwese, M.; Van Damme, E.; Dvorak, J.; Van Loock, M.; Li, G.; Tachezy, R.; Busson, L.; Aerssens, J.; et al. High-throughput analysis of human cytomegalovirus genome diversity highlights the widespread occurrence of gene-disrupting mutations and pervasive recombination. J. Virol. 2015, 89, 7673–7695. [Google Scholar] [CrossRef]
- Suarez, N.M.; Wilkie, G.S.; Hage, E.; Camiolo, S.; Holton, M.; Hughes, J.; Maabar, M.; Vattipally, S.B.; Dhingra, A.; Gompels, U.A.; et al. Human Cytomegalovirus Genomes Sequenced Directly From Clinical Material: Variation, Multiple-Strain Infection, Recombination, and Gene Loss. J. Infect. Dis. 2019, 220, 781–791. [Google Scholar] [CrossRef]
- Semmes, E.C.; Miller, I.G.; Wimberly, C.E.; Phan, C.T.; Jenks, J.A.; Harnois, M.J.; Berendam, S.J.; Webster, H.; Hurst, J.H.; Kurtzberg, J.; et al. Maternal Fc-mediated non-neutralizing antibody responses correlate with protection against congenital human cytomegalovirus infection. J. Clin. Investig. 2022, 132, e156827. [Google Scholar] [CrossRef]
- Baraniak, I.; Kropff, B.; Ambrose, L.; McIntosh, M.; McLean, G.R.; Pichon, S.; Atkinson, C.; Milne, R.S.B.; Mach, M.; Griffiths, P.D.; et al. Protection from cytomegalovirus viremia following glycoprotein B vaccination is not dependent on neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2018, 115, 6273–6278. [Google Scholar] [CrossRef] [PubMed]
- Semmes, E.C.; Permar, S.R. Human Cytomegalovirus Infection Primes Fetal Natural Killer Cells for Fc-Mediated Antiviral Defense. J. Infect. Dis. 2023, 227, 739–741. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HCMV-Encoded Protein/microRNA | Cellular Target | Cellular Receptor | Mechanism | References |
|---|---|---|---|---|
| gpUL18 | LIR-1/LILRB1 | LIR-1/LILRB1 | Direct binding to transmit an inhibitory signal | [98] |
| spUL40 | HLA-E | CD94/NKG2A (CD94/NKG2C) | Up-regulation of surface HLA-E to transmit an inhibitory signal (NKG2C is the paired activating receptor) | [111,112,113] |
| UL16 | MICB, ULBP1, ULBP2, ULBP5 | NKG2D | Retains specific NKG2D ligands in the ER | [128,129,130,131] |
| UL142 | MICA, ULBP3 | NKG2D | Retains specific NKG2D ligands in the trans-Golgi | [137,138,139,140] |
| miR-UL112 | MICB | NKG2D | Prevents MICB mRNA translation | [132] |
| US18 and US20 | MICA | NKG2D | Targets MICA for lysosomal degradation | [126] |
| US12 | ULBP2 | NKG2D | Targets ULBP2 for lysosomal degradation, involvement of UL16? | [141] |
| UL148A | MICA | NKG2D | Targets MICA for lysosomal degradation | [142] |
| US9 | MICA*008 | NKG2D | Targets MICA*008 through its GPI anchor | [143] |
| UL147A | MICA*008 | NKG2D | Inhibits MICA*008 maturation and proteasomal degradation | [146] |
| UL141 | CD155 | DNAM1 | Retains CD155 within the ER | [149] |
| UL141 and US2 | CD112 | DNAM1 | Targets CD112 for proteasomal degradation | [150,151] |
| US18 and US20 | B7-H6 | NKp30 | Target B7-H6 for proteasomal degradation | [141,157] |
| UL83 | NKp30 | NKp30 | Direct binding to inhibit the activating signal | [155] |
| UL141 | TRAIL-R | TRAIL-R | Retains TRAIL-R within the ER to prevent the induction of apoptosis | [159] |
| UL4 | TRAIL | TRAIL-R | Binds to TRAIL with high affinity to prevent pro-apoptotic signalling and NK activation | [161] |
| Unknown | CD95 | CD95 | Removes CD95 from the cell surface | [163] |
| UL36 | Inhibitor of apoptosis | [160] | ||
| UL135 | Actin polymerization | N/A | Prevents actin polymerization in the infected cell, thereby inhibiting the immunological synapse | [182] |
| UL148 | CD58 | CD2 | Retains CD58 within the ER, preventing immunological synapse formation | [183] |
| UL141 and US2 | Integrins | Targets various integrins for proteasomal degradation, inhibiting cell adhesion | [151] | |
| Unknown | HLA-C-peptide | KIR2DL1 | Contains a peptide which, when loaded onto HLA-C, triggers KIR2DL1 signalling | [180] |
| RL11 | Fc portion of antibody | Anti-HCMV antibodies/CD16 | Binds to Fc portion of antibody and prevents Fc-mediated signalling | [186] |
| RL12 | Fc portion of antibody | Anti-HCMV antibodies/CD16 | Binds to Fc portion of antibody and prevents Fc-mediated signalling | [185] |
| RL13 | Fc portion of antibody | Anti-HCMV antibodies/CD16 | Binds to Fc portion of antibody and prevents Fc-mediated signalling | [185] |
| UL119-UL118 | Fc portion of antibody | Anti-HCMV antibodies/CD16 | Binds to Fc portion of antibody and prevents Fc-mediated signalling | [186] |
| UL141 | Viral glycoproteins | Anti-glycoprotein antibodies/CD16 | Restricts cell surface expression of glycoprotein complexes, limiting the effect of anti-glycoprotein antibodies | [187] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preston, H.; Casey, R.; Ferris, E.; Kerr-Jones, L.; Jones, L.; Latif, F.; Clement, M.; Aicheler, R.J.; Wang, E.C.Y.; Stanton, R.J.; et al. Human Cytomegalovirus Immune Evasion of Natural Killer Cells: A Virus for All Seasons? Pathogens 2025, 14, 629. https://doi.org/10.3390/pathogens14070629
Preston H, Casey R, Ferris E, Kerr-Jones L, Jones L, Latif F, Clement M, Aicheler RJ, Wang ECY, Stanton RJ, et al. Human Cytomegalovirus Immune Evasion of Natural Killer Cells: A Virus for All Seasons? Pathogens. 2025; 14(7):629. https://doi.org/10.3390/pathogens14070629
Chicago/Turabian StylePreston, Hannah, Rowan Casey, Elizabeth Ferris, Lauren Kerr-Jones, Lauren Jones, Farah Latif, Mathew Clement, Rebecca J. Aicheler, Eddie C. Y. Wang, Richard J. Stanton, and et al. 2025. "Human Cytomegalovirus Immune Evasion of Natural Killer Cells: A Virus for All Seasons?" Pathogens 14, no. 7: 629. https://doi.org/10.3390/pathogens14070629
APA StylePreston, H., Casey, R., Ferris, E., Kerr-Jones, L., Jones, L., Latif, F., Clement, M., Aicheler, R. J., Wang, E. C. Y., Stanton, R. J., & Fielding, C. A. (2025). Human Cytomegalovirus Immune Evasion of Natural Killer Cells: A Virus for All Seasons? Pathogens, 14(7), 629. https://doi.org/10.3390/pathogens14070629