
3,3′-((3-Hydroxyphenyl)azanediyl)dipropionic Acid Derivatives as a Promising Scaffold Against Drug-Resistant Pathogens and Chemotherapy-Resistant Cancer


 , ,
, ,  , ,
, ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemical Synthesis
2.1.1. 3,3′-((3-Hydroxyphenyl)azanediyl)dipropionic Acid (2)
2.1.2. Dimethyl 3,3′-((3-hydroxyphenyl)azanediyl)dipropionate (3)
2.1.3. 3,3′-((3-Hydroxyphenyl)azanediyl)di(propanehydrazide) (4)
2.1.4. 1-(3-Hydroxyphenyl)dihydropyrimidine-2,4(1H,3H)-dione (5)
2.1.5. 1-(3-Hydroxyphenyl)-2-thioxotetrahydropyrimidin-4(1H)-one (6)
2.1.6. 3-(7-Hydroxy-4-oxo-3,4-dihydroquinolin-1(2H)-yl)propanoic Acid (7)
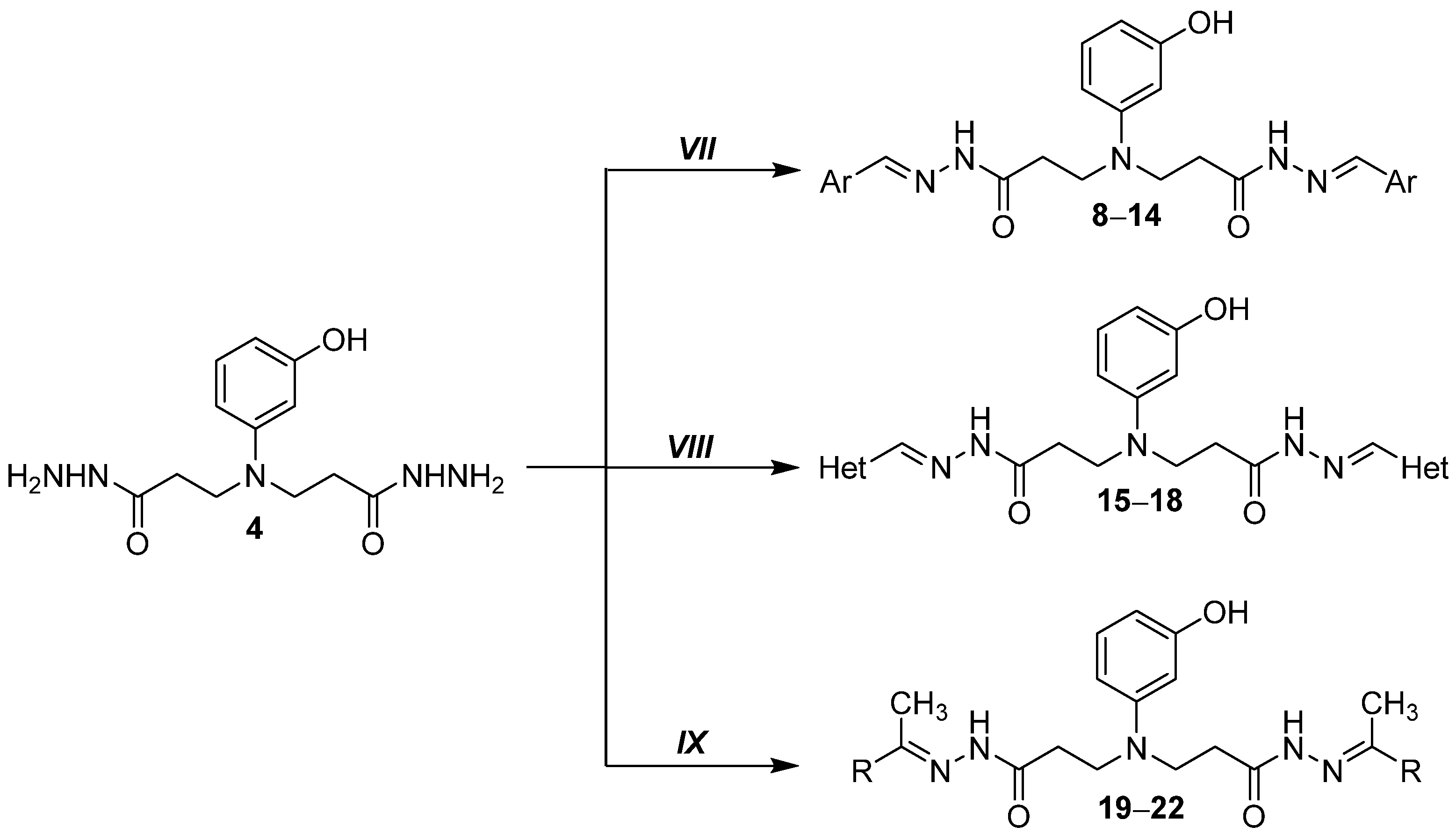
2.1.7. General Procedure for Preparation of Hydrazones 8–18
2.1.8. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(benzylidene)propanehydrazide) (8)
2.1.9. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(2,4-difluorobenzylidene)propanehydrazide) (9)
2.1.10. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(4-nitrobenzylidene)propanehydrazide) (10)
2.1.11. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(4-chlorobenzylidene)propanehydrazide) (11)
2.1.12. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(4-dimethylamino)benzylidene)propanehydrazide) (12)
2.1.13. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(3,4,5-trimethoxybenzylidene)propanehydrazide) (13)
2.1.14. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(naphthalen-1-ylmethylene)propanehydrazide) (14)
2.1.15. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(furan-2-ylmethylene)benzylidene)propanehydrazide) (15)
2.1.16. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-((5-nitrothiophen-2-yl)methylene)propanehydrazide) (16)
2.1.17. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(5-nitrofuran-2-yl)methylene)benzylidene)propanehydrazide) (17)
2.1.18. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(thiophen-3-yl)methylene)benzylidene)propanehydrazide) (18)
2.1.19. General Procedure for Preparation of Hydrazones 19–22
2.1.20. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(propan-2-ylidene)propanehydrazide) (19)
2.1.21. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(butan-2-ylidene)propanehydrazide) (20)
2.1.22. 3-((3-Hydroxyphenyl)(3-oxo-3-(2-(1-phenylethylidene)hydrazineyl)propyl)amino)-N′-(1-phenylethylidene)propanehydrazide (21)
2.1.23. 4,4′-(((3,3′-((3-Hydroxyphenyl)azanediyl)bis(propanoyl))bis(hydrazin-2-yl-1-ylidene))bis(ethan-1-yl-1-ylidene))dibenzenesulfonamide (22)
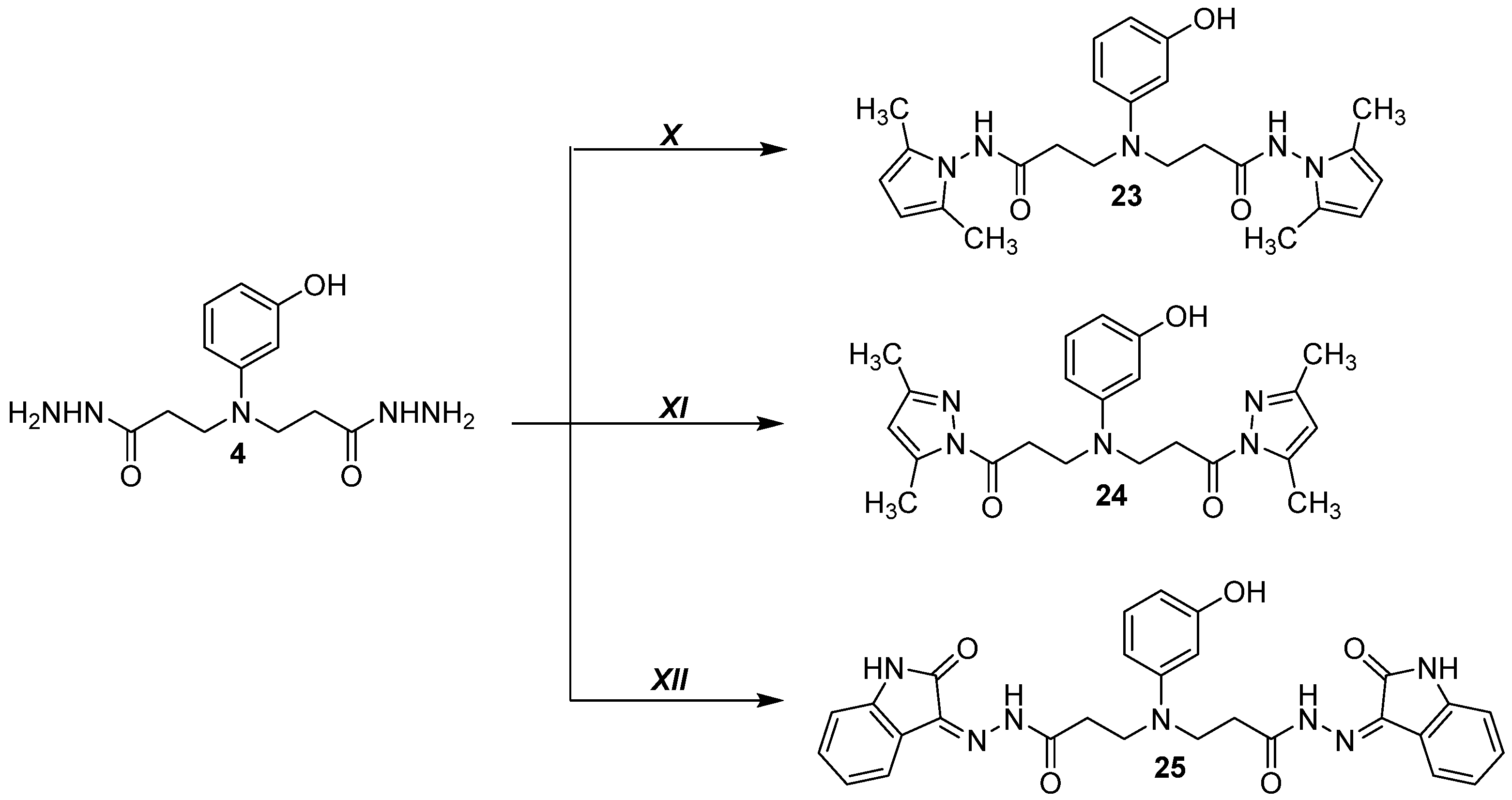
2.1.24. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N-(2,5-dimethyl-1H-pyrrol-1-yl)propanamide) (23)
2.1.25. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(1-(3,5-dimethyl-1H-pyrazol-1-yl)propan-1-one) (24)
2.1.26. 3,3′-((3-Hydroxyphenyl)azanediyl)bis(N′-(2-oxoindolin-3-ylidene)propanehydrazide (25)
2.2. Microbial Strains and Culture Conditions
2.3. Minimal Inhibitory Concentration Determination
2.4. Preparation of Test Compounds and Screening Libraries
2.5. Cell Lines and Culture Conditions
2.6. MTT-Based Cell Viability Assay
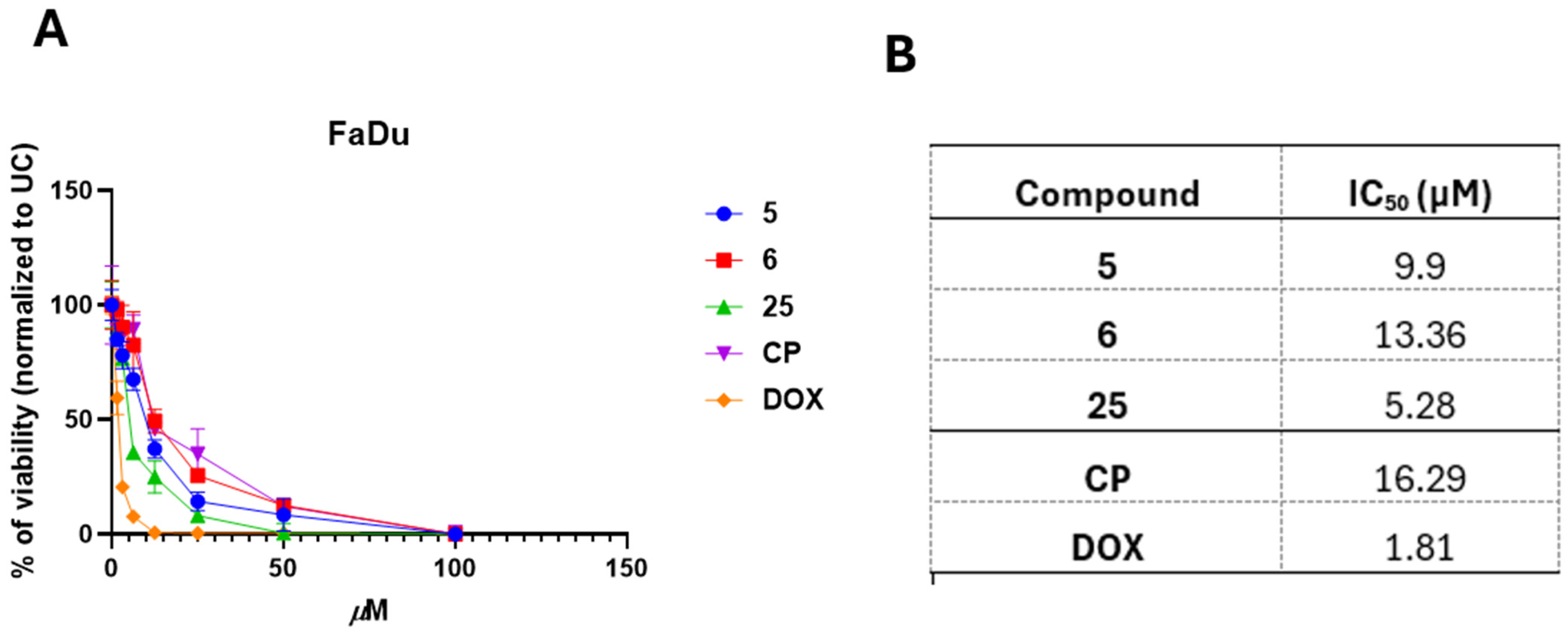
2.7. IC50 Determination
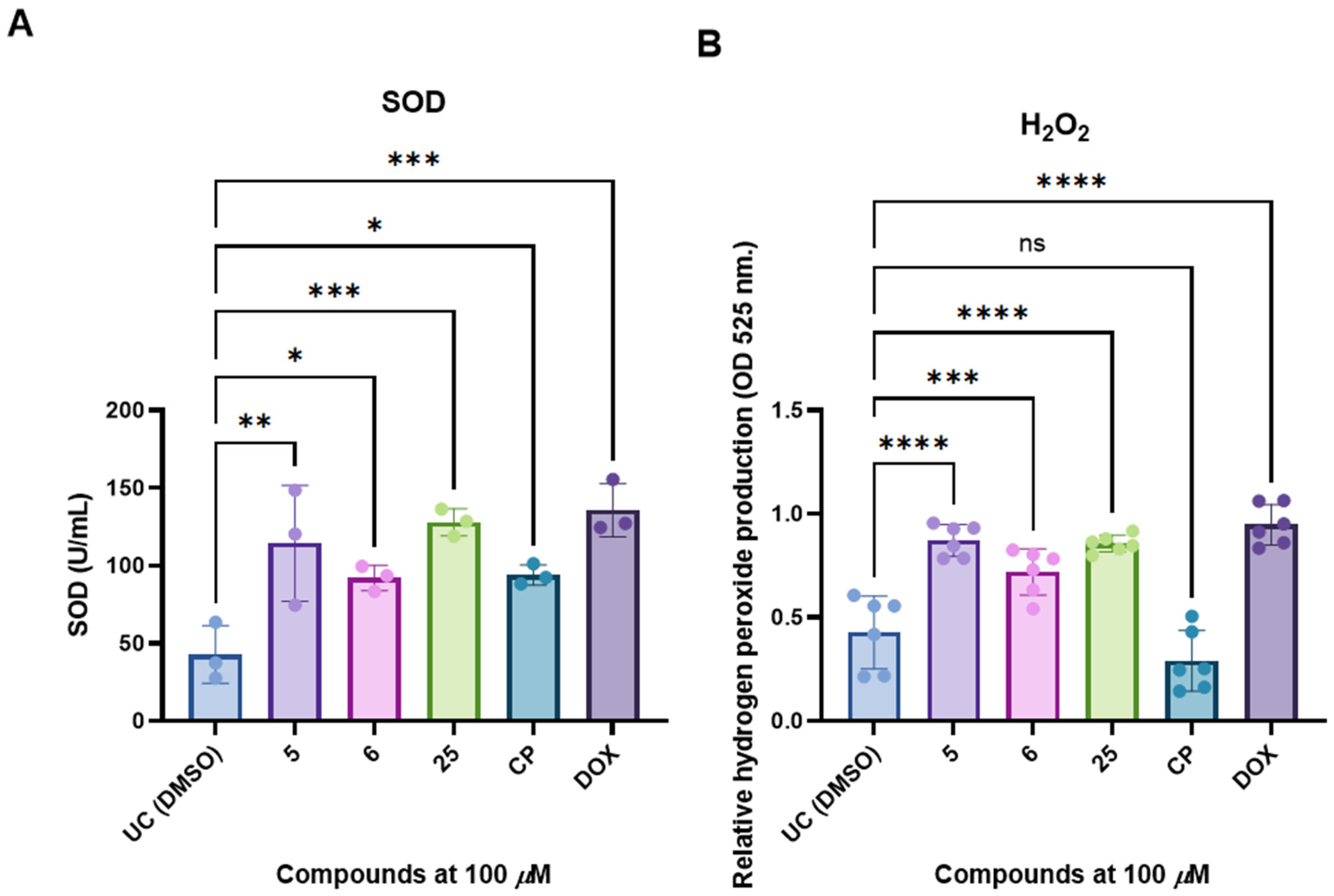
2.8. Superoxide Dismutase (SOD) Activity Determination
2.9. Hydrogen Peroxide Production Assay
2.10. In Silico Molecular Modeling
2.10.1. Receptor Preparation
2.10.2. Ligand Preparation
2.10.3. Docking of Ligand–Protein Interaction
2.11. Statistical Analysis
3. Results
3.1. Synthesis and Characterization of N-Aryl-Substituted β-Amino Acid Derivatives
3.2. 3,3′-((3-Hydroxyphenyl)azanediyl)dipropionic Acid Derivatives 2–25 Show Antimicrobial Activity Against Multidrug-Resistant Bacterial Pathogens
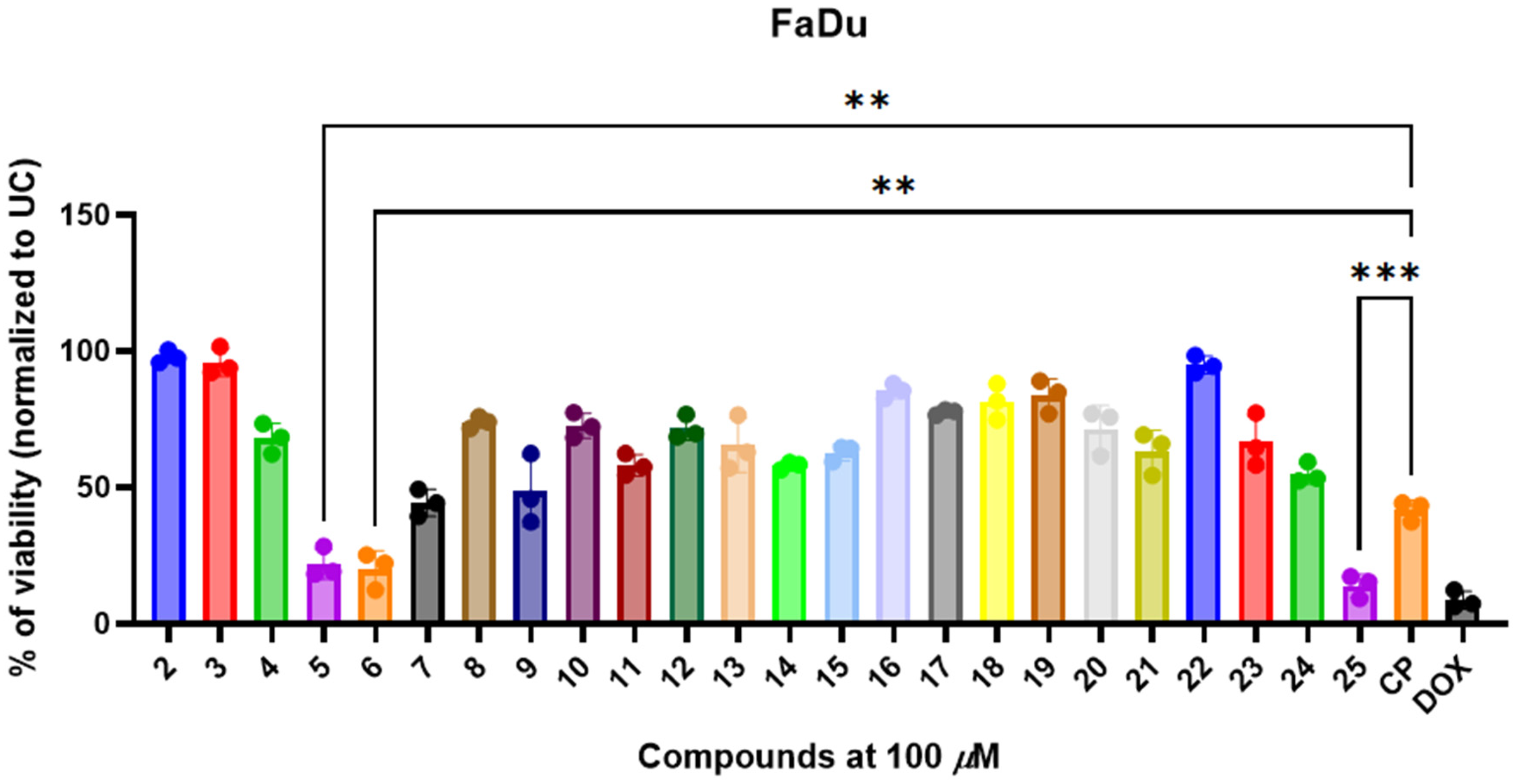
3.3. 3,3′-((3-Hydroxyphenyl)azanediyl)dipropionic Acid Derivatives 2–25 Demonstrate Structure-Dependent Anticancer Activity
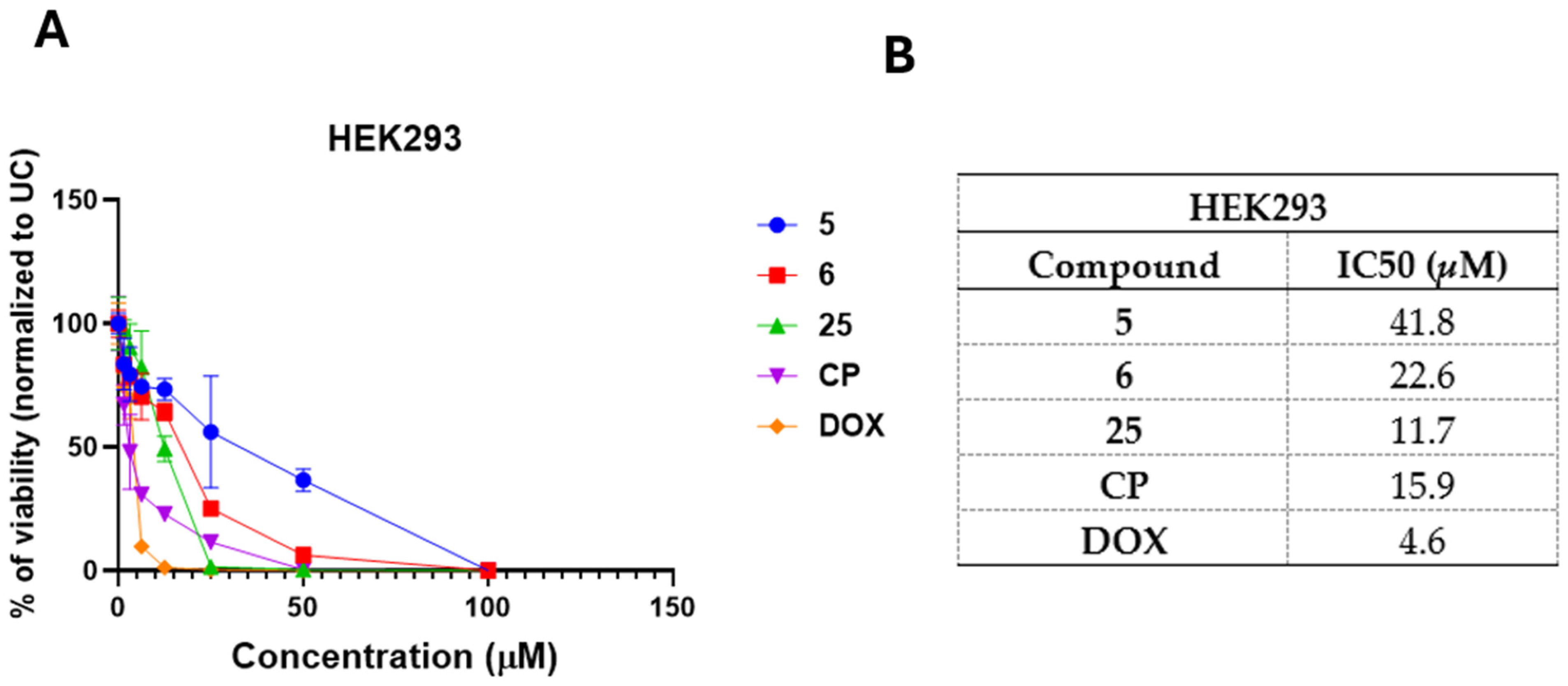
3.4. Selected and Most Promising 3,3′-((3-Hydroxyphenyl)azanediyl)dipropionic Acid Derivatives 5, 6, and 25 Are Able to Induce Cell Death and Oxidative Stress in FaDu Cells
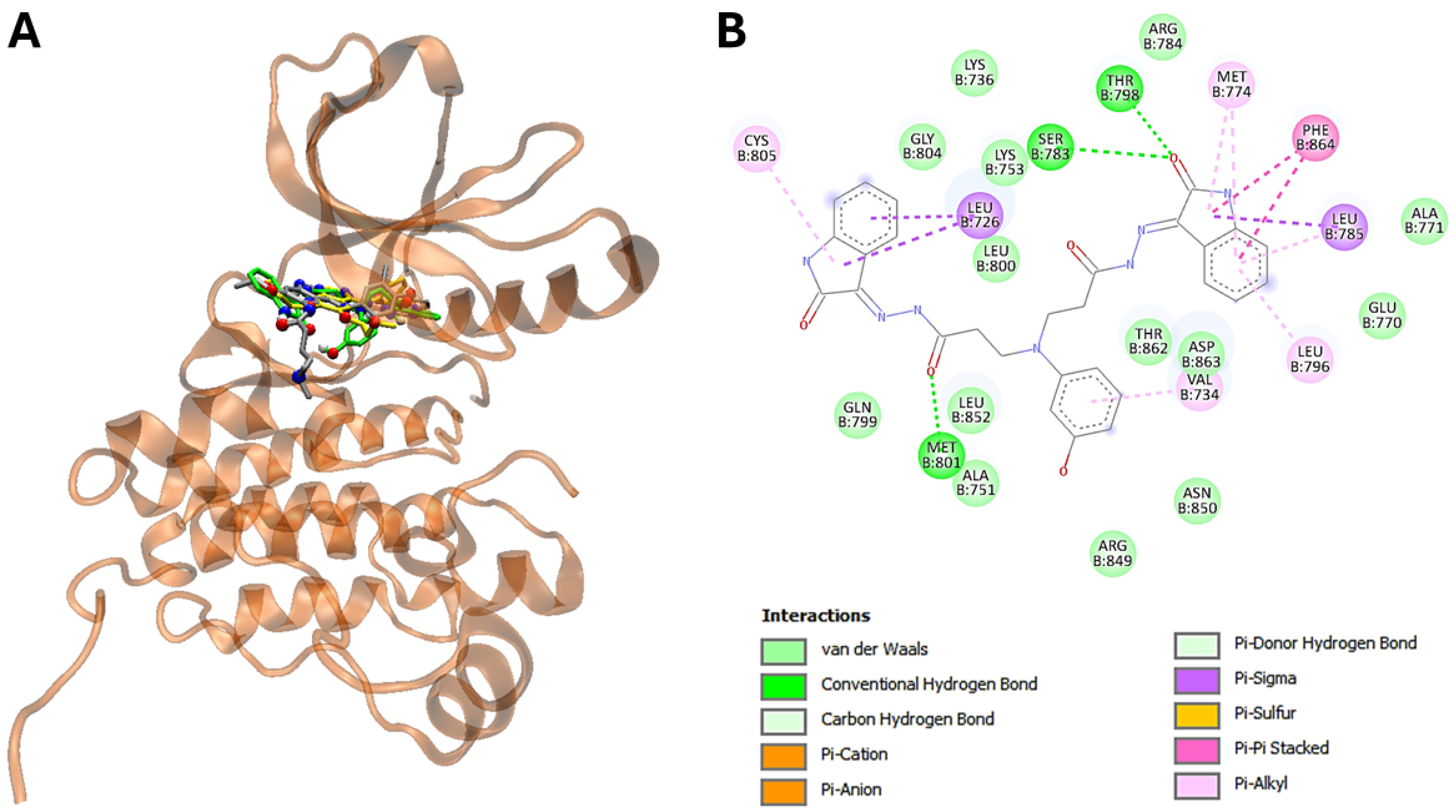
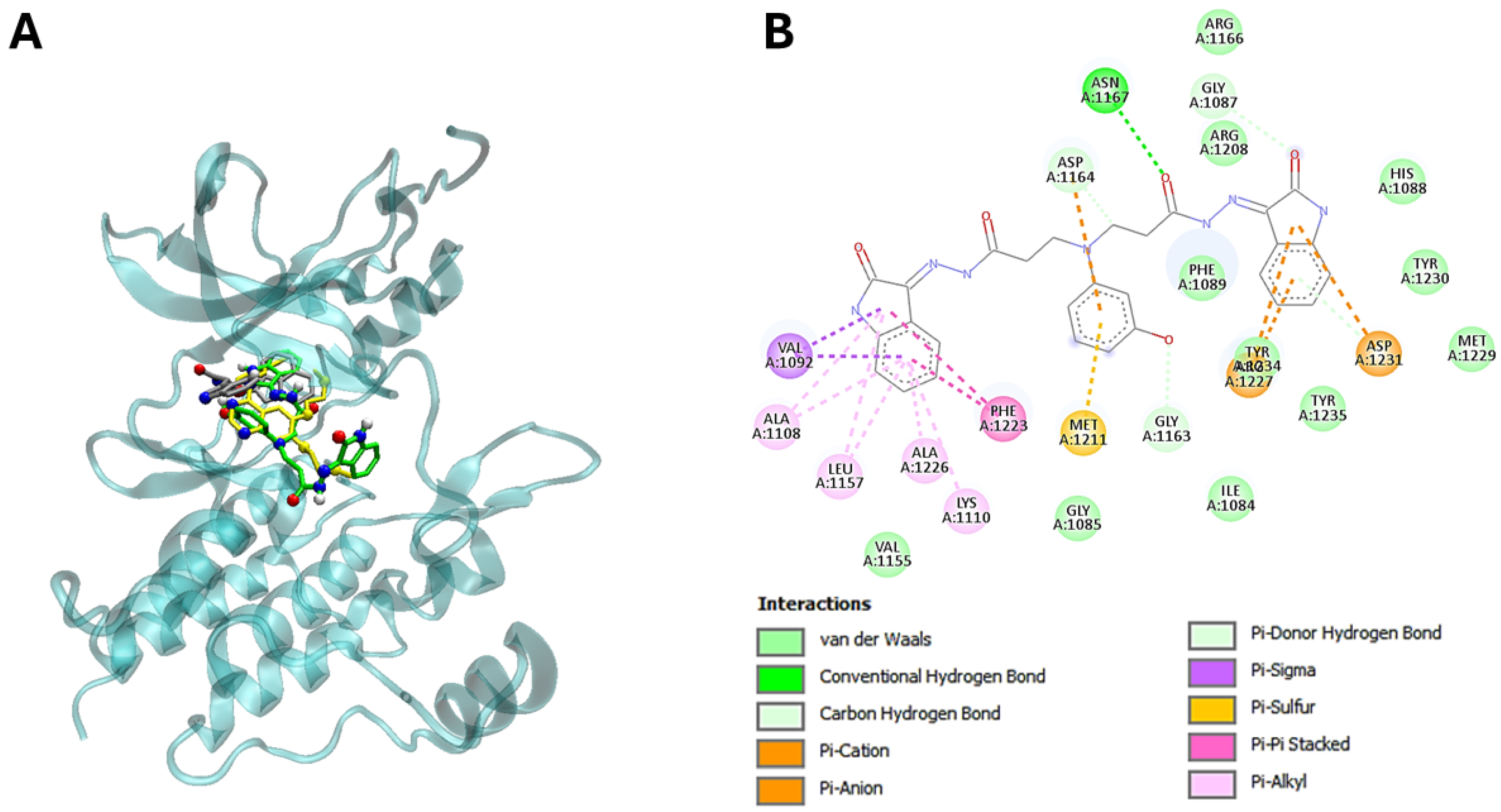
3.5. The Molecular Modeling of the Most Potent Compound (25) with Major Overexpressed Head and Neck Cancer Targets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Yang, L.; Huang, Y.; Jia, J.; Dou, T. Antibiotic resistance genes in bacteria: Occurrence, spread, and control. J. Basic Microbiol. 2021, 61, 1049–1070. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4, 485–511. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial resistance in Eskape pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Escolà-Vergé, L.; Los-Arcos, I.; Almirante, B. New antibiotics for the treatment of infections by multidrug-resistant microorganisms. Med. Clin. (Barc.) 2020, 154, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Li, J.; Sun, Z. The combination of antibiotic and non-antibiotic compounds improves antibiotic efficacy against multidrug-resistant bacteria. Int. J. Mol. Sci. 2023, 24, 15493. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Raj, S.; Kesari, K.K.; Kumar, A.; Rathi, B.; Sharma, A.; Gupta, P.K.; Jha, S.K.; Jha, N.K.; Slama, P.; Roychoudhury, S.; et al. Molecular mechanism(s) of regulation(s) of c-MET/HGF signaling in head and neck cancer. Mol. Cancer 2022, 21, 31. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, H. Induction chemotherapy in locally advanced head and neck cancers, is there a best choice? Crit. Rev. Oncol. Hematol. 2023, 186, 103986. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox. Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, T.; Yang, Y.; Chen, Z.; Wang, J.; Wang, X.; Zheng, Y.; Wang, C.; Wang, Y.; Zhu, Z.; Ding, X.; et al. TNFAIP2 confers cisplatin resistance in head and neck squamous cell carcinoma via KEAP1/NRF2 signaling. J. Exp. Clin. Cancer Res. 2023, 42, 190. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Griso, A.B.; Acero-Riaguas, L.; Castelo, B.; Cebrián-Carretero, J.L.; Sastre-Perona, A. Mechanisms of cisplatin resistance in HPV negative head and neck squamous cell carcinomas. Cells 2022, 11, 561. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hu, H.; Li, B.; Wang, J.; Tan, Y.; Xu, M.; Xu, W.; Lu, H. New advances into cisplatin resistance in head and neck squamous carcinoma: Mechanisms and therapeutic aspects. Biomed. Pharmacother. 2023, 163, 114778. [Google Scholar] [CrossRef] [PubMed]
- Otero, J.M.; Estévez, A.M.; Estévez, J.C.; Fleet, G.W.; Estévez, R.J. Highly functionalized cyclic and bicyclic β-amino acids from sugar β-nitroesters. Tetrahedron 2020, 76, 130837. [Google Scholar] [CrossRef]
- Fu, H.; Katsumura, Y.; Lin, M.; Muroya, Y.; Hata, K.; Fujii, K.; Yokoya, A.; Hatano, Y. Free radical scavenging and radioprotective effects of carnosine and anserine. Radiat. Phys. Chem. 2009, 78, 1192–1197. [Google Scholar] [CrossRef]
- Anusevičius, K.; Mickevičius, V.; Stasevych, M.; Zvarych, V.; Komarovska-Porokhnyavets, O.; Novikov, V.; Tarasova, O.; Gloriozova, T.; Poroikov, V. Synthesis and chemoinformatics analysis of N-aryl-β-alanine derivatives. Res. Chem. Intermed. 2015, 41, 7517–7540. [Google Scholar] [CrossRef]
- Escalante, J.; Hernández, A.L.; Juaristi, E. Highly diastereoselective addition of a racemic β-alanine enolate derivative to electrophiles. Rev. Soc. Quím. Méx. 2001, 45, 177–182. [Google Scholar]
- Adrover-Castellano, M.L.; Schmidt, J.J.; Sherman, D.H. biosynthetic cyclization catalysts for the assembly of peptide and polyketide natural products. Chem. Cat. Chem. 2021, 13, 2095–2116. [Google Scholar] [CrossRef]
- Cordero, F.M.; Giomi, D.; Machetti, F. Synthesis of 2-azetidinones via cycloaddition approaches: An update. Reactions 2024, 5, 492–566. [Google Scholar] [CrossRef]
- Li, Q.; Liu, Y.; Li, C. Enantioselective synthesis of unprotected 2-quinolinone-based cyclic amino acids via sequential palladium-catalyzed asymmetric allylation/desymmetrization. Chin. J. Biol. 2023, 47, 222–228. [Google Scholar] [CrossRef]
- Dangolani, S.K.; Niknam, E.; Shahraki, O.; Khalafi-Nezhad, A. Unexpected dihydropyridinium derivatives using a multicomponent reaction containing unprotected amino acids. J. Mol. Struct. 2021, 1245, 131061. [Google Scholar] [CrossRef]
- Amaghnouje, A.; Bohza, S.; Bohdan, N.; Es-Safi, I.; Kyrylchuk, A.; Achour, S.; Fatemi, H.E.; Bousta, D.; Grafov, A. New 2, 3-benzodiazepine derivative: Synthesis, activity on central nervous system, and toxicity study in mice. Pharmaceuticals 2021, 14, 814. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.E.F.; Moustafa, M.S.; El-Wahed, A.A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [PubMed]
- Barelli, L.; Behie, S.W.; Hu, S.; Bidochka, M.J. Profiling destruxin synthesis by specialist and generalist metarhizium insect pathogens during coculture with plants. Appl. Environ. Microbiol. 2022, 88, e02474-21. [Google Scholar] [CrossRef] [PubMed]
- Alvariño, R.; Alonso, E.; Tabudravu, J.N.; Pérez-Fuentes, N.; Alfonso, A.; Botana, L.M. Tavarua deoxyriboside A and jasplakinolide as potential neuroprotective agents: Effects on cellular models of oxidative stress and neuroinflammation. ACS Chem. Neurosci. 2021, 12, 150–162. [Google Scholar] [CrossRef]
- Berillo, D.A.; Dyusebaeva, M.A. Synthesis of hydrazides of heterocyclic amines and their antimicrobial and spasmolytic activity. Saudi Pharm. J. 2022, 30, 1036–1043. [Google Scholar] [CrossRef]
- Mali, S.N.; Thorat, B.R.; Gupta, D.R.; Pandey, A. Mini-review of the importance of hydrazides and their derivatives—Synthesis and biological activity. Eng. Proc. 2021, 11, 21. [Google Scholar]
- Rusu, A.; Moga, I.M.; Uncu, L. The role of five-membered heterocycles in the molecular structure of antibacterial drugs used in therapy. Pharmaceutics 2023, 15, 2554. [Google Scholar] [CrossRef]
- Petrova, K.T.; Potewar, T.M.; Correia-da-Silva, P.; Barros, M.T.; Calhelha, R.C.; Ćiric, A.; Ferreira, I.C. Antimicrobial and cytotoxic activities of 1, 2, 3-triazole-sucrose derivatives. Carbohydr. Res. 2015, 417, 66–71. [Google Scholar] [CrossRef] [PubMed]
- El Sadek, M.M.; El-Dayem, N.S.A.; Hassan, S.Y.; Mostafa, M.A.; Yacout, G.A. Antioxidant and antitumor activities of new synthesized aromatic C-nucleoside derivatives. Molecules 2014, 19, 5163–5190. [Google Scholar] [CrossRef]
- Thakkar, S.S.; Thakor, P.; Doshi, H.; Ray, A. 1, 2, 4-Triazole and 1, 3, 4-oxadiazole analogues: Synthesis, MO studies, in silico molecular docking studies, antimalarial as DHFR inhibitor and antimicrobial activities. Bioorg. Med. Chem. 2017, 25, 4064–4075. [Google Scholar] [CrossRef]
- Sahu, J.K.; Ganguly, S.; Kaushik, A. Triazoles: A valuable insight into recent developments and biological activities. Chin. J. Nat. Med. 2013, 11, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Grybaitė, B.; Vaickelionienė, R.; Stasevych, M.; Komarovska-Porokhnyavets, O.; Novikov, V.; Mickevičius, V. Synthesis, transformation of 3-[(4-arylthiazol-2-yl)-(p-tolyl)amino]propanoic acids, bis(thiazol-5-yl)phenyl-, bis(thiazol-5-yl)methane derivatives, and their antimicrobial activity. Heterocycles 2018, 96, 86–105. [Google Scholar]
- Abdel-Raheem, S.A.A.; El-Dean, A.M.K.; Abdul-Malik, M.A.; Marae, I.S.; Bakhite, E.A.; Hassanien, R.; Abd-Ella, A.A. Facile synthesis and pesticidal activity of substituted heterocyclic pyridine compounds. Rev. Roum. Chim. 2022, 67, 305–309. [Google Scholar]
- Voskienė, A.; Sapijanskaitė, B.; Mickevičius, V.; Kantminienė, K.; Stasevych, M.; Komarovska-Porokhnyavets, O.; Musyanovych, R.; Novikov, V. Synthesis, chemical properties, and antimicrobial activity of 2- and 2, 3-substituted [(tetrahydro-2, 4-dioxopyrimidin-1(2H)-yl)-phenoxy]naphthalene-1, 4-diones. Monatsh. Chem. 2011, 142, 529–537. [Google Scholar] [CrossRef]
- Mickevičius, V. Synthesis of 2, 3-dihydro-4(1H)-quinolinone derivatives. Chem. Heterocycl. Compd. 1996, 4, 523–526. [Google Scholar]
- Petraitis, V.; Petraitiene, R.; Kavaliauskas, P.; Naing, E.; Garcia, A.; Sutherland, C.; Kau, A.Y.; Goldner, N.; Bulow, C.; Nicolau, D.P.; et al. Pharmacokinetics, tissue distribution, and efficacy of VIO-001 (meropenem/piperacillin/tazobactam) for treatment of methicillin-resistant staphylococcus aureus bacteremia in immunocompetent rabbits with chronic indwelling vascular catheters. Antimicrob. Agents Chemother. 2021, 65, e0116821. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kavaliauskas, P.; Grybaite, B.; Mickevicius, V.; Petraitiene, R.; Grigaleviciute, R.; Planciuniene, R.; Gialanella, P.; Pockevicius, A.; Petraitis, V. Synthesis, ADMET properties, and In Vitro antimicrobial and antibiofilm activity of 5-Nitro-2-thiophenecarbaldehyde N-((E)-(5-Nitrothienyl)methylidene)hydrazone (KTU-286) against Staphylococcus aureus with Defined Resistance Mechanisms. Antibiotics 2020, 9, 612. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of cell viability by the MTT assay. Cold Spring Harb. Protoc. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 8, 235–242. [Google Scholar] [CrossRef]
- Fennessy, B.G.; Woolley, J.F.; Cotter, T.G. Head and neck surgery bFGF redox signaling in hypopharyngeal cancer. Otolaryngol.-Head Neck Surg. 2012, 147, 147–148. [Google Scholar] [CrossRef]
- Jing, P.; Zhou, S.; Xu, P.; Cui, P.; Liu, X.; Liu, X.; Liu, X.; Wang, H.; Xu, W. PDK1 promotes metastasis by inducing epithelial–mesenchymal transition in hypopharyngeal carcinoma via the Notch1 signaling pathway. Exp. Cell Res. 2020, 386, 111746. [Google Scholar] [CrossRef] [PubMed]
- Koenigs, M.B.; Lefranc-Torres, A.; Bonilla-Velez, J.; Patel, K.B.; Hayes, D.N.; Glomski, K.; Busse, P.M.; Chan, A.W.; Clark, J.R.; Deschler, D.G.; et al. Association of estrogen receptor alpha expression with survival in oropharyngeal cancer following chemoradiation therapy. J. Natl. Cancer Inst. 2019, 111, 933–942. [Google Scholar] [CrossRef]
- Peng, J.P.; Su, C.Y.; Chang, H.C.; Chai, C.Y.; Hung, W.C. Overexpression of cyclo-oxygenase 2 in squamous cell carcinoma of the hypopharynx. Hum. Pathol. 2002, 33, 100–104. [Google Scholar] [CrossRef]
- Peng, X.; Liu, Y.; Zhu, S.; Peng, X.; Li, H.; Jiao, W.; Lin, P.; Zhang, Z.; Qiu, Y.; Jin, M.; et al. Co-targeting PI3K/Akt and MAPK/ERK pathways leads to an enhanced antitumor effect on human hypopharyngeal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2019, 145, 2921–2936. [Google Scholar] [CrossRef] [PubMed]
- Pollock, N.I.; Grandis, J.R. HER2 as a Therapeutic Target in Head and Neck Squamous Cell Carcinoma Netanya. Clin. Cancer Res. 2015, 21, 526–533. [Google Scholar] [CrossRef]
- Ravi, D.K.; Kumar, V.; Kumar, M.; Singh, G.; Rai, S.B.; Saxena, A.K.; Pandey, M. Expression of vascular endothelial growth factors (VEGF) in head and neck squamous cell carcinoma and adjacent normal tissue. World J. Pathol. 2012, 1. [Google Scholar]
- Song, P.N.; Lynch, S.E.; DeMellier, C.T.; Mansur, A.; Gallegos, C.A.; Wright, B.D.; Hartman, Y.E.; Minton, L.E.; Lapi, S.E.; Warram, J.M.; et al. Dual anti-HER2/EGFR inhibition synergistically increases therapeutic effects and alters tumor oxygenation in HNSCC. Sci. Rep. 2024, 14, 3771. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.M.; Kim, S.-A.; Park, Y.-L.; Lee, K.-H.; Sung, M.-W.; Lee, J.K.; Lim, S.C.; Chung, I.-J.; Joo, Y.-E. Expression of the receptor tyrosine kinase recepteur d’origine nantais and its association with tumor progression in hypopharyngeal cancer. Head Neck 2013, 35, 1106–1113. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2014, 4, 17. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | S. aureus TCH 1516 1 | E. faecalis AR-0671 2 | E. coli AR-0001 3 | K. pneumoniae AR-0003 4 | P. aeruginosa AR-1114 5 | A. baumannii AR-0273 6 |
|---|---|---|---|---|---|---|
| 2 | >64 | >64 | >64 | >64 | >64 | >64 |
| 3 | >64 | >64 | 32 | >64 | >64 | >64 |
| 4 | 8 | >64 | >64 | >64 | >64 | >64 |
| 5 | 16 | 32 | >64 | >64 | >64 | >64 |
| 6 | 16 | 32 | >64 | >64 | >64 | >64 |
| 7 | >64 | >64 | >64 | >64 | >64 | >64 |
| 8 | >64 | >64 | >64 | >64 | >64 | >64 |
| 9 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10 | 4 | 16 | >64 | >64 | >64 | >64 |
| 11 | 64 | >64 | >64 | >64 | >64 | >64 |
| 12 | 64 | >64 | >64 | 64 | 64 | 64 |
| 13 | >64 | >64 | >64 | >64 | >64 | >64 |
| 14 | 2 | 8 | 64 | >64 | >64 | >64 |
| 15 | >64 | >64 | >64 | >64 | 64 | 64 |
| 16 | 1 | 4 | 32 | >64 | >64 | >64 |
| 17 | 16 | >64 | >64 | >64 | >64 | >64 |
| 18 | >64 | >64 | >64 | >64 | >64 | >64 |
| 19 | >64 | >64 | >64 | >64 | >64 | >64 |
| 20 | 16 | 32 | >64 | >64 | >64 | >64 |
| 21 | 32 | 32 | >64 | >64 | >64 | >64 |
| 22 | 8 | 32 | 16 | >64 | >64 | >64 |
| 23 | 64 | >64 | >64 | >64 | >64 | >64 |
| 24 | >64 | >64 | >64 | >64 | >64 | >64 |
| 25 | 32 | 16 | 64 | >64 | >64 | >64 |
| Vancomycin | 2 | 4 | N/A | N/A | N/A | N/A |
| Cefazolin | 32 | 8 | >64 | >64 | >64 | >64 |
| Meropenem | 2 | 2 | 8 | 64 | 32 | >64 |
| Compound | Target Proteins | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| COX-2 | FGFR-2 | VEGRF-2 | NR3A1 | NR3A2 | EGFR | HER2 | c-MET | ERK2 | MEK1 | CK4 | TPK | TopoII | |
| 5 | −7,7 | −7,0 | −7,5 | −7,2 | −7,2 | −7,9 | −7,8 | −7,4 | −7,2 | −7,0 | −6,5 | −7,5 | −7,7 |
| 6 | −7,3 | −6,7 | −7,1 | −6,9 | −6,9 | −7,3 | −7,8 | −7,2 | −7,2 | −6,8 | −6,4 | −7,6 | −7,3 |
| 16 | −8,3 | −8,0 | −6,5 | −8,3 | −7,4 | −8,4 | −9,6 | −8,9 | −8,0 | −8,8 | −7,1 | −8,1 | −8,3 |
| 25 | −9,8 | −11,3 | −7,6 | −10,9 | −10,8 | −9,1 | −11,7 | −11,6 | −10,3 | −10,5 | −10,2 | −10,1 | −9,8 |
| P avge. | −8,29 | −8,23 | −7,19 | −8,34 | −8,09 | −8,16 | −9,24 | −8,76 | −8,16 | −8,27 | −7,54 | −8,34 | −7,54 |
| Compound | ΔGbin (kcal/mol) | H Bonds and Hydrophobic Contacts in Binding Site |
|---|---|---|
| HER2 | ||
| 25 | −11,7 | Leu726, Val734, Lys736, Ala751, Lys753, Glu770, Ala771, Met774, Ser783, Arg784, Leu785, Leu796, Thr798, Gln799, Leu800, Met801, Gly804, Arg849, Asn850, Leu852, Thr862, Asp863, Phe864 |
| Ligand 1 [a] | −14,6 | Leu726, Val734, Ala751, Ile752, Lys753, Met774, Ser783, Leu785, Leu796, Thr798, Gln799, Leu800, Met801, Glu804, Cys805, Asp808, Leu852, Thr862, Asp863, Phe864 |
| Erlotinib [b] | −8,1 | Leu726, Gly727, Ser728, Val734, Ala751, Lys753, Met774, Ser783, Leu785, Leu796, Thr798, Gln799, Leu800, Met801, Gly804, Cys805, Leu852, Thr862, Asp863, Phe864 |
| c-MET | ||
| 25 | −11,6 | Ile1084, Gly1085, Gly1087, His1088, Phe1089, Val1092, Ala1108, Lys1110, Val1155, Leu1157, Gly1163, Asp1164, Asn1167, Arg1208, Met1211, Phe1223, Ala1226, Arg1227, Met1229, Tyr1230, Asp1231, Tyr1234, Tyr1235, Gly1163 |
| Ligand 2 [a] | −14,6 | Ile1084, Gly1085, Phe1089, Val1092, Ala1108, Lys1110, Leu1140, Leu1157, Pro1158, Tyr1159, Met1160, Gly1163, Met1211, Phe1223, Ala1226, Arg1227 |
| Erlotinib [b] | −9,0 | Ile1084, Phe1089, Val1092, Ala1108, Lys1110, Val1155, Leu1157, Met1160, Gly1163, Asp1164, Asn1167, Arg1208, Met1211, Phe1223, Ala1226, Arg1227, Asp1231, Tyr1234, Tyr1235 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kavaliauskas, P.; Acevedo, W.; Mickevičiūtė, E.; Grigalevičiūtė, R.; Grybaitė, B.; Sapijanskaitė-Banevič, B.; Pranaitytė, G.; Petraitis, V.; Petraitienė, R.; Mickevičius, V. 3,3′-((3-Hydroxyphenyl)azanediyl)dipropionic Acid Derivatives as a Promising Scaffold Against Drug-Resistant Pathogens and Chemotherapy-Resistant Cancer. Pathogens 2025, 14, 484. https://doi.org/10.3390/pathogens14050484
Kavaliauskas P, Acevedo W, Mickevičiūtė E, Grigalevičiūtė R, Grybaitė B, Sapijanskaitė-Banevič B, Pranaitytė G, Petraitis V, Petraitienė R, Mickevičius V. 3,3′-((3-Hydroxyphenyl)azanediyl)dipropionic Acid Derivatives as a Promising Scaffold Against Drug-Resistant Pathogens and Chemotherapy-Resistant Cancer. Pathogens. 2025; 14(5):484. https://doi.org/10.3390/pathogens14050484
Chicago/Turabian StyleKavaliauskas, Povilas, Waldo Acevedo, Eglė Mickevičiūtė, Ramunė Grigalevičiūtė, Birutė Grybaitė, Birutė Sapijanskaitė-Banevič, Guoda Pranaitytė, Vidmantas Petraitis, Rūta Petraitienė, and Vytautas Mickevičius. 2025. "3,3′-((3-Hydroxyphenyl)azanediyl)dipropionic Acid Derivatives as a Promising Scaffold Against Drug-Resistant Pathogens and Chemotherapy-Resistant Cancer" Pathogens 14, no. 5: 484. https://doi.org/10.3390/pathogens14050484
APA StyleKavaliauskas, P., Acevedo, W., Mickevičiūtė, E., Grigalevičiūtė, R., Grybaitė, B., Sapijanskaitė-Banevič, B., Pranaitytė, G., Petraitis, V., Petraitienė, R., & Mickevičius, V. (2025). 3,3′-((3-Hydroxyphenyl)azanediyl)dipropionic Acid Derivatives as a Promising Scaffold Against Drug-Resistant Pathogens and Chemotherapy-Resistant Cancer. Pathogens, 14(5), 484. https://doi.org/10.3390/pathogens14050484