
In Silico Targeting and Immunological Profiling of PpiA in Mycobacterium tuberculosis: A Computational Approach



Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Sequence Retrieval and Analysis
2.2. Prediction of Antigenicity, Allergenicity, and Physicochemical Properties
2.2.1. Antigenicity Prediction
2.2.2. Allergenicity Prediction
2.2.3. Physicochemical Properties
2.3. Secondary and Tertiary Structure Prediction
2.3.1. Secondary Structure Prediction
2.3.2. Tertiary Structure Prediction
2.3.3. Model Validation
2.4. B-Cell Epitope Mapping
2.5. T-Cell Epitope Prediction
2.5.1. MHC-I Epitope Prediction
2.5.2. MHC-II Epitope Prediction
3. Results
3.1. Protein Sequence Retrieval and Analysis
3.2. Antigenicity, Allergenicity, and Physicochemical Properties
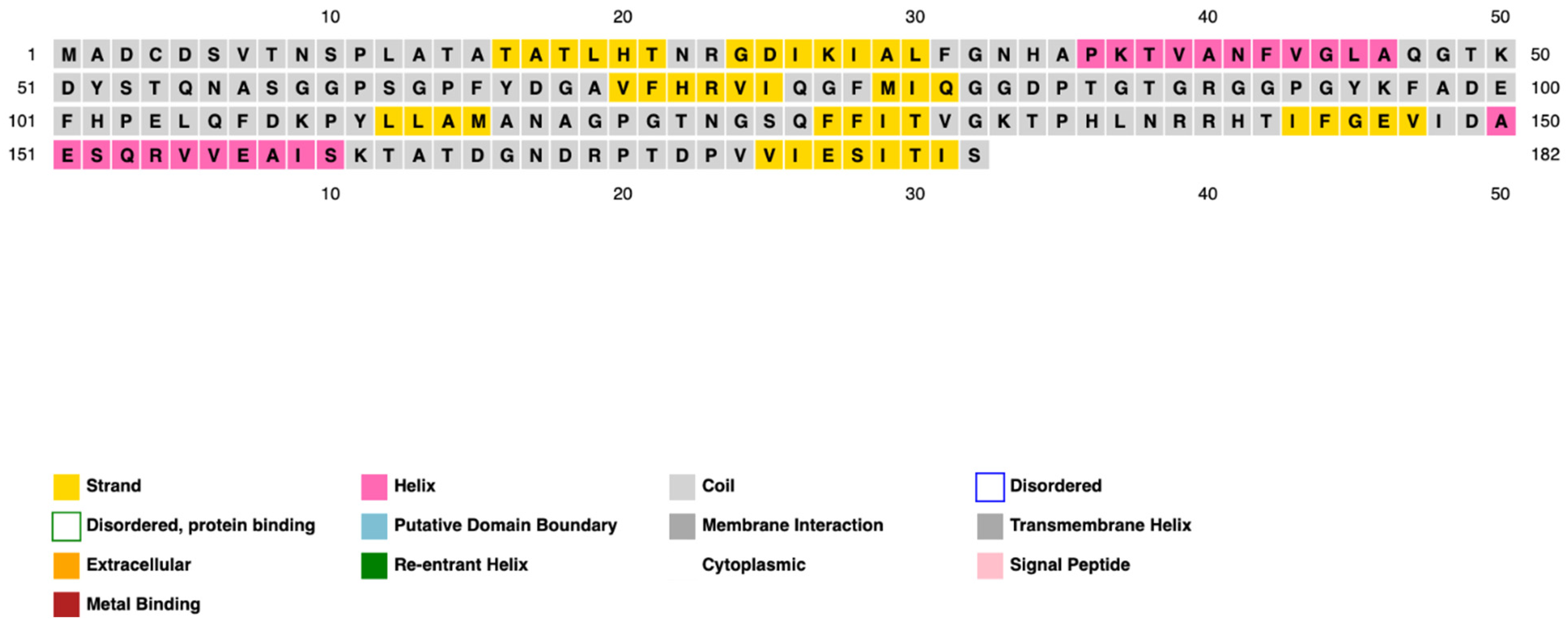
3.3. Prediction of PpiA Tertiary and Secondary Structures
3.3.1. Secondary Structure Prediction
3.3.2. Tertiary Structure Prediction
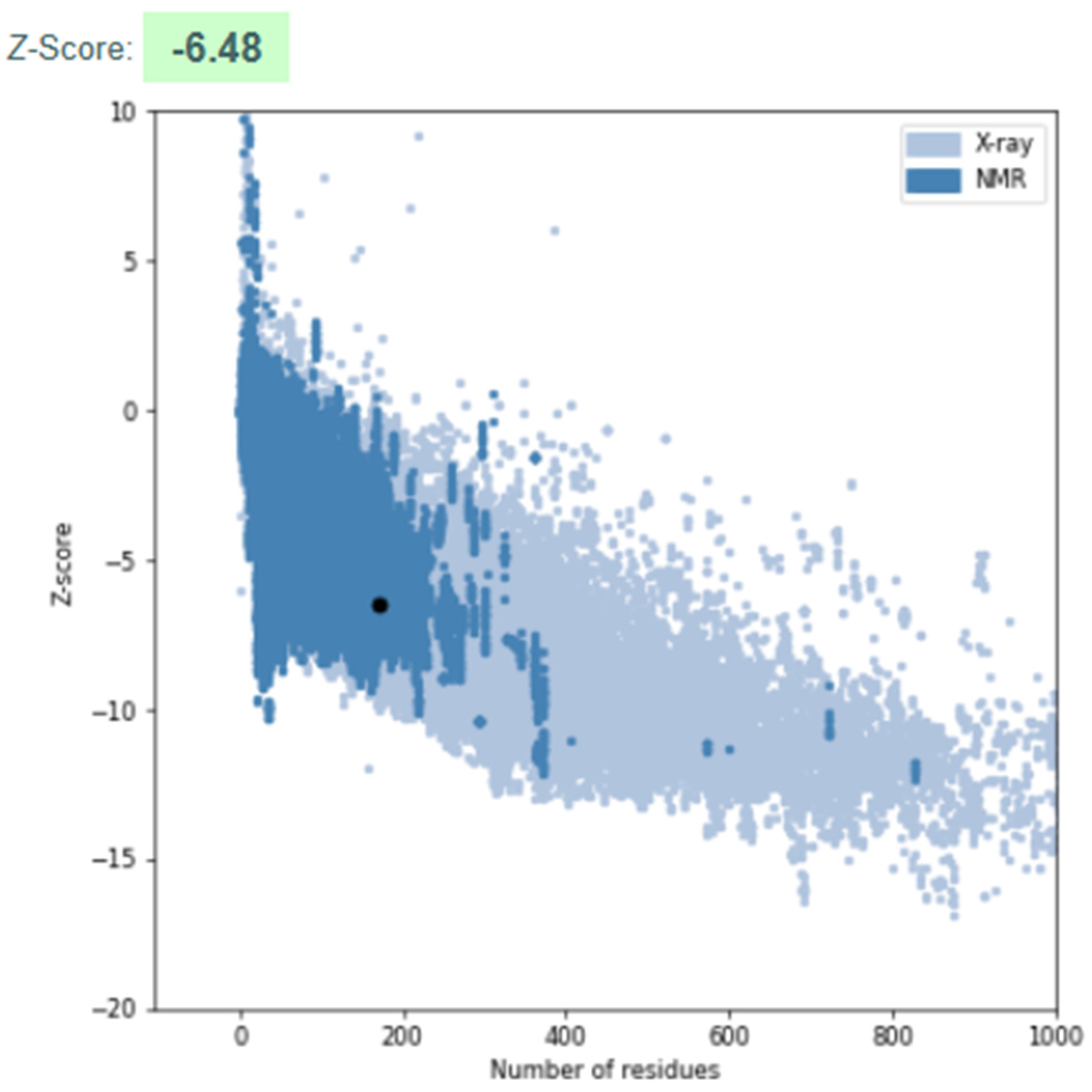
3.3.3. Model Validation
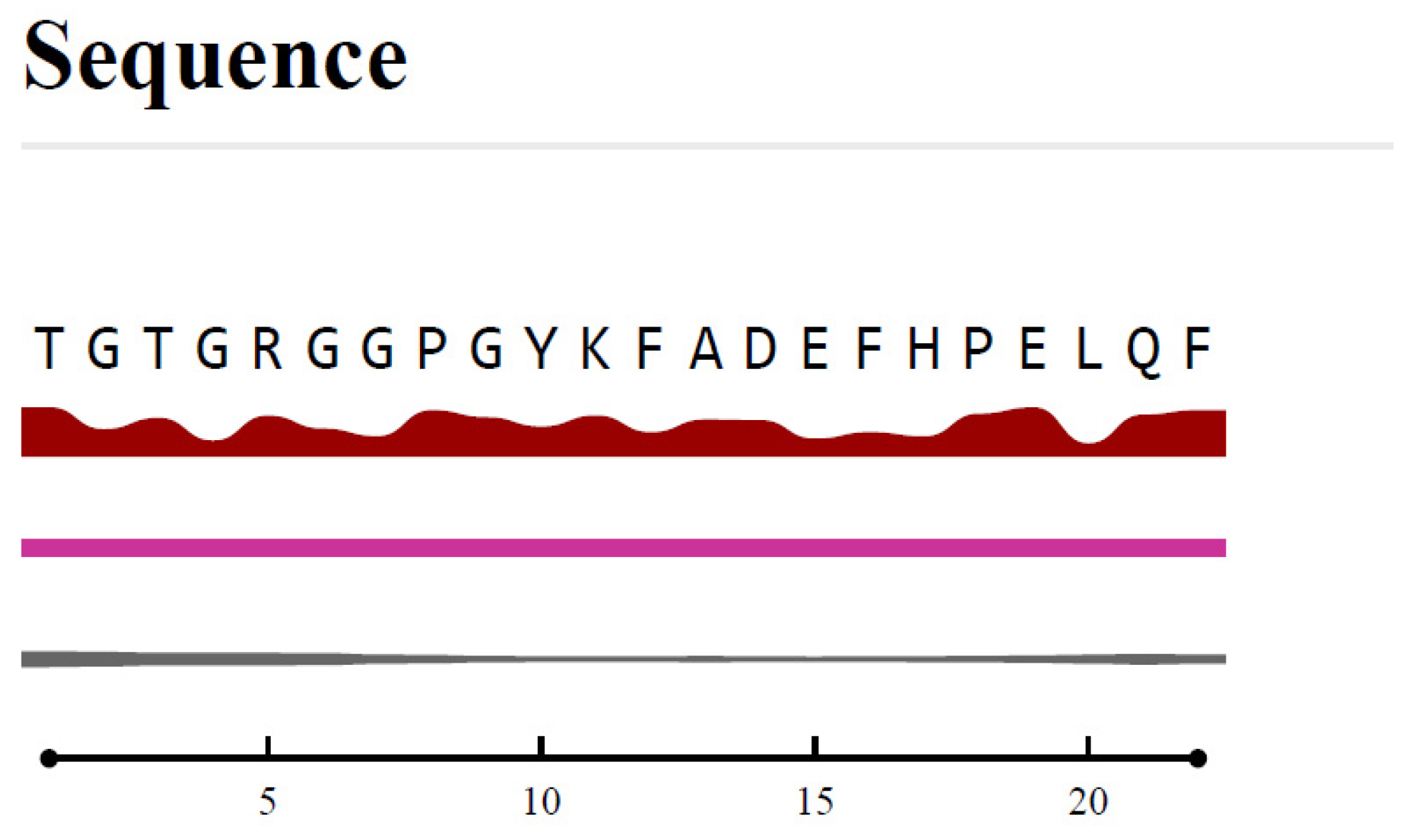
3.4. B-Cell Epitope Prediction
3.5. MHC Class I and Class II Epitope Prediction
3.6. Immunogenicity and Selection of Candidate Peptides
4. Discussion
4.1. Immunogenic Potential and Epitope Accessibility
4.2. Comparison with Existing Vaccine Strategies
4.3. Future Directions and Challenges
4.4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PpiA | Peptidyl-prolyl isomerase A |
| Mtb | Mycobacterium tuberculosis |
| TB | Tuberculosis |
| MHC | Major histocompatibility complex |
References
- World Health Organization. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2024.
- Dheda, K.; Mirzayev, F.; Cirillo, D.M.; Udwadia, Z.; Dooley, K.E.; Chang, K.-C.; Omar, S.V.; Reuter, A.; Perumal, T.; Horsburgh, C.R., Jr. Multidrug-resistant tuberculosis. Nat. Rev. Dis. Primers 2024, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, X.; Zhang, X.; Bai, J.; You, S.; Li, X.; Li, S.; Wang, Y.; Zhang, W.; Xu, Y. Global prevalence and burden of multidrug-resistant tuberculosis from 1990 to 2019. BMC Infect. Dis. 2024, 24, 243. [Google Scholar] [CrossRef]
- Nasiri, M.J.; Zangiabadian, M.; Arabpour, E.; Amini, S.; Khalili, F.; Centis, R.; D’Ambrosio, L.; Denholm, J.T.; Schaaf, H.S.; van den Boom, M. Delamanid-containing regimens and multidrug-resistant tuberculosis: A systematic review and meta-analysis. Int. J. Infect. Dis. 2022, 124, S90–S103. [Google Scholar] [CrossRef]
- Nasiri, M.J.; Lutfy, K.; Venketaraman, V. Challenges of Multidrug-Resistant Tuberculosis Meningitis: Current Treatments and the Role of Glutathione as an Adjunct Therapy. Vaccines 2024, 12, 1397. [Google Scholar] [CrossRef] [PubMed]
- Seeberg, J. An epidemic of drug resistance: Tuberculosis in the twenty-first century. Pathogens 2023, 12, 652. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, M.J.; Venketaraman, V. Advances in Host–Pathogen Interactions in Tuberculosis: Emerging Strategies for Therapeutic Intervention. Int. J. Mol. Sci. 2025, 26, 1621. [Google Scholar] [CrossRef]
- Sharma, A.K.; Mal, S.; Sahu, S.K.; Bagchi, S.; Majumder, D.; Chakravorty, D.; Saha, S.; Kundu, M.; Basu, J. Mycobacterial peptidyl prolyl isomerase A activates STING-TBK1-IRF3 signaling to promote IFNβ release in macrophages. FEBS J. 2025, 292, 94–114. [Google Scholar] [CrossRef]
- Dubey, N.; Khan, M.Z.; Kumar, S.; Sharma, A.; Das, L.; Bhaduri, A.; Singh, Y.; Nandicoori, V.K. Mycobacterium tuberculosis peptidyl prolyl isomerase a interacts with host integrin receptor to exacerbate disease progression. J. Infect. Dis. 2021, 224, 1383–1393. [Google Scholar] [CrossRef]
- Pandey, S.; Tripathi, D.; Khubaib, M.; Kumar, A.; Sheikh, J.A.; Sumanlatha, G.; Ehtesham, N.Z.; Hasnain, S.E. Mycobacterium tuberculosis peptidyl-prolyl isomerases are immunogenic, alter cytokine profile and aid in intracellular survival. Front. Cell. Infect. Microbiol. 2017, 7, 38. [Google Scholar] [CrossRef]
- Pandey, S.; Sharma, A.; Tripathi, D.; Kumar, A.; Khubaib, M.; Bhuwan, M.; Chaudhuri, T.K.; Hasnain, S.E.; Ehtesham, N.Z. Mycobacterium tuberculosis peptidyl-prolyl isomerases also exhibit chaperone like activity in-vitro and in-vivo. PLoS ONE 2016, 11, e0150288. [Google Scholar] [CrossRef]
- Nugraha, M.F.; Changestu, D.A.; Ramadhan, R.; Salsabila, T.; Nurizati, A.; Pratiwi, S.E.; Ysrafil, Y. Novel prophylactic and therapeutic multi-epitope vaccine based on Ag85A, Ag85B, ESAT-6, and CFP-10 of Mycobacterium tuberculosis using an immunoinformatics approach. Osong Public Health Res. Perspect. 2024, 15, 286–306. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Dockrell, H.M.; Ottenhoff, T.H.M.; Evans, T.G.; Zhang, Y. Tuberculosis vaccines: Opportunities and challenges. Respirology 2018, 23, 359–368. [Google Scholar] [CrossRef]
- Ouyang, J.; Guo, S.; Hu, Z.; Cao, T.; Mou, J.; Gu, X.; Huang, C.; Liu, J. Recombinant protein Ag85B-Rv2660c-MPT70 promotes quality of BCG-induced immune response against Mycobacterium tuberculosis H37Ra. Front. Immunol. 2025, 16, 1430808. [Google Scholar] [CrossRef]
- Phogat, S.; Yadav, J.; Chaudhary, D.; Jaiwal, R.; Jaiwal, P.K. Synthesis of an Adjuvant-Free Single Polypeptide-Based Tuberculosis Subunit Vaccine that Elicits In Vivo Immunogenicity in Rats. In Molecular Biotechnology; Springer: Berlin/Heidelberg, Germany, 2025. [Google Scholar] [CrossRef]
- Cardona, P.J. Pathogenesis of tuberculosis and other mycobacteriosis. Enferm. Infecc. Microbiol. Clin. 2018, 36, 38–46. [Google Scholar] [CrossRef]
- Chirani, A.S.; Majidzadeh, R.; Pouriran, R.; Heidary, M.; Nasiri, M.J.; Gholami, M.; Goudarzi, M.; Omrani, V.F. The effect of in silico targeting Pseudomonas aeruginosa patatin-like protein D, for immunogenic administration. Comput. Biol. Chem. 2018, 74, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Rahman, M.K.; Saha, S.; Kaykobad, M.; Rahman, M.S. Antigenic: An improved prediction model of protective antigens. Artif. Intell. Med. 2019, 94, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Safari, N.; Ardakan, A.K.; Hamedi, E.; Kalantarzadeh, F.; Kaveh, P.; Rahmanian, P.; Ghiabi, S.; Hosseini, S.A.; Siamian, D.; Gorgipour, M. An in silico approach to decipher immunogenic epitopes in Toxoplasma gondii GRA1 and GRA3. Inform. Med. Unlocked 2024, 44, 101435. [Google Scholar] [CrossRef]
- Azimi, R.; Ozgul, M.; Kenney, M.C.; Kuppermann, B.D. Bioinformatic analysis of small humanin like peptides using AlfaFold-2 and Expasy ProtParam. Investig. Ophthalmol. Vis. Sci. 2024, 65, 1320. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.e.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Morita, R.; Shigeta, Y.; Harada, R. Comprehensive predictions of secondary structures for comparative analysis in different species. J. Struct. Biol. 2021, 213, 107735. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Carrascoza, F.; Zaric, S.; Silaghi-Dumitrescu, R. Computational study of protein secondary structure elements: Ramachandran plots revisited. J. Mol. Graph. Model. 2014, 50, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Kim, Y.; Ponomarenko, J.; Zhu, Z.; Tamang, D.; Wang, P.; Greenbaum, J.; Lundegaard, C.; Sette, A.; Lund, O.; Bourne, P.E. Immune epitope database analysis resource. Nucleic Acids Res. 2012, 40, W525–W530. [Google Scholar] [CrossRef]
- Vita, R.; Zarebski, L.; Greenbaum, J.A.; Emami, H.; Hoof, I.; Salimi, N.; Damle, R.; Sette, A.; Peters, B. The immune epitope database 2.0. Nucleic Acids Res. 2010, 38, D854–D862. [Google Scholar] [CrossRef]
- Potocnakova, L.; Bhide, M.; Pulzova, L.B. An introduction to B-cell epitope mapping and in silico epitope prediction. J. Immunol. Res. 2016, 2016, 6760830. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Yun, J.S.; Kim, A.R.; Kim, S.M.; Shin, E.; Ha, S.J.; Kim, D.; Jeong, H.S. In silico analysis for the development of multi-epitope vaccines against Mycobacterium tuberculosis. Front. Immunol. 2024, 15, 1474346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.-c.; Hu, Z.-d.; Fan, X.-y. Advances in protein subunit vaccines against tuberculosis. Front. Immunol. 2023, 14, 1238586. [Google Scholar] [CrossRef]
- Kim, H.; Choi, H.-G.; Shin, S.J. Bridging the gaps to overcome major hurdles in the development of next-generation tuberculosis vaccines. Front. Immunol. 2023, 14, 1193058. [Google Scholar] [CrossRef]
- Romano, M.; Squeglia, F.; Kramarska, E.; Barra, G.; Choi, H.-G.; Kim, H.-J.; Ruggiero, A.; Berisio, R. A structural view at vaccine development against M. tuberculosis. Cells 2023, 12, 317. [Google Scholar] [CrossRef]
- Jurczak, M.; Druszczynska, M. Beyond Tuberculosis: The Surprising Immunological Benefits of the Bacillus Calmette–Guérin (BCG) Vaccine in Infectious, Auto-Immune, and Inflammatory Diseases. Pathogens 2025, 14, 196. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Programme; New TB Vaccine Research. Available online: https://www.who.int/teams/global-tuberculosis-programme/research-innovation/vaccines?utm_source=chatgpt.com (accessed on 1 January 2025).
- Sagawa, Z.K.; Goman, C.; Frevol, A.; Blazevic, A.; Tennant, J.; Fisher, B.; Day, T.; Jackson, S.; Lemiale, F.; Toussaint, L. Safety and immunogenicity of a thermostable ID93+ GLA-SE tuberculosis vaccine candidate in healthy adults. Nat. Commun. 2023, 14, 1138. [Google Scholar] [CrossRef]
- Pal, R.; Bisht, M.K.; Mukhopadhyay, S. Secretory proteins of Mycobacterium tuberculosis and their roles in modulation of host immune responses: Focus on therapeutic targets. FEBS J. 2022, 289, 4146–4171. [Google Scholar] [CrossRef] [PubMed]
- Passos, B.B.; Araújo-Pereira, M.; Vinhaes, C.L.; Amaral, E.P.; Andrade, B.B. The role of ESAT-6 in tuberculosis immunopathology. Front. Immunol. 2024, 15, 1383098. [Google Scholar] [CrossRef]
- Shi, S.; Yu, L.; Sun, D.; Liu, J.; Hickey, A.J. Rational design of multiple TB antigens TB10. 4 and TB10. 4-Ag85B as subunit vaccine candidates against Mycobacterium tuberculosis. Pharm. Res. 2010, 27, 224–234. [Google Scholar] [CrossRef]
- Kudryavtsev, I.; Zinchenko, Y.; Serebriakova, M.; Akisheva, T.; Rubinstein, A.; Savchenko, A.; Borisov, A.; Belenjuk, V.; Malkova, A.; Yablonskiy, P. A key role of cd8+ t cells in controlling of tuberculosis infection. Diagnostics 2023, 13, 2961. [Google Scholar] [CrossRef]
- Liu, H.; Gui, X.; Chen, S.; Fu, W.; Li, X.; Xiao, T.; Hou, J.; Jiang, T. Structural Variability of Lipoarabinomannan Modulates Innate Immune Responses within Infected Alveolar Epithelial Cells. Cells 2022, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Vergne, I.; Chua, J.; Singh, S.B.; Deretic, V. Cell biology of Mycobacterium tuberculosis phagosome. Annu. Rev. Cell Dev. Biol. 2004, 20, 367–394. [Google Scholar] [CrossRef] [PubMed]
- Chugh, S.; Bahal, R.K.; Dhiman, R.; Singh, R. Antigen identification strategies and preclinical evaluation models for advancing tuberculosis vaccine development. NPJ Vaccines 2024, 9, 57. [Google Scholar] [CrossRef]
- Yun, J.-S.; Shin, E.; Lee, Y.-R.; Lee, J.-A.; Lee, H.; Kim, J.-S.; Shin, S.J.; Ha, S.-J.; Lee, S.-W.; Kim, D. Immunogenicity and protective efficacy of a multi-antigenic adenovirus-based vaccine candidate against Mycobacterium tuberculosis. Front. Microbiol. 2025, 16, 1492268. [Google Scholar] [CrossRef] [PubMed]
- Pillay, K.; Chiliza, T.E.; Senzani, S.; Pillay, B.; Pillay, M. In silico design of Mycobacterium tuberculosis multi-epitope adhesin protein vaccines. Heliyon 2024, 10, e37536. [Google Scholar] [CrossRef]
- An, Y.; Ni, R.; Zhuang, L.; Yang, L.; Ye, Z.; Li, L.; Parkkila, S.; Aspatwar, A.; Gong, W. Tuberculosis vaccines and therapeutic drug: Challenges and future directions. Mol. Biomed. 2025, 6, 1–51. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Start | End | Peptide | Length | VaxiJen Score |
|---|---|---|---|---|---|
| 1 | 86 | 107 | TGTGRGGPGYKFADEFHPELQF | 22 | 0.760 |
| 2 | 47 | 66 | QGTKDYSTQNASGGPSGPFY | 20 | 0.750 |
| 3 | 164 | 169 | TDGNDR | 6 | 0.535 |
| 4 | 5 | 11 | DSVTNSP | 7 | 0.475 |
| 5 | 135 | 137 | PHL | 3 | 0.392 |
| No. | Start | End | Peptide | Length | Core Sequence | Score | Percentile Rank |
|---|---|---|---|---|---|---|---|
| 1 | 97 | 105 | FADEFHPEL | 9 | FADEFHPEL | 0.865 | 0.05 |
| 2 | 105 | 113 | LQFDKPYLL | 9 | LQFDKPYLL | 0.862 | 0.05 |
| 3 | 29 | 39 | ALFGNHAPKTV | 11 | ALFGAPKTV | 0.535 | 0.24 |
| 4 | 147 | 155 | VIDAESQRV | 9 | VIDAESQRV | 0.377035 | 0.4 |
| No. | Start | End | Peptide Sequence | Length | Core Sequence | Score | Percentile Rank |
|---|---|---|---|---|---|---|---|
| 1 | 108 | 122 | DKPYLLAMANAGPGT | 15 | YLLAMANAG | 0.9713 | 0.08 |
| 2 | 107 | 121 | FDKPYLLAMANAGPG | 15 | YLLAMANAG | 0.9630 | 0.12 |
| 3 | 106 | 120 | QFDKPYLLAMANAGP | 15 | YLLAMANAG | 0.8837 | 0.55 |
| 4 | 109 | 123 | KPYLLAMANAGPGTN | 15 | YLLAMANAG | 0.8551 | 0.64 |
| Peptide | Residues | Type of Immunity | VaxiJen Score/Binding Score | Percentile Rank | Target Immune Cell | Additional Characteristics |
|---|---|---|---|---|---|---|
| TGTGRGGPGYKFADEFHPELQF | 86–107 | B-cell immunity | 0.760 | N/A | B-cells | Surface-exposed, flexible secondary structure (helix + coil), high immunogenicity potential |
| FADEFHPEL | 97–105 | Cytotoxic T-cell (CD8+) | 0.865 | 0.05% | CD8+ T-cells | Strong MHC Class I binding, low percentile rank, high potential for inducing cytotoxic T-cell response |
| DKPYLLAMANAGPGT | 108–122 | Helper T-cell (CD4+) | 0.9713 | 0.08% | CD4+ T-cells | Strong MHC Class II binding, low percentile rank, excellent candidate for CD4+ T-cell activation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasiri, M.J.; Rogowski, L.; Venketaraman, V. In Silico Targeting and Immunological Profiling of PpiA in Mycobacterium tuberculosis: A Computational Approach. Pathogens 2025, 14, 370. https://doi.org/10.3390/pathogens14040370
Nasiri MJ, Rogowski L, Venketaraman V. In Silico Targeting and Immunological Profiling of PpiA in Mycobacterium tuberculosis: A Computational Approach. Pathogens. 2025; 14(4):370. https://doi.org/10.3390/pathogens14040370
Chicago/Turabian StyleNasiri, Mohammad J., Lily Rogowski, and Vishwanath Venketaraman. 2025. "In Silico Targeting and Immunological Profiling of PpiA in Mycobacterium tuberculosis: A Computational Approach" Pathogens 14, no. 4: 370. https://doi.org/10.3390/pathogens14040370
APA StyleNasiri, M. J., Rogowski, L., & Venketaraman, V. (2025). In Silico Targeting and Immunological Profiling of PpiA in Mycobacterium tuberculosis: A Computational Approach. Pathogens, 14(4), 370. https://doi.org/10.3390/pathogens14040370