

Genomic Insights into Vector–Pathogen Adaptation in Haemaphysalis longicornis and Rhipicephalus microplus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Alignment
2.2. Alignment, Variant Calling, and Annotation
2.3. Genetic Structure Analyses
2.4. Detection of Genome-Wide Selection Signals
2.5. Association of Microbial Composition with Tick SNPs
2.6. Functional Enrichments and Differential Expression Analysis of Genes
3. Results
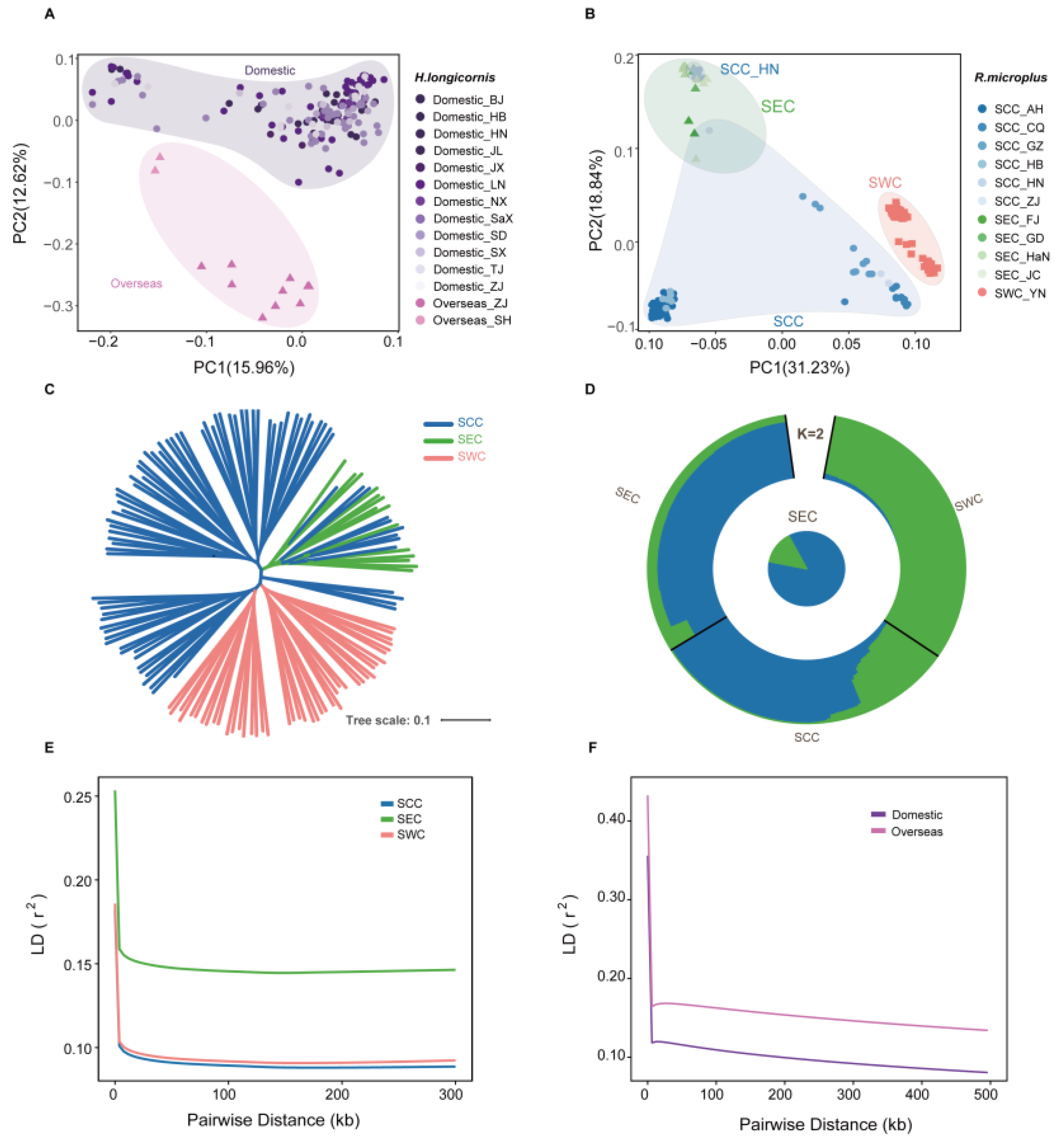
3.1. Distinct Population Structures in R. microplus and H. longicornis
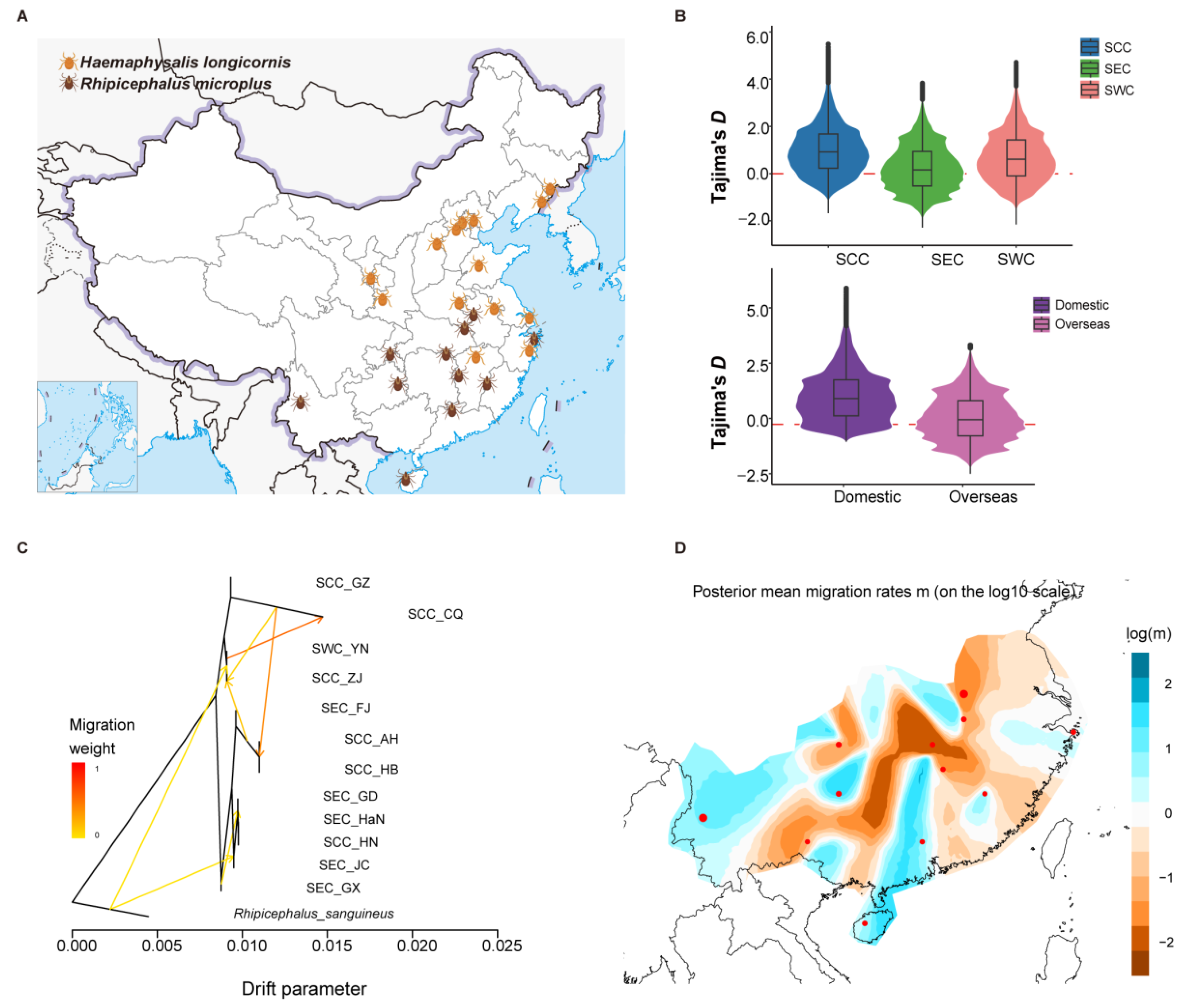
3.2. Genomic Diversity and Population Migration
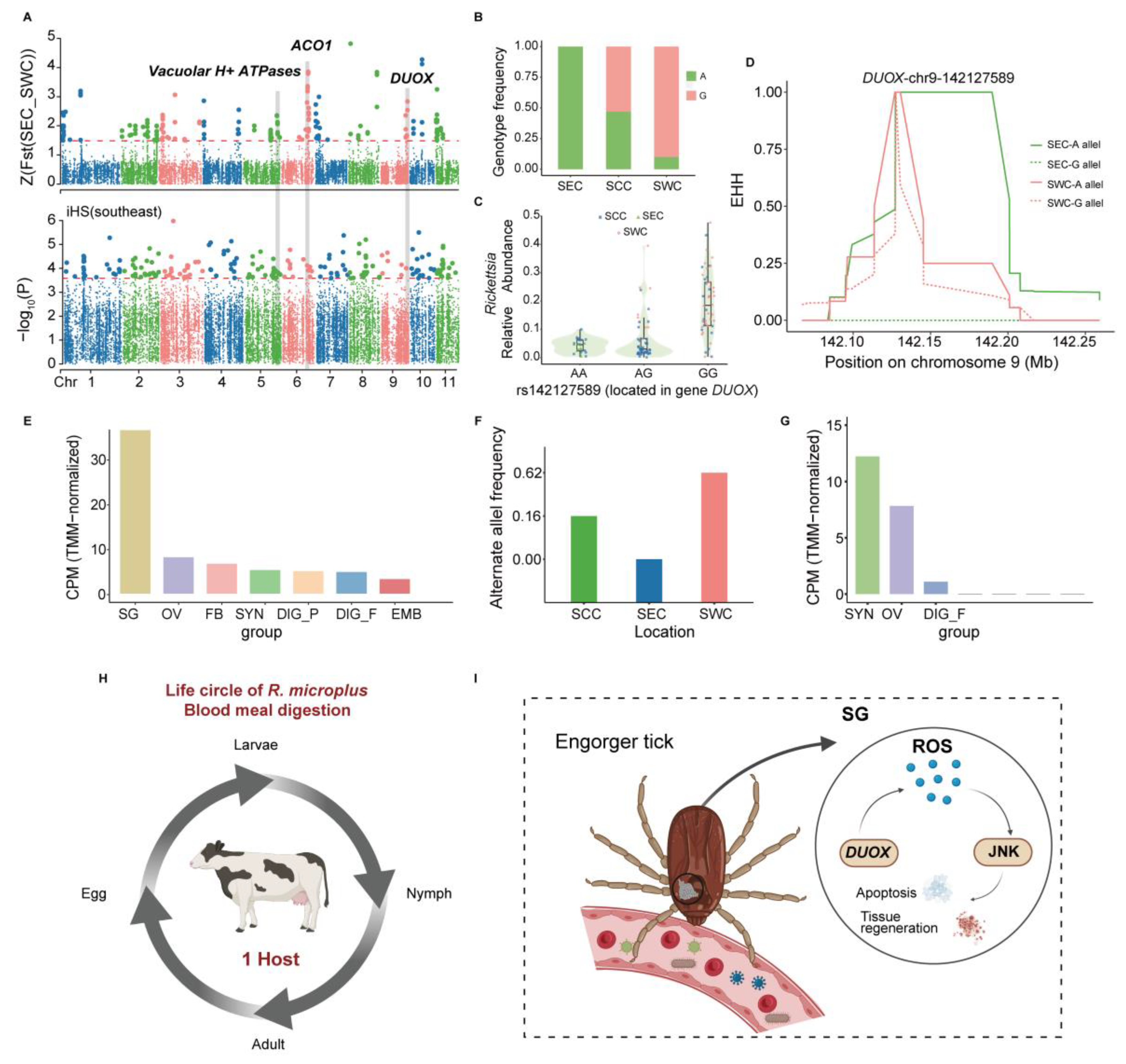
3.3. Genetic Variations Contribute to Blood Meal Digestion and Vector–Pathogen Adaptation in R. microplus
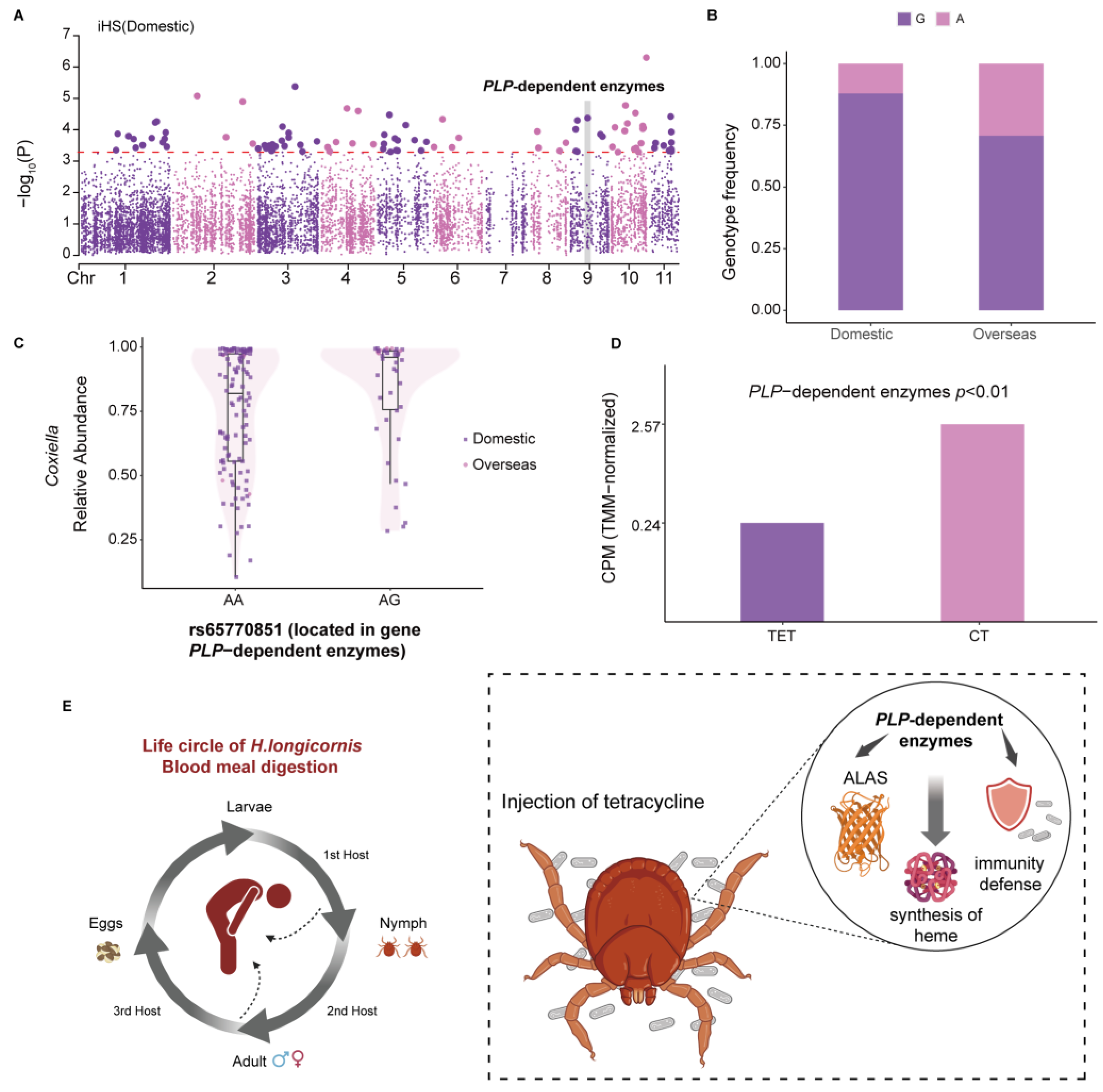
3.4. Genetic Variations of H. longicornis Contribute to Heme Synthesis and Correlate with Coxiella Abundance
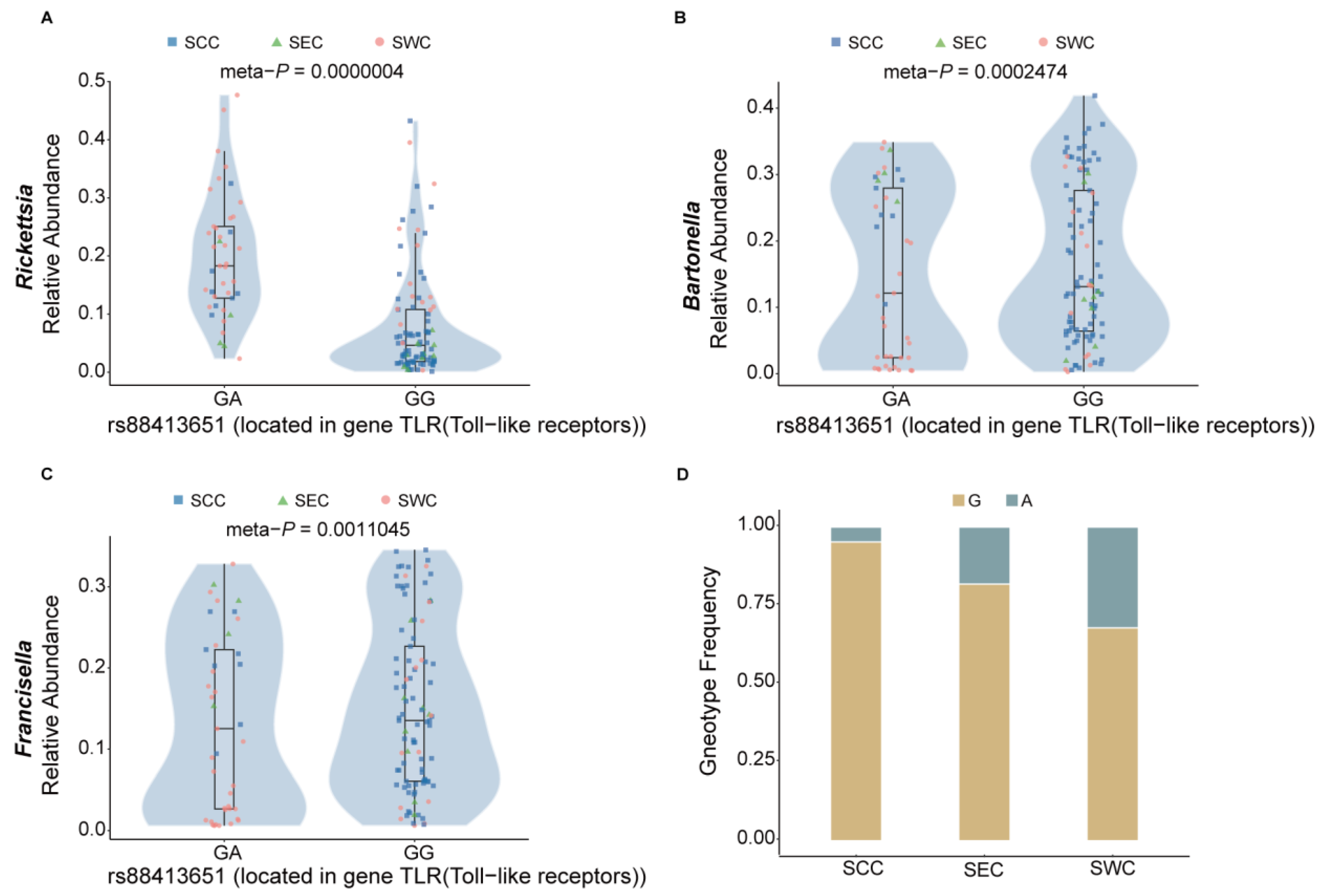
3.5. Correlation Between Genetic Variants of Ticks and Tick-Borne Pathogen Abundance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dantas-Torres, F.; Chomel, B.B.; Otranto, D. Ticks and Tick-Borne Diseases: A One Health Perspective. Trends Parasitol. 2012, 28, 437–446. [Google Scholar] [CrossRef]
- Pfäffle, M.; Littwin, N.; Muders, S.V.; Petney, T.N. The Ecology of Tick-Borne Diseases. Int. J. Parasitol. 2013, 43, 1059–1077. [Google Scholar] [CrossRef] [PubMed]
- Guglielmone, A.A.; Robbins, R.G.; Apanaskevich, D.A.; Petney, T.N.; Estrada-Peña, A.; Horak, I.G.; Shao, R.; Barker, S.C. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida) of the World: A List of Valid Species Names. Zootaxa 2010, 2528, 1. [Google Scholar] [CrossRef]
- Baneth, G. Tick-Borne Infections of Animals and Humans: A Common Ground. Int. J. Parasitol. 2014, 44, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Brites-Neto, J.; Duarte, K.M.R.; Martins, T.F. Tick-Borne Infections in Human and Animal Population Worldwide. Vet. World 2015, 8, 301–315. [Google Scholar] [CrossRef]
- Rochlin, I.; Toledo, A. Emerging Tick-Borne Pathogens of Public Health Importance: A Mini-Review. J. Med. Microbiol. 2020, 69, 781–791. [Google Scholar] [CrossRef]
- Marques, A.R.; Strle, F.; Wormser, G.P. Comparison of Lyme Disease in the United States and Europe. Emerg. Infect. Dis. 2021, 27, 2017–2024. [Google Scholar] [CrossRef]
- Kugeler, K.J.; Schwartz, A.M.; Delorey, M.J.; Mead, P.S.; Hinckley, A.F. Estimating the Frequency of Lyme Disease Diagnoses, United States, 2010–2018. Emerg. Infect. Dis. 2021, 27, 616–619. [Google Scholar] [CrossRef]
- Radolf, J.D.; Caimano, M.J.; Stevenson, B.; Hu, L.T. Of Ticks, Mice and Men: Understanding the Dual-Host Lifestyle of Lyme Disease Spirochaetes. Nat. Rev. Microbiol. 2012, 10, 87–99. [Google Scholar] [CrossRef]
- Wu, X.-B.; Na, R.-H.; Wei, S.-S.; Zhu, J.-S.; Peng, H.-J. Distribution of Tick-Borne Diseases in China. Parasites Vectors 2013, 6, 119. [Google Scholar] [CrossRef]
- Zhang, X.; Meltzer, M.I.; Peña, C.A.; Hopkins, A.B.; Wroth, L.; Fix, A.D. Economic Impact of Lyme Disease. Emerg. Infect. Dis. 2006, 12, 653–660. [Google Scholar] [CrossRef] [PubMed]
- de Castro, J.J. Sustainable Tick and Tickborne Disease Control in Livestock Improvement in Developing Countries. Vet. Parasitol. 1997, 71, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Kivaria, F.M. Estimated Direct Economic Costs Associated with Tick-Borne Diseases on Cattle in Tanzania. Trop. Anim. Health Prod. 2006, 38, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Ocaido, M.; Muwazi, R.T.; Opuda, J.A. Economic Impact of Ticks and Tick-Borne Diseases on Cattle Production Systems around Lake Mburo National Park in South Western Uganda. Trop. Anim. Health Prod. 2009, 41, 731–739. [Google Scholar] [CrossRef]
- de Castro, J.J.; James, A.D.; Minjauw, B.; Di Giulio, G.U.; Permin, A.; Pegram, R.G.; Chizyuka, G.B.; Sinyangwe, P. Long-Term Studies on the Economic Impact of Ticks on Sanga Cattle in Zambia. Exp. Appl. Acarol. 1997, 21, 3–19. [Google Scholar] [CrossRef]
- Heath, A.C.G. Implications for New Zealand of Potentially Invasive Ticks Sympatric with Haemaphysalis longicornis Neumann, 1901 (Acari: Ixodidae). Syst. Appl. Acarol. 2013, 18, 1–26. [Google Scholar] [CrossRef]
- Zhao, L.; Li, J.; Cui, X.; Jia, N.; Wei, J.; Xia, L.; Wang, H.; Zhou, Y.; Wang, Q.; Liu, X.; et al. Distribution of Haemaphysalis longicornis and Associated Pathogens: Analysis of Pooled Data from a China Field Survey and Global Published Data. Lancet Planet. Health 2020, 4, e320–e329. [Google Scholar] [CrossRef]
- Fang, L.-Q.; Liu, K.; Li, X.-L.; Liang, S.; Yang, Y.; Yao, H.-W.; Sun, R.-X.; Sun, Y.; Chen, W.-J.; Zuo, S.-Q.; et al. Emerging Tick-Borne Infections in Mainland China: An Increasing Public Health Threat. Lancet Infect. Dis. 2015, 15, 1467–1479. [Google Scholar] [CrossRef]
- Yin, H.; Luo, J. Ticks of Small Ruminants in China. Parasitol. Res. 2007, 101 (Suppl. 2), S187–S189. [Google Scholar] [CrossRef]
- Jia, N.; Wang, J.; Shi, W.; Du, L.; Sun, Y.; Zhan, W.; Jiang, J.-F.; Wang, Q.; Zhang, B.; Ji, P.; et al. Large-Scale Comparative Analyses of Tick Genomes Elucidate Their Genetic Diversity and Vector Capacities. Cell 2020, 182, 1328–1340.e13. [Google Scholar] [CrossRef]
- de la Fuente, J.; Estrada-Peña, A.; Venzal, J.; Kocan, K.; Sonenshine, D. Overview: Ticks as Vectors of Pathogens That Cause Disease in Humans and Animals. Front. Biosci. 2008, 13, 6938–6946. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Peña, A.; Bouattour, A.; Camicas, J.-L.; Guglielmone, A.; Horak, I.; Jongejan, F.; Latif, A.; Pegram, R.; Walker, A.R. The Known Distribution and Ecological Preferences of the Tick Subgenus Boophilus (Acari: Ixodidae) in Africa and Latin America. Exp. Appl. Acarol. 2006, 38, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Peter, R.J.; Van den Bossche, P.; Penzhorn, B.L.; Sharp, B. Tick, Fly, and Mosquito Control--Lessons from the Past, Solutions for the Future. Vet. Parasitol. 2005, 132, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Kumar, D.; Budachetri, K. Recent Advances in Understanding Tick and Rickettsiae Interactions. Parasite Immunol. 2021, 43, e12830. [Google Scholar] [CrossRef]
- Frischknecht, F. The Skin as Interface in the Transmission of Arthropod-borne Pathogens. Cell. Microbiol. 2007, 9, 1630–1640. [Google Scholar] [CrossRef]
- Paesen, G.C.; Adams, P.L.; Nuttall, P.A.; Stuart, D.L. Tick Histamine-Binding Proteins: Lipocalins with a Second Binding Cavity. Biochim. Et. Biophys. Acta 2000, 1482, 92–101. [Google Scholar] [CrossRef]
- Ribeiro, J.M.C.; Mather, T.N. Ixodes Scapularis: Salivary Kininase Activity Is a Metallo Dipeptidyl Carboxypeptidase. Exp. Parasitol. 1998, 89, 213–221. [Google Scholar] [CrossRef]
- Francischetti, I.M.B. Platelet Aggregation Inhibitors from Hematophagous Animals. Biochim. Biophys. Acta 2010, 1482, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Theilgaard-Monch, K.; Knudsen, S.; Follin, P.; Borregaard, N. The Transcriptional Activation Program of Human Neutrophils in Skin Lesions Supports Their Important Role in Wound Healing. J. Immunol. 2004, 172, 7684–7693. [Google Scholar] [CrossRef]
- Fukumoto, S.; Sakaguchi, T.; You, M.; Xuan, X.; Fujisaki, K. Tick Troponin I-like Molecule Is a Potent Inhibitor for Angiogenesis. Microvasc. Res. 2006, 3, 43. [Google Scholar] [CrossRef]
- Kramer, C.; Nahmias, Z.; Norman, D.D.; Mulvihill, T.A.; Coons, L.B.; Cole, J.A. Dermacentor Variabilis: Regulation of FIbroblast Migration by Tick Salivary Gland Extract and Saliva. Exp. Parasitol. 2008, 119, 391–397. [Google Scholar] [CrossRef]
- Hovius, J.W.R. Spitting Image: Tick Saliva Assists the Causative Agent of Lyme Disease in Evading Host Skin’s Innate Immune Response. J. Invest. Dermatol. 2009, 129, 2337–2339. [Google Scholar] [CrossRef]
- Wikel, S.K. Tick Modulation of Host Immunity: An Important Factor in Pathogen Transmission. Int. J. Parasitol. 1999, 29, 851–859. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, J.; Villar, M.; Cabezas-Cruz, A.; Estrada-Peña, A.; Ayllón, N.; Alberdi, P. Tick–Host–Pathogen Interactions: Conflict and Cooperation. PLoS Pathog. 2016, 12, e1005488. [Google Scholar] [CrossRef] [PubMed]
- Hajdusek, O.; Sima, R.; Ayllon, N.; Jalovecka, M.; Perner, J.; De La Fuente, J.; Kopacek, P. Interaction of the Tick Immune System with Transmitted Pathogens. Front. Cell. Infect. Microbiol. 2013, 3, 26. [Google Scholar]
- Núñez, G.; Sakamoto, K.; Soares, M.P. Innate Nutritional Immunity. J. Immunol. 2019, 201, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Galay, R.L.; Umemiya-Shirafuji, R.; Mochizuki, M.; Fujisaki, K.; Tanaka, T. Iron Metabolism in Hard Ticks (Acari: Ixodidae): The Antidote to Their Toxic Diet. Parasitol. Int. 2015, 64, 182–189. [Google Scholar] [CrossRef]
- Cabezas-Cruz, A.; Espinosa, P.; Alberdi, P.; Fuente, J. de la Tick–Pathogen Interactions: The Metabolic Perspective. Trends Parasitol. 2019, 35, 316–328. [Google Scholar] [CrossRef]
- Busby, A.; Ayllón, N.; Kocan, K.; Blouin, E.; de la Fuente, G.; Galindo, R.; Villar, M.; de la Fuente, J. Expression of Heat Shock Proteins and Subolesin Affects Stress Responses, Anaplasma phagocytophilum Infection and Questing Behaviour in the Tick, Ixodes Scapularis. Med. Vet. Entomol. 2011, 26, 92–102. [Google Scholar] [CrossRef]
- Cabezas-Cruz, A.; Estrada-Peña, A.; Rego, R.O.M.; De la Fuente, J. Tick-Pathogen Ensembles: Do Molecular Interactions Lead Ecological Innovation? Front. Cell Infect. Microbiol. 2017, 7, 74. [Google Scholar] [CrossRef]
- Shaw, D.K.; Tate, A.T.; Schneider, D.S.; Levashina, E.A.; Kagan, J.C.; Pal, U.; Fikrig, E.; Pedra, J.H.F. Vector Immunity and Evolutionary Ecology: The Harmonious Dissonance. Trends Immunol. 2018, 39, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Duron, O.; Binetruy, F.; Noël, V.; Cremaschi, J.; McCoy, K.D.; Arnathau, C.; Plantard, O.; Goolsby, J.; Pérez de León, A.A.; Heylen, D.J.A.; et al. Evolutionary Changes in Symbiont Community Structure in Ticks. Mol. Ecol. 2017, 26, 2905–2921. [Google Scholar] [CrossRef] [PubMed]
- Shastry, B.S. SNPs: Impact on Gene Function and Phenotype. Methods Mol. Biol. 2009, 578, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, D.; Donnelly, P. The International HapMap Consortium A Haplotype Map of the Human Genome. Nature 2005, 437, 1299–1320. [Google Scholar] [CrossRef]
- Brookes, A.J. The Essence of SNPs. Gene 1999, 234, 177–186. [Google Scholar] [CrossRef]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Genome-Wide Association Study of 14,000 Cases of Seven Common Diseases and 3,000 Shared Controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Francioli, L.C.; Menelaou, A.; Pulit, S.L.; van Dijk, F.; Palamara, P.F.; Elbers, C.C.; Neerincx, P.B.T.; Ye, K.; Guryev, V.; Kloosterman, W.P.; et al. Whole-Genome Sequence Variation, Population Structure and Demographic History of the Dutch Population. Nat. Genet. 2014, 46, 818–825. [Google Scholar] [CrossRef]
- Vignal, A.; Milan, D.; SanCristobal, M.; Eggen, A. A Review on SNP and Other Types of Molecular Markers and Their Use in Animal Genetics. Genet. Sel. Evol. 2002, 34, 275–305. [Google Scholar] [CrossRef]
- Tirloni, L.; Braz, G.; Nunes, R.D.; Gandara, A.C.P.; Vieira, L.R.; Assumpcao, T.C.; Sabadin, G.A.; Da Silva, R.M.; Guizzo, M.G.; Machado, J.A.; et al. A Physiologic Overview of the Organ-Specific Transcriptome of the Cattle Tick Rhipicephalus microplus. Sci. Rep. 2020, 10, 18296. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaSci 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Alexander, D.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Feng, Q.; Lu, D.; Xu, S. AncestryPainter: A Graphic Program for Displaying Ancestry Composition of Populations and Individuals. Genom. Proteom. Bioinform. 2018, 16, 382–385. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 2018, 35, 1786–1788. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Pritchard, J.K. Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef]
- Petkova, D.; Novembre, J.; Stephens, M. Visualizing Spatial Population Structure with Estimated Effective Migration Surfaces. Nat. Genet. 2016, 48, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Tian, X.; Zhou, Y.; Browning, S.R. Fast Two-Stage Phasing of Large-Scale Sequence Data. Am. J. Human. Genet. 2021, 108, 1880–1890. [Google Scholar] [CrossRef]
- Szpiech, Z.; Hernandez, R. Selscan: An Efficient Multithreaded Program to Perform EHH-Based Scans for Positive Selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef]
- Hämälä, T.; Savolainen, O. Genomic Patterns of Local Adaptation under Gene Flow in Arabidopsis lyrata. Mol. Biol. Evol. 2019, 36, 2557–2571. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and Visualization of LD and Haplotype Maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2015, 428, 726–731. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; Mcanulla, C.; Mcwilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Feng, J.; Meyer, C.A.; Wang, Q.; Liu, J.S.; Shirley Liu, X.; Zhang, Y. GFOLD: A Generalized Fold Change for Ranking Differentially Expressed Genes from RNA-Seq Data. Bioinformatics 2012, 28, 2782–2788. [Google Scholar] [CrossRef]
- Lee, M.R.; Kim, J.C.; Park, S.E.; Lee, S.J.; Kim, W.J.; Lee, D.-H.; Kim, J.S. Interactive Gene Expression Between Metarhizium Anisopliae JEF-290 and Longhorned Tick Haemaphysalis Longicornis at Early Stage of Infection. Front. Physiol. 2021, 12, 643389. [Google Scholar] [CrossRef]
- Paulino, P.; Vitari, G.; Rezende, A.; Couto, J.; Antunes, S.; Domingos, A.; Peckle, M.; Massard, C.; Araújo, F.; Santos, H. Characterization of the Rhipicephalus (Boophilus) Microplus Sialotranscriptome Profile in Response to Theileria equi Infection. Pathogens 2021, 10, 167. [Google Scholar] [CrossRef]
- Ye, J.; McGinnis, S.; Madden, T.L. BLAST: Improvements for Better Sequence Analysis. Nucleic Acids Res. 2006, 34, W6–W9. [Google Scholar] [CrossRef]
- Stutzer, C. Gene Expression Profiling of Adult Female Tissues in Feeding Rhipicephalus microplus Cattle Ticks. Int. J. Parasitol. 2013, 43, 541–554. [Google Scholar] [CrossRef]
- Mishra, S.; Sharma, P.; Singh, R.; Tiwari, R.; Singh, G. Genome-Wide Identification and Expression Analysis of Sucrose Nonfermenting-1-Related Protein Kinase (SnRK) Genes in Triticum Aestivum in Response to Abiotic Stress. Sci. Rep. 2021, 11, 22477. [Google Scholar] [CrossRef]
- Morand, S.; Ueyama, T.; Tsujibe, S.; Saito, N.; Korzeniowska, A.; Leto, T. Duox Maturation Factors Form Cell Surface Complexes with Duox Affecting the Specificity of Reactive Oxygen Species Generation. FASEB J. 2009, 23, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-S.; Wu, S.-B.; Lee, W.-Y.; Cheng, J.-S.; Wei, Y.-H. Response to the Increase of Oxidative Stress and Mutation of Mitochondrial DNA in Aging. Biochim. Biophys. Acta 2009, 1790, 1021–1029. [Google Scholar] [CrossRef]
- Koerver, L.; Papadopoulos, C.; Liu, B.; Kravic, B.; Rota, G.; Brecht, L.; Veenendaal, T.; Polajnar, M.; Bluemke, A.; Ehrmann, M.; et al. The Ubiquitin-conjugating Enzyme UBE2QL1 Coordinates Lysophagy in Response to Endolysosomal Damage. EMBO Rep. 2019, 20, e48014. [Google Scholar] [CrossRef] [PubMed]
- McGuire, M.J.; Lipsky, P.; Thiele, D. Generation of Active Myeloid and Lymphoid Granule Serine Proteases Requires Processing by the Granule Thiol Protease Dipeptidyl Peptidase I. J. Biol. Chem. 1993, 268, 2458–2467. [Google Scholar] [CrossRef]
- Nishi, T.; Forgac, M. The Vacuolar (H+)-ATPases--Nature’s Most Versatile Proton Pumps. Nat. Rev. Mol. Cell Biol. 2002, 3, 94–103. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Gibson, T.B.; Xu, B.-E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef]
- Omar, S.A.; Webb, A.J. Nitrite Reduction and Cardiovascular Protection. J. Mol. Cell. Cardiol. 2014, 73, 57–69. [Google Scholar] [CrossRef]
- de Macario, E.C.; Yohda, M.; Macario, A.J.L.; Robb, F.T. Bridging Human Chaperonopathies and Microbial Chaperonins. Commun. Biol. 2019, 2, 103. [Google Scholar] [CrossRef]
- Kumar, D. Investigating the Functional Role of Tick Antioxidants in Hematophagy and Vector Competence. Ph.D. Thesis, University of Southern Mississippi, Hattiesburg, MS, USA, 2016; p. 905. [Google Scholar]
- Donkó, Á.; Péterfi, Z.; Sum, A.; Leto, T.; Geiszt, M. Dual Oxidases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2301–2308. [Google Scholar] [CrossRef]
- Galay, R.L.; Aung, K.M.; Umemiya-Shirafuji, R.; Maeda, H.; Matsuo, T.; Kawaguchi, H.; Miyoshi, N.; Suzuki, H.; Xuan, X.; Mochizuki, M.; et al. Multiple Ferritins Are Vital to Successful Blood Feeding and Reproduction of the Hard Tick Haemaphysalis longicornis. J. Exp. Biol. 2013, 216, 1905–1915. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of Cellular Iron Metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef]
- Zhou, G.; Kohlhepp, P.; Geiser, D.; Frasquillo, M.d.C.; Vazquez-Moreno, L.; Winzerling, J.J. Fate of Blood Meal Iron in Mosquitoes. J. Insect Physiol. 2007, 53, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, X.; Wang, J.; Huang, R.; Wan, D. Regulation of Iron Homeostasis and Related Diseases. Mediat. Inflamm. 2020, 2020, 6062094. [Google Scholar] [CrossRef]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian Iron Metabolism and Its Control by Iron Regulatory Proteins. Biochim. Et. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 1468–1483. [Google Scholar] [CrossRef]
- Cairo, G.; Recalcati, S.; Pietrangelo, A.; Minotti, G. The Iron Regulatory Proteins: Targets and Modulators of Free Radical Reactions and Oxidative Damage. Free Radic. Biol. Med. 2002, 32, 1237–1243. [Google Scholar] [CrossRef]
- Cairo, G.; Ronchi, R.; Recalcati, S.; Campanella, A.; Minotti, G. Nitric Oxide and Peroxynitrite Activate the Iron Regulatory Protein-1 of J774A.1 Macrophages by Direct Disassembly of the Fe-S Cluster of Cytoplasmic Aconitase. Biochemistry 2002, 41, 7435–7442. [Google Scholar] [CrossRef]
- Hentze, M.W.; Kühn, L.C. Molecular Control of Vertebrate Iron Metabolism: mRNA-Based Regulatory Circuits Operated by Iron, Nitric Oxide, and Oxidative Stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z. Morphological, Biological and Molecular Characteristics of Bisexual and Parthenogenetic Haemaphysalis longicornis. Vet. Parasitol. 2012, 189, 344–352. [Google Scholar] [CrossRef]
- Hunter, G.A.; Ferreira, G.C. 5-Aminolevulinate Synthase: Catalysis of the First Step of Heme Biosynthesis. Cell. Mol. Biol. 2009, 55, 102–110. [Google Scholar]
- Percudani, R.; Peracchi, A. A Genomic Overview of Pyridoxal-Phosphate-Dependent Enzymes. EMBO Rep. 2003, 4, 850–854. [Google Scholar] [CrossRef]
- Zhong, Z.; Zhong, T.; Peng, Y.; Zhou, X.; Wang, Z.; Tang, H.; Wang, J. Symbiont-Regulated Serotonin Biosynthesis Modulates Tick Feeding Activity. Cell Host Microbe 2021, 29, 1545–1557.e4. [Google Scholar] [CrossRef] [PubMed]
- Fogaça, A.C.; Sousa, G.; Pavanelo, D.B.; Esteves, E.; Martins, L.A.; Urbanová, V.; Kopáček, P.; Daffre, S. Tick Immune System: What Is Known, the Interconnections, the Gaps, and the Challenges. Front. Immunol. 2021, 12, 628054. [Google Scholar] [CrossRef]
- Quevedo-Diaz, M.A.; Song, C.; Xiong, Y.; Chen, H.; Wahl, L.M.; Radulovic, S.; Medvedev, A.E. Involvement of TLR2 and TLR4 in Cell Responses to Rickettsia akari. J. Leukoc. Biol. 2010, 88, 675–685. [Google Scholar] [CrossRef]
- Feldhaar, H.; Gross, R. Immune Reactions of Insects on Bacterial Pathogens and Mutualists. Microbes Infect. 2008, 10, 1082–1088. [Google Scholar] [CrossRef]
- Pan, X.; Tamilselvam, B.; Hansen, E.J.; Daefler, S. Modulation of Iron Homeostasis in Macrophages by Bacterial Intracellular Pathogens. BMC Microbiol. 2010, 10, 64. [Google Scholar] [CrossRef]
- Fadahunsi, A.I. Biochemical Characterisation of Cytochrome P450 Oxidoreductase from the Cattle Tick, Rhipicephalus microplus, Highlights Potential New Acaricide Target. Ticks Tick. Borne Dis. 2023, 14, 102148. [Google Scholar] [CrossRef] [PubMed]
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. The Iron Chaperone Poly(rC)-Binding Protein 2 Forms a Metabolon with the Heme Oxygenase 1/Cytochrome P450 Reductase Complex for Heme Catabolism and Iron Transfer. J. Biol. Chem. 2017, 292, 13205–13229. [Google Scholar] [CrossRef]
- Ouyang, Z.; Deka, R.K.; Norgard, M.V. BosR (BB0647) Controls the RpoN-RpoS Regulatory Pathway and Virulence Expression in Borrelia Burgdorferi by a Novel DNA-Binding Mechanism. PLoS Pathog. 2011, 7, e1001272. [Google Scholar] [CrossRef]
- Deng, K.; Blick, R.J.; Liu, W.; Hansen, E.J. Identification of Francisella Tularensis Genes Affected by Iron Limitation. Infect. Immun. 2006, 74, 4224–4236. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, K.; Xie, B.; Gao, Y.; Xu, S.; Lu, Y. Mapping Structural Variations in Haemaphysalis longicornis and Rhipicephalus microplus Reveals Vector-Pathogen Adaptation. iScience 2023, 26, 106398. [Google Scholar] [CrossRef]
- Zhao, C.; Cai, G.; Zhang, X.; Liu, X.; Wang, P.; Zheng, A. Comparative Analysis of Bisexual and Parthenogenetic Populations in Haemaphysalis longicornis. Microorganisms 2024, 12, 823. [Google Scholar] [CrossRef]
- Chu, C.; Wang, Y.; Liu, Y.; Jiang, L.; He, F. Advances in Species Coexistence Theory. Biodivers. Sci. 2017, 25, 345. [Google Scholar] [CrossRef]
- Jia, N.; Wang, J.; Shi, W.; Du, L.; Ye, R.-Z.; Zhao, F.; Cao, W.-C. Haemaphysalis Longicornis. Trends Genet. 2021, 37, 292–293. [Google Scholar] [CrossRef] [PubMed]
- Heath, A. Biology, Ecology and Distribution of the Tick, Haemaphysalis longicornis Neumann (Acari: Ixodidae) in New Zealand. Vet. J. 2016, 64, 10–20. [Google Scholar] [CrossRef]
- Chen, X.; Xu, S.; Yu, Z.; Guo, L.; Yang, S.; Liu, L.; Yang, X.; Liu, J. Multiple Lines of Evidence on the Genetic Relatedness of the Parthenogenetic and Bisexual Haemaphysalis longicornis (Acari: Ixodidae). Infect. Genet. Evol. 2014, 21, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, C.; Cheng, C.; Zhang, G.; Yu, T.; Lawrence, K.; Li, H.; Sun, J.; Yang, Z.; Ye, L.; et al. Rapid Spread of Severe Fever with Thrombocytopenia Syndrome Virus by Parthenogenetic Asian Longhorned Ticks. Emerg. Infect. Dis. 2022, 28, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Sungirai, M.; Moyo, D.; De Clercq, P.; Madder, M.; Vanwambeke, S.; De Clercq, E. Modelling the Distribution of Rhipicephalus microplus and R. decoloratus in Zimbabwe. Vet. Parasitol. 2018, 14, 41–49. [Google Scholar] [CrossRef]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-Genome Resequencing Reveals World-Wide Ancestry and Adaptive Introgression Events of Domesticated Cattle in East Asia. Nat. Commun. 2018, 9, 2337. [Google Scholar] [CrossRef]
- Yu, Y.; Lian, L.-S.; Wen, J.-K.; Shi, X.-W.; Zhu, F.-X.; Nie, L.; Zhang, Y.-P. Genetic Diversity and Relationship of Yunnan Native Cattle Breeds and Introduced Beef Cattle Breeds. Biochem. Genet. 2004, 42, 1–9. [Google Scholar] [CrossRef]
- Mccoy, K.; Léger, E.; Dietrich, M. Host Specialization in Ticks and Transmission of Tick-Borne Diseases: A Review. Front. Cell. Infect. Microbiol. 2013, 3, 57. [Google Scholar]
- Layer, G.; Reichelt, J.; Jahn, D.; Heinz, D.W. Structure and Function of Enzymes in Heme Biosynthesis. Protein Sci. 2010, 19, 1137–1161. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.U.; Brown, B.L. Structural Basis for Allostery in PLP-Dependent Enzymes. Front. Mol. Biosci. 2022, 9, 884281. [Google Scholar] [CrossRef] [PubMed]
- Ha, E.-M.; Lee, K.-A.; Seo, Y.Y.; Kim, S.-H.; Lim, J.-H.; Oh, B.-H.; Kim, J.; Lee, W.-J. Coordination of Multiple Dual Oxidase–Regulatory Pathways in Responses to Commensal and Infectious Microbes in Drosophila Gut. Nat. Immunol. 2009, 10, 949–957. [Google Scholar] [CrossRef]
- Kumar, S.; Molina-Cruz, A.; Gupta, L.; Rodrigues, J.; Barillas-Mury, C. A Peroxidase/Dual Oxidase System Modulates Midgut Epithelial Immunity in Anopheles Gambiae. Science 2010, 327, 1644–1648. [Google Scholar] [CrossRef]
- Yang, X.; Smith, A.A.; Williams, M.S.; Pal, U. A Dityrosine Network Mediated by Dual Oxidase and Peroxidase Influences the Persistence of Lyme Disease Pathogens within the Vector. J. Biol. Chem. 2014, 289, 12813–12822. [Google Scholar] [CrossRef]
- Bowman, A.S.; Sauer, J.R. Tick Salivary Glands: Function, Physiology and Future. Parasitology 2004, 129, S67–S81. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Renneker, S.; Beyer, D.; Kullmann, B.; Seitzer, U.; Ahmed, J.; Bakheit, M.A. Identification and Partial Characterization of a Salp15 Homolog from Ixodes Ricinus. Ticks Tick. Borne Dis. 2014, 5, 318–322. [Google Scholar] [CrossRef]
- Mans, B.J. Evolution of Vertebrate Hemostatic and Inflammatory Control Mechanisms in Blood-Feeding Arthropods. J. Innate Immun. 2011, 3, 41–51. [Google Scholar] [CrossRef]
- Šimo, L.; Kazimirova, M.; Richardson, J.; Bonnet, S.I. The Essential Role of Tick Salivary Glands and Saliva in Tick Feeding and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 281. [Google Scholar] [CrossRef]
- Sabadin, G.A.; Salomon, T.B.; Leite, M.S.; Benfato, M.S.; Oliveira, P.L.; da Silva Vaz, I. An Insight into the Functional Role of Antioxidant and Detoxification Enzymes in Adult Rhipicephalus microplus Female Ticks. Parasitol. Int. 2021, 81, 102274. [Google Scholar] [CrossRef]
- Freitas, D.R.J.; Rosa, R.M.; Moura, D.J.; Seitz, A.L.; Colodel, E.M.; Driemeier, D. Cell Death during Preoviposition Period in Boophilus microplus Tick. Vet. Parasitol. 2007, 144, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Becker, S.; Eberle, A.; Grether-Beck, S.; Häussinger, D. Involvement of NADPH Oxidase Isoforms and Src Family Kinases in CD95-Dependent Hepatocyte Apoptosis. J. Biol. Chem. 2005, 280, 27179–27194. [Google Scholar] [CrossRef]
- Khan, S.J.; Abidi, S.N.F.; Skinner, A.; Tian, Y.; Smith-Bolton, R.K. The Drosophila Duox Maturation Factor Is a Key Component of a Positive Feedback Loop That Sustains Regeneration Signaling. PLoS Genet. 2017, 13, e1006937. [Google Scholar] [CrossRef]
- Palmer, W.J.; Jiggins, F.M. Comparative Genomics Reveals the Origins and Diversity of Arthropod Immune Systems. Mol. Biol. Evol. 2015, 32, 2111–2129. [Google Scholar] [CrossRef]
- Guizzo, M.G.; Budachetri, K.; Adegoke, A.; Ribeiro, J.M.C.; Karim, S. Rickettsia Parkeri Infection Modulates the Sialome and Ovariome of the Gulf Coast Tick, Amblyomma maculatum. Front. Microbiol. 2022, 13, 1023980. [Google Scholar] [CrossRef]
- Hajdusek, O.; Sojka, D.; Kopacek, P.; Buresova, V.; Franta, Z.; Sauman, I.; Winzerling, J.; Grubhoffer, L. Knockdown of Proteins Involved in Iron Metabolism Limits Tick Reproduction and Development. Proc. Natl. Acad. Sci. USA 2009, 106, 1033–1038. [Google Scholar] [CrossRef]
- Tabor, A.E.; Ali, A.; Rehman, G.; Rocha Garcia, G.; Zangirolamo, A.F.; Malardo, T.; Jonsson, N.N. Cattle Tick Rhipicephalus microplus-Host Interface: A Review of Resistant and Susceptible Host Responses. Front. Cell Infect. Microbiol. 2017, 7, 506. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.I.; Nijhof, A.M.; de la Fuente, J. Editorial: Tick-Host-Pathogen Interactions. Front. Cell Infect. Microbiol. 2018, 8, 194. [Google Scholar] [CrossRef]
- Artigas-Jerónimo, S.; Villar, M.; Cabezas-Cruz, A.; Valdés, J.J.; Estrada-Peña, A.; Alberdi, P. Functional Evolution of Subolesin/Akirin. Front. Physiol. 2018, 9, 1612. [Google Scholar] [CrossRef]
- Ali, A.; Mulenga, A.; Vaz, I.S.J. Editorial: Tick and Tick-Borne Pathogens: Molecular and Immune Targets for Control Strategies. Front. Physiol. 2020, 11, 744. [Google Scholar] [CrossRef]
- Kiewra, D.; Krysmann, A. Interactions between Hard Ticks (Ixodidae) and Bacterial Tick-Borne Pathogens. Ann. Parasitol. 2023, 69, 7–16. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, J.; Antunes, S.; Bonnet, S.; Cabezas-Cruz, A.; Domingos, A.G.; Estrada-Peña, A.; Johnson, N.; Kocan, K.M.; Mansfield, K.L.; Nijhof, A.M.; et al. Tick-Pathogen Interactions and Vector Competence: Identification of Molecular Drivers for Tick-Borne Diseases. Front. Cell. Infect. Microbiol. 2017, 7, 114. [Google Scholar] [CrossRef]
- Ouedraogo, A.S. Cattle Ticks and Associated Tick-Borne Pathogens in Burkina Faso and Benin: Apparent Northern Spread of Rhipicephalus Microplus in Benin and First Evidence of Theileria velifera and Theileria annulata. Ticks Tick-Borne Dis. 2021, 12, 101733. [Google Scholar] [CrossRef]
- Bickerton, M. A Life Stage-Targeted Acaricide Application Approach for the Control of Haemaphysalis longicornis. Ticks Tick-Borne Dis. 2021, 12, 101581. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, N.; Piper, P.; Constantinoiu, C. Host Resistance in Cattle to Infestation with the Cattle Tick Rhipicephalus microplus. Parasite Immunol. 2014, 36, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Toure, A.; Sanogo, M.; Sghiri, A.; Sahibi, H. Incidences of Rhipicephalus (Boophilus) Microplus (Canestrini, 1888) Transmitted Pathogens in Cattle in West Africa. Acta Parasitol 2022, 67, 1282–1289. [Google Scholar] [CrossRef]
- Kim, H.K. Rickettsia-Host-Tick Interactions: Knowledge Advances and Gaps. Infect. Immun. 2022, 90, e00621-21. [Google Scholar] [CrossRef]
- Jiang, N.; Tian, H.; Li, C.; Ma, R.; Liu, M.; Wang, S.; Zhou, Q.; Wei, X.; Mo, J.; Chen, Z.; et al. Species Composition, Genetic Structure, and Pathogen Prevalence in Tick Populations in Guangxi, China. Zoonoses 2025, 5, 998. [Google Scholar] [CrossRef]
- Bonnet, S.I. The Tick Microbiome: Why Non-Pathogenic Microorganisms Matter in Tick Biology and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 236. [Google Scholar] [CrossRef]
- Barillas-Mury, C.; Ribeiro, J.M.C.; Valenzuela, J.G. Understanding Pathogen Survival and Transmission by Arthropod Vectors to Prevent Human Disease. Science 2022, 377, eabc2757. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Zhou, A.; Liu, Q.; Gao, Y.; Xu, S.; Lu, Y. Genomic Insights into Vector–Pathogen Adaptation in Haemaphysalis longicornis and Rhipicephalus microplus. Pathogens 2025, 14, 306. https://doi.org/10.3390/pathogens14040306
Liu J, Zhou A, Liu Q, Gao Y, Xu S, Lu Y. Genomic Insights into Vector–Pathogen Adaptation in Haemaphysalis longicornis and Rhipicephalus microplus. Pathogens. 2025; 14(4):306. https://doi.org/10.3390/pathogens14040306
Chicago/Turabian StyleLiu, Jin, An Zhou, Qi Liu, Yang Gao, Shuhua Xu, and Yan Lu. 2025. "Genomic Insights into Vector–Pathogen Adaptation in Haemaphysalis longicornis and Rhipicephalus microplus" Pathogens 14, no. 4: 306. https://doi.org/10.3390/pathogens14040306
APA StyleLiu, J., Zhou, A., Liu, Q., Gao, Y., Xu, S., & Lu, Y. (2025). Genomic Insights into Vector–Pathogen Adaptation in Haemaphysalis longicornis and Rhipicephalus microplus. Pathogens, 14(4), 306. https://doi.org/10.3390/pathogens14040306