
Genotypic Characterization of Human Parvovirus B19 Circulating in the 2024 Outbreak in Tuscany, Italy



Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Serological and Molecular Analysis
2.3. Next Generation Sequencing
2.4. Phylogenetic Analysis
2.5. Evolutionary Rate and Selection Analysis
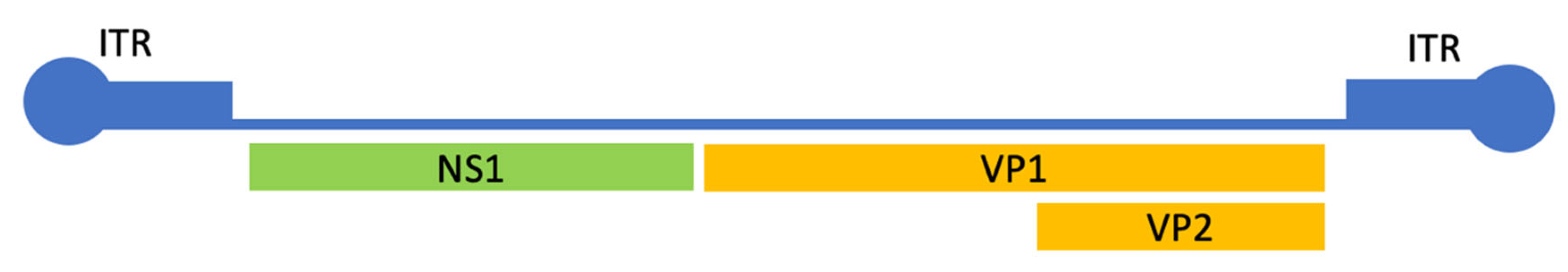
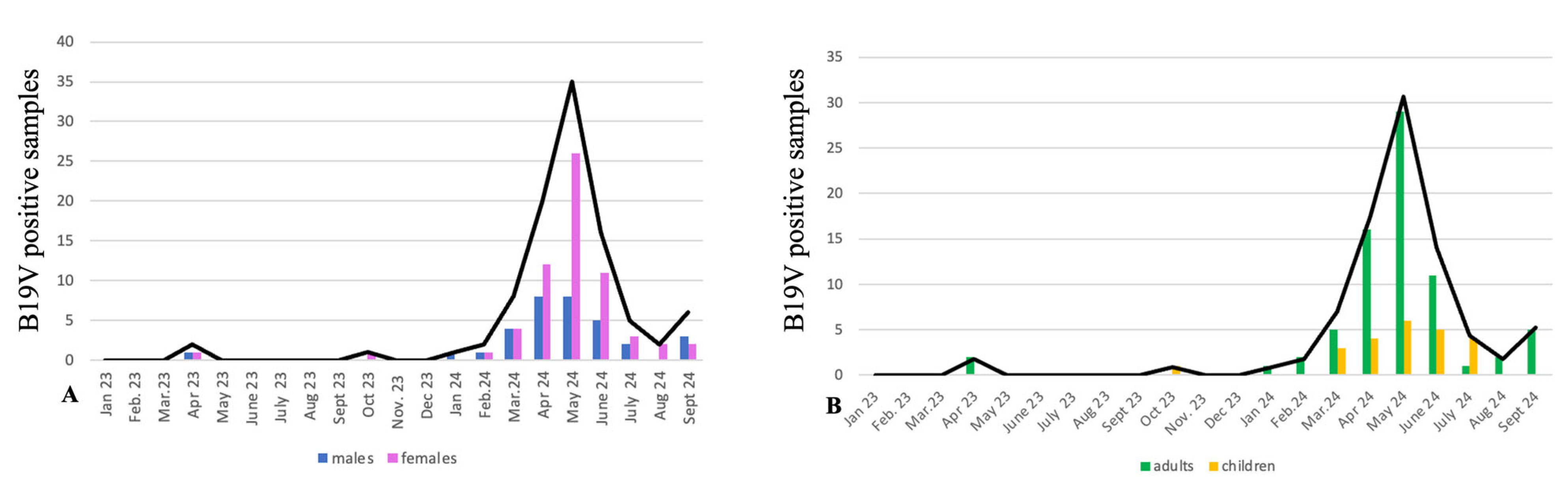
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Servey, J.T.; Reamy, B.V.; Hodge, J. Clinical presentations of parvovirus B19 infection. Am. Fam. Physician 2007, 75, 373–376. [Google Scholar] [PubMed]
- Badrinath, A.; Gardere, A.; Samantha, L.; Palermo, S.L.; Campbell, K.S.; Kloc, A. Analysis of Parvovirus B19 persistence and reactivation in human heart layers. Front. Virol. 2024, 4, 1304779. [Google Scholar] [CrossRef]
- Lehmann, T.; von Landenberg, P.; Modrow, S. Parvovirus B19 and Autoimmunity: A Review of Current Findings in Adult Infections. Autoimmun. Rev. 2003, 2, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, F.P.; De Moura Guimarães, C.; Alberto Borges Peixoto, A.; Monezi Pontes, K.F.; Bonasoni, M.P.; Tonni, G.; Araujo Júnior, E. Parvovirus B19 Infection and Pregnancy: Review of the Current Knowledge. J. Pers. Med. 2024, 14, 139. [Google Scholar] [CrossRef] [PubMed]
- De Cnc Garcia, R.; Leon, L.A. Human Parvovirus B19: A Review of Clinical and Epidemiologial Aspects in Brazil. Future Microbiol. 2021, 16, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Gallinella, G. Parvoviridae. In Encyclopedia of Infection and Immunity; Rezaei, N., Ed.; Elsevier: Oxford, UK, 2022; pp. 259–277. [Google Scholar]
- Enders, M.; Weidner, A.; Enders, G. Current epidemiological aspects of human parvovirus B19 infection during pregnancy and childhood in the western part of Germany. Epidemiol. Infect. 2007, 135, 563–569. [Google Scholar] [CrossRef]
- Vyse, A.J.; Andrews, N.J.; Hesketh, L.M.; Pebody, R. The burden of parvovirus B19 infection in women of childbearing age in England and Wales. Epidemiol. Infect. 2007, 135, 1354–1362. [Google Scholar] [CrossRef]
- d’Humières, C.; Fouillet, A.; Verdurme, L.; Lakoussan, S.-B.; Gallien, Y.; Coignard, C.; Hervo, M.; Ebel, A.; Visseaux, B.; Maire, B.; et al. An unusual outbreak of parvovirus B19 infections, France, 2023 to 2024. Euro Surveill. 2024, 29, 2400339. [Google Scholar] [CrossRef]
- Tassis, B.; Testa, L.; Pampo, F.; Boito, S.; Tiso, G.; Accurti, V.; Cetin, I.; Persico, N. The 2024 Outbreak of Parvovirus B19 as a Global Obstetrical Threat Insights From an Obstetrics Referral Center in Northern Italy. J. Med. Virol. 2024, 11, e70046. [Google Scholar] [CrossRef]
- Makhlouf, M.M.; Elwakil, S.G.; Ibrahim, N. Molecular and serological assessment of parvovirus B-19 infection in Egyptian children with sickle cell disease. J. Microbiol. Immunol. Infect. 2017, 50, 565–569. [Google Scholar] [CrossRef]
- Zou, W.; Wang, Z.; Xiong, M.; Chen, A.Y.; Xu, P.; Ganaie, S.S.; Badawi, Y.; Kleiboeker, S.; Nishimune, H.; Ye, S.Q.; et al. Human Parvovirus B19 Utilizes Cellular DNA Replication Machinery for Viral DNA Replication. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Ganaie Safder, S.; Qiu, J. Recent Advances in Replication and Infection of Human Parvovirus B19. Front. Cell. Infect. Microbiol. 2018, 8, 166. [Google Scholar]
- Luo, Y.; Qiu, J. Human parvovirus B19: A mechanistic overview of infection and DNA replication. Future Virol. 2015, 10, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, G.; Branda, F.; Ciccozzi, A.; Romano, C.; Sanna, D.; Casu, M.; Albanese, M.; Alessandri, F.; d’Ettorre, G.; Ciccozzi, M.; et al. Reassessing the Risk of Severe Parvovirus B19 Infection in the Immunocompetent Population: A Call for Vigilance in the Wake of Resurgence. Viruses 2024, 16, 1352. [Google Scholar] [CrossRef]
- Servant, A.; Laperche, S.; Lallemand, F.; Marinho, V.; De Saint Maur, G.; Meritet, J.F.; Garbarg-Chenon, A. Genetic diversity within human erythroviruses: Identification of three genotypes. J. Virol. 2002, 76, 9124–9134. [Google Scholar] [CrossRef]
- Huübschen, J.M.; Mihneva, Z.; Mentis, A.F.; Schneider, F.; Aboudy, Y.; Grossman, Z.; Rudich, H.; Kasymbekova, K.; Sarv, I.; Nedeljkovic, J.; et al. Phylogenetic analysis of human parvovirus B19 sequences from eleven different countries confirms the predominance of genotype 1 and suggests the spread of genotype 3b. J. Clin. Microbiol. 2009, 47, 3735–3738. [Google Scholar] [CrossRef]
- Gallinella, G.; Venturoli, S.; Manaresi, E.; Musiani, M.; Zerbini, M. B19 virus genome diversity: Epidemiological and clinical correlations. J. Clin. Virol. 2003, 28, 1–13. [Google Scholar] [CrossRef]
- Eis-Hübinger, A.M.; Reber, U.; Edelmann, A.; Kalus, U.; Hofmann, J. Parvovirus B19 genotype 2 in blood donations. Transfusion 2014, 54, 1682–1684. [Google Scholar] [CrossRef]
- Cong, B.; Koç, U.; Bandeira, T.; Bassat, Q.; Bont, L.; Chakhunashvili, G.; Cohen, C.; Desnoyers, C.; Hammitt, L.L.; Heikkinen, T.; et al. Changes in the global hospitalisation burden of respiratory syncytial virus in young children during the COVID-19 pandemic: A systematic analysis. Lancet Infect. Dis. 2024, 46, 361–374. [Google Scholar] [CrossRef]
- Gates, S.; Andreani, J.; Dewar, R.; Smith, D.B.; Templeton, K.; Child, H.T.; Bruer, J.; Golubchik, T.; Wade, M.J.; Jeffries, A.R.; et al. Postpandemic rebound of adeno-associated virus type 2 (AAV2) infections temporally associated with an outbreak of unexplained severe acute hepatitis in children in the United Kingdom. J. Med. Virol. 2023, 95, e28921. [Google Scholar] [CrossRef]
- Forero, E.L.; Knoester, M.; Gard, L.; Ott, A.; Brandenburg, A.H.; McCall, M.B.; Niesters, H.G.; Van Leer-Buter, C. Changes in enterovirus epidemiology after easing of lockdown measures. J. Clin. Virol. 2023, 169, 105617. [Google Scholar] [CrossRef] [PubMed]
- Molenaar-de Backer, M.W.; Hogema, B.M.; Koppelman, M.H.; van de Laar, T.J.; Slot, E.; Zaaijer, H.L. Lower Incidence of Parvovirus-B19 Infections in Dutch Blood Donors during SARS-CoV-2 Pandemic. Microbiol. Spectr. 2021, 9, e00253-21. [Google Scholar] [CrossRef] [PubMed]
- Russcher, A.; van Boven, M.; Benincà, E.; Verweij, E.J.T.; Molenaar-de Backer, M.W.; Zaaijer, H.L.; Vossen, A.C.T.M.; Kroes, A.C.M. Changing epidemiology of parvovirus B19 in the Netherlands since 1990, including its re-emergence after the COVID-19 pandemic. Sci Rep. 2024, 14, 9630. [Google Scholar] [CrossRef] [PubMed]
- Fourgeaud, J.; Allali, S.; Toubiana, J.; Pinhas, Y.; Frange, P.; Leruez-Ville, M.; Cohen, J.F. Post-COVID-19 pandemic outbreak of severe Parvovirus B19 primary infections in Paris, France: 10-year interrupted time-series analysis (2012-2023). J. Clin. Virol. 2023, 167, 105576. [Google Scholar] [CrossRef] [PubMed]
- de Jong, E.P.; Walther, F.J.; Kroes, A.C.M.; Oepkes, D. Parvovirus B19 infection in pregnancy: New insights and management. Prenat. Diagn. 2011, 31, 419–425. [Google Scholar] [CrossRef]
- Jain, A.; Kant, R. Genotypes of erythrovirus B19, their geographical distribution & circulation in cases with various clinical manifestations. Indian J. Med. Res. 2018, 147, 239–247. [Google Scholar]
- Karlin, D.G. Parvovirus B19 and Human Parvovirus 4 Encode Similar Proteins in a Reading Frame Overlapping the VP1 Capsid Gene. Viruses 2024, 16, 191. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Holmes, E.C. Phylogenetic evidence for the rapid evolution of human B19 erythrovirus. J. Virol. 2006, 80, 3666–3669. [Google Scholar] [CrossRef]
- Stamenković, G.; Ćirković, V.; Šiljić, M.M.; Blagojević, J.V.; Knežević, A.M.; Joksić, I.D.; Stanojević, M.P. Substitution rate and natural selection in parvovirus B19. Sci. Rep. 2016, 6, 35759. [Google Scholar] [CrossRef]
- Jiang, H.; Qiu, Q.; Zhou, Y.; Xu, W.; Cui, A.; Li, X. The epidemiological and genetic characteristics of human parvovirus B19 in patients with febrile rash illnesses in China. Sci. Rep. 2023, 13, 15913. [Google Scholar] [CrossRef]
- Saikawa, T.; Anderson, S.; Momoeda, M.; Kajigaya, S.; Young, N.S. Neutralizing linear epitopes of B19 parvovirus cluster in the VP1 unique and VP1-VP2 junction regions. J. Virol. 1993, 67, 3004–3009. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Momoeda, M.; Kawase, M.; Kajigaya, S.; Young, N.S. Peptides derived from the unique region of B19 parvovirus minor capsid protein elicit neutralizing antibodies in rabbits. Virology 1995, 206, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Kurtzman, G.J.; Cohen, B.J.; Field, A.M.; Oseas, R.; Blaese, R.M.; Young, N.S. Immune response to B19 parvovirus and an antibody defect in persistent viral infection. J. Clin. Investig. 1989, 84, 1114–1123. [Google Scholar] [CrossRef]
- Kurtzman, G.; Frickhofen, N.; Kimball, J.; Jenkins, D.W.; Nienhuis, A.W.; Young, N.S. Pure Red-Cell Aplasia of 10 Years’ Duration Due to Persistent Parvovirus B19 Infection and Its Cure with Immunoglobulin Therapy. N. Engl. J. Med. 1989, 321, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Alidjinou, E.K.; De Coninck, L.; Swinnen, J.; Lazrek, M.; Hober, D.; Matthijnssens, J. Four complete genomes of human parvovirus B19 from amniotic fluid specimens. Microbiol. Resour. Announc. 2023, 12, e0055623. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Gene | Mutations |
|---|---|
| NS1 | 803G>A; 1175 C>T; 1227 A>T, 1430 A>T; 1140 C>T; 1529A>G; 1928 A>C; 2286 T>C |
| VP1/VP2 | 2754 C>T; 3351T>G; 3444 T>A; 3489 C>T; 3531 A>C; 3752 C>G; 3795 A>G; 3894 C>T; 4264C>T; 43069A>T; 4345 T>C; 4346 C>A. |
| Gene | Mutation | Frequency |
|---|---|---|
| NS1 | C17S | 9/23 |
| NS1 | F57L | 4/23 |
| NS1 | V71A | 5/23 |
| NS1 | L111F | 6/23 |
| NS1 | F554S | 9/23 |
| VP1u | K14E | 7/23 |
| VP1 | V30L | 18/23 |
| VP1u | S98N | 17/23 |
| VP1/VP2 | A260T-A33T | 6/23 |
| VP1/VP2 | N533S-N306S | 17/23 |
| VP1/VP2 | G537A-G310A | 9/23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beligni, G.; Alessandri, G.; Cusi, M.G. Genotypic Characterization of Human Parvovirus B19 Circulating in the 2024 Outbreak in Tuscany, Italy. Pathogens 2025, 14, 121. https://doi.org/10.3390/pathogens14020121
Beligni G, Alessandri G, Cusi MG. Genotypic Characterization of Human Parvovirus B19 Circulating in the 2024 Outbreak in Tuscany, Italy. Pathogens. 2025; 14(2):121. https://doi.org/10.3390/pathogens14020121
Chicago/Turabian StyleBeligni, Giada, Giulia Alessandri, and Maria Grazia Cusi. 2025. "Genotypic Characterization of Human Parvovirus B19 Circulating in the 2024 Outbreak in Tuscany, Italy" Pathogens 14, no. 2: 121. https://doi.org/10.3390/pathogens14020121
APA StyleBeligni, G., Alessandri, G., & Cusi, M. G. (2025). Genotypic Characterization of Human Parvovirus B19 Circulating in the 2024 Outbreak in Tuscany, Italy. Pathogens, 14(2), 121. https://doi.org/10.3390/pathogens14020121