
An Investigation of Severe Influenza Cases in Russia during the 2022–2023 Epidemic Season and an Analysis of HA-D222G/N Polymorphism in Newly Emerged and Dominant Clade 6B.1A.5a.2a A(H1N1)pdm09 Viruses
 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Influenza Diagnostics
2.2. Sequence Analysis of Influenza Viruses
2.3. Virus Isolation and Antigenic Characterization
2.4. Phenotypic Analysis of Neuraminidase Inhibitors Susceptibility
3. Results
3.1. Detection of Influenza Cases in Russia during the 2022–2023 Flu Season
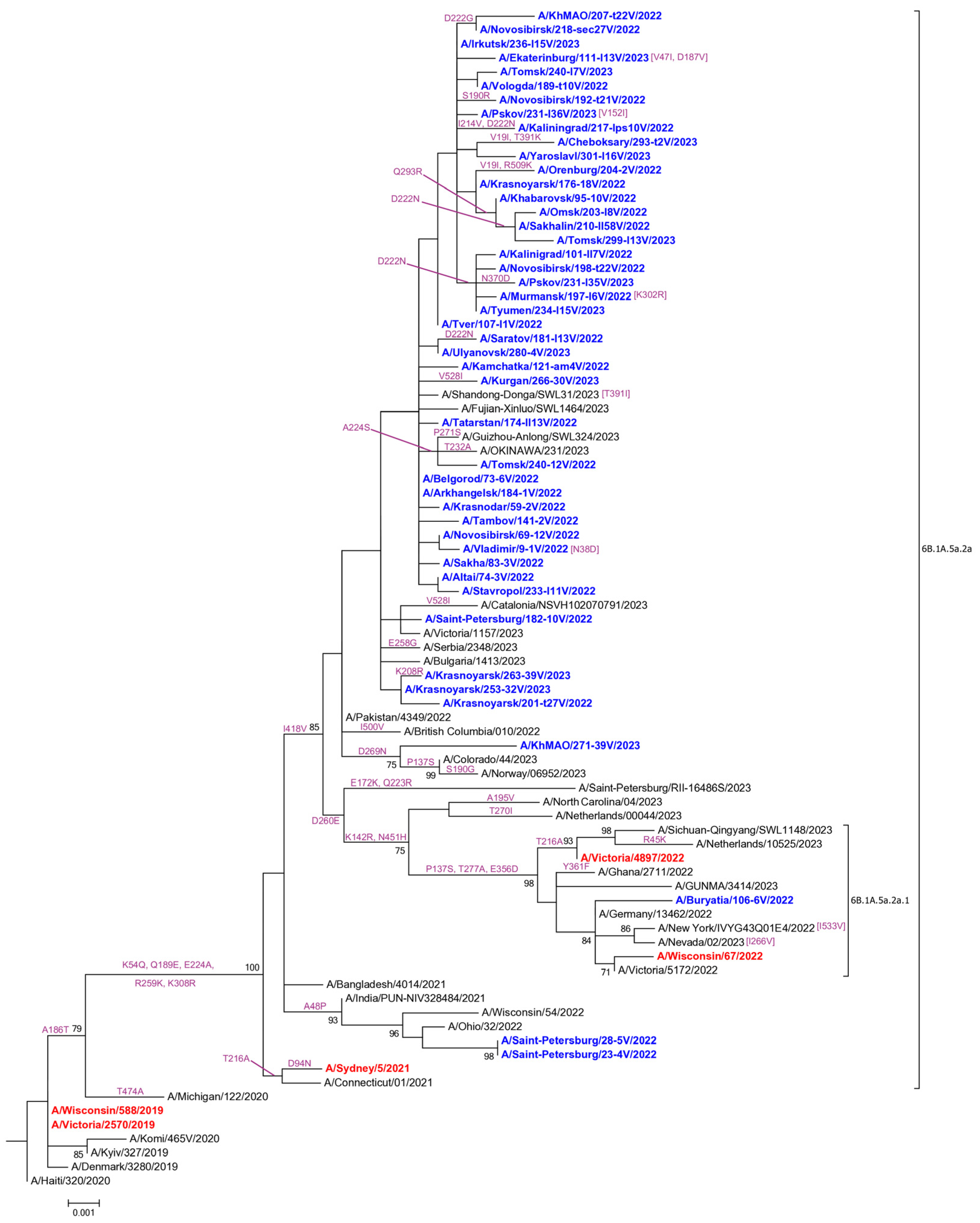
3.2. Genetic and Virological Analysis of Influenza A(H1N1)pdm09 Viruses
3.3. Genetic and Virological Analysis of Influenza A(H3N2) Viruses
3.4. Genetic and Virological Analysis of Influenza B Viruses
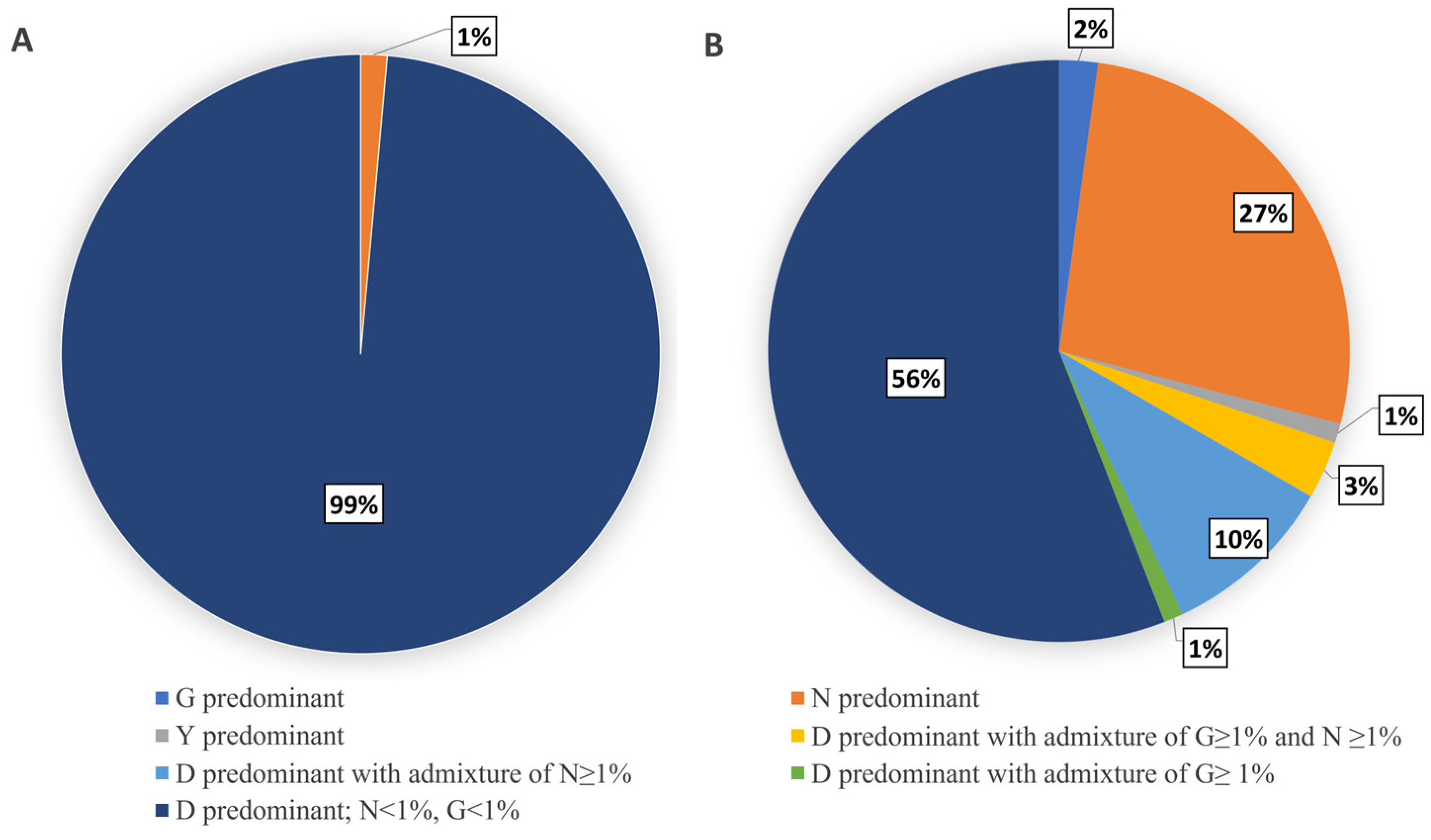
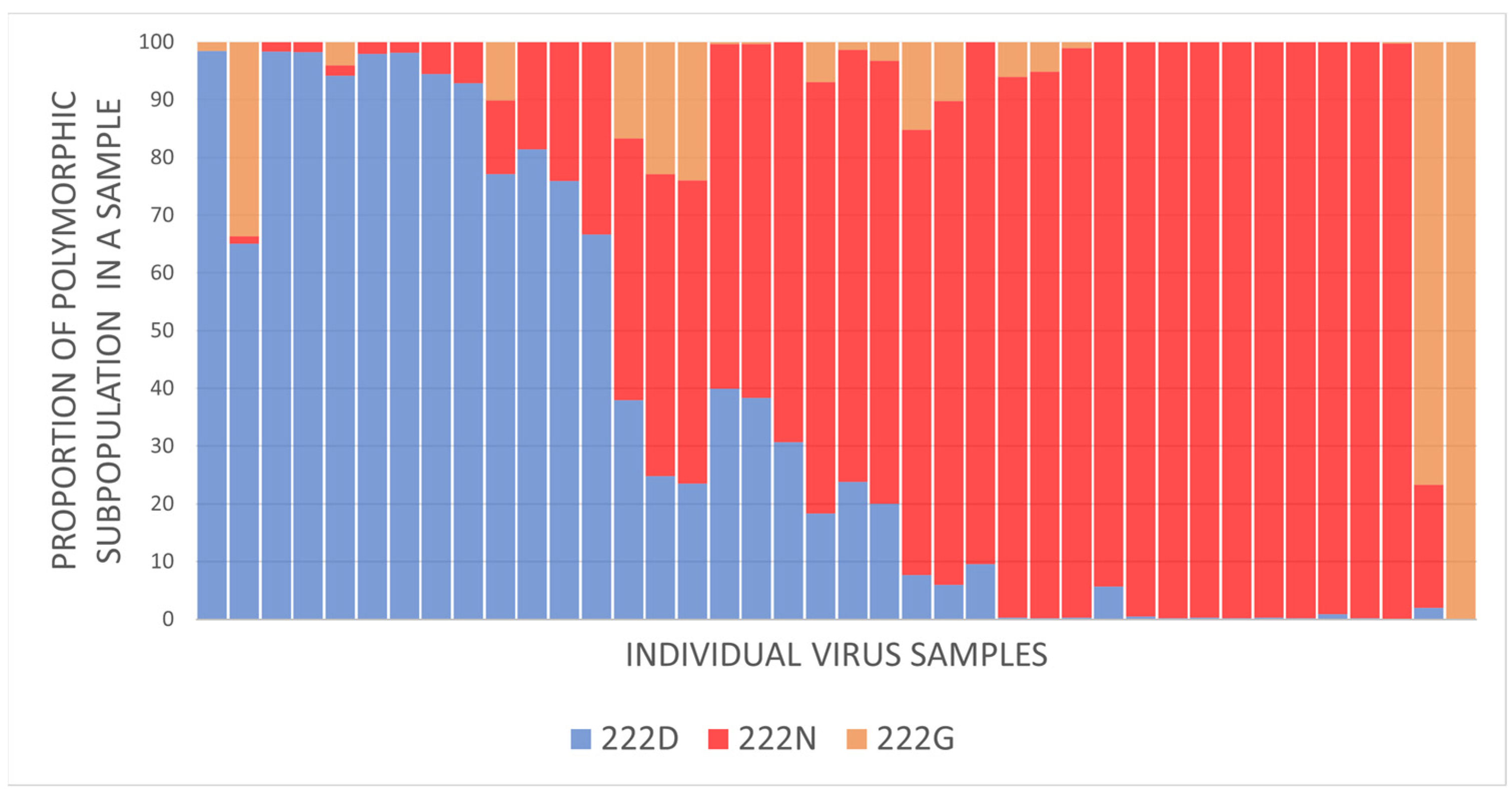
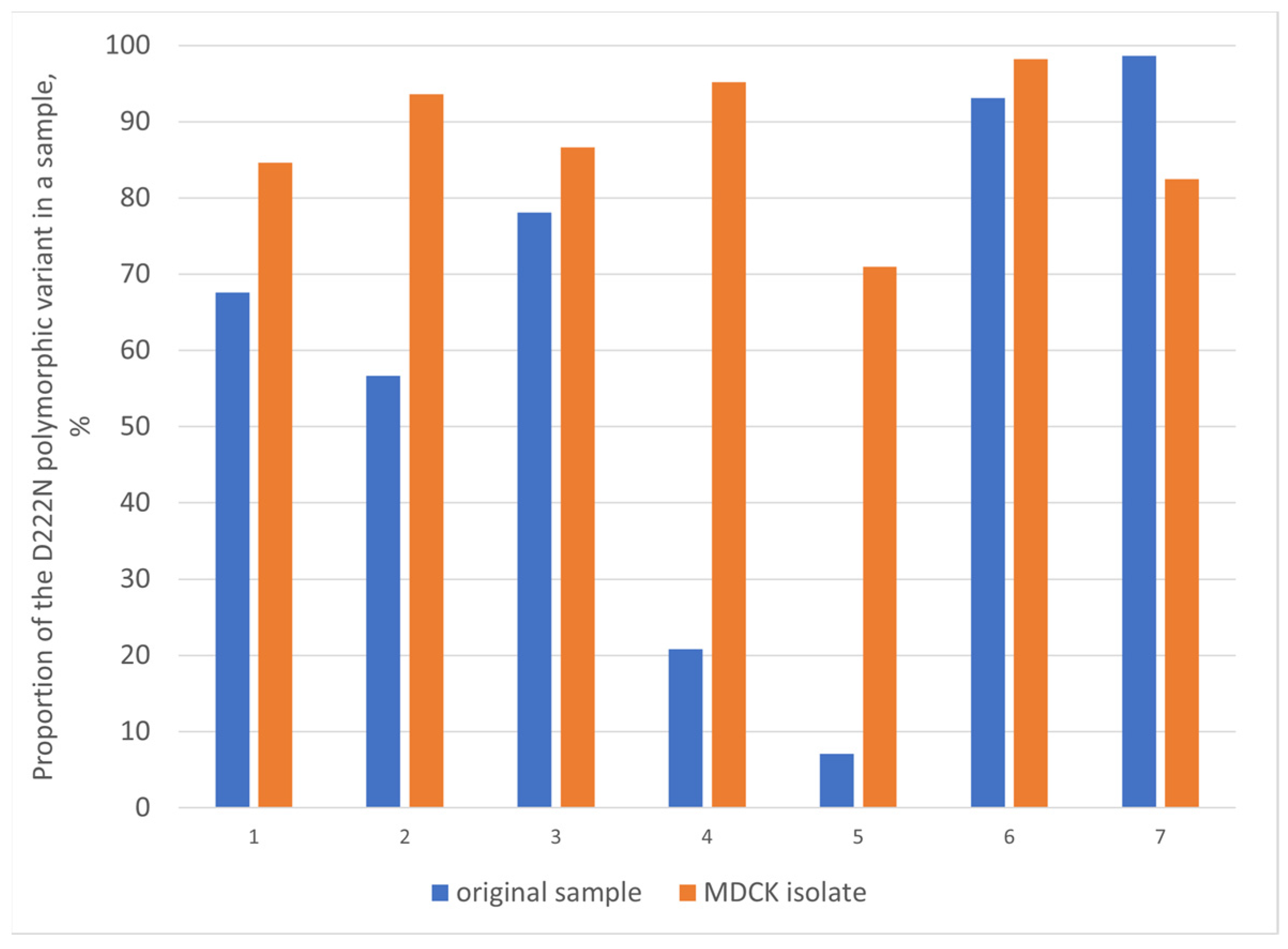
3.5. Evaluation of HA-D222G/N Polymorphism in Influenza A(H1N1)pdm09 Viruses
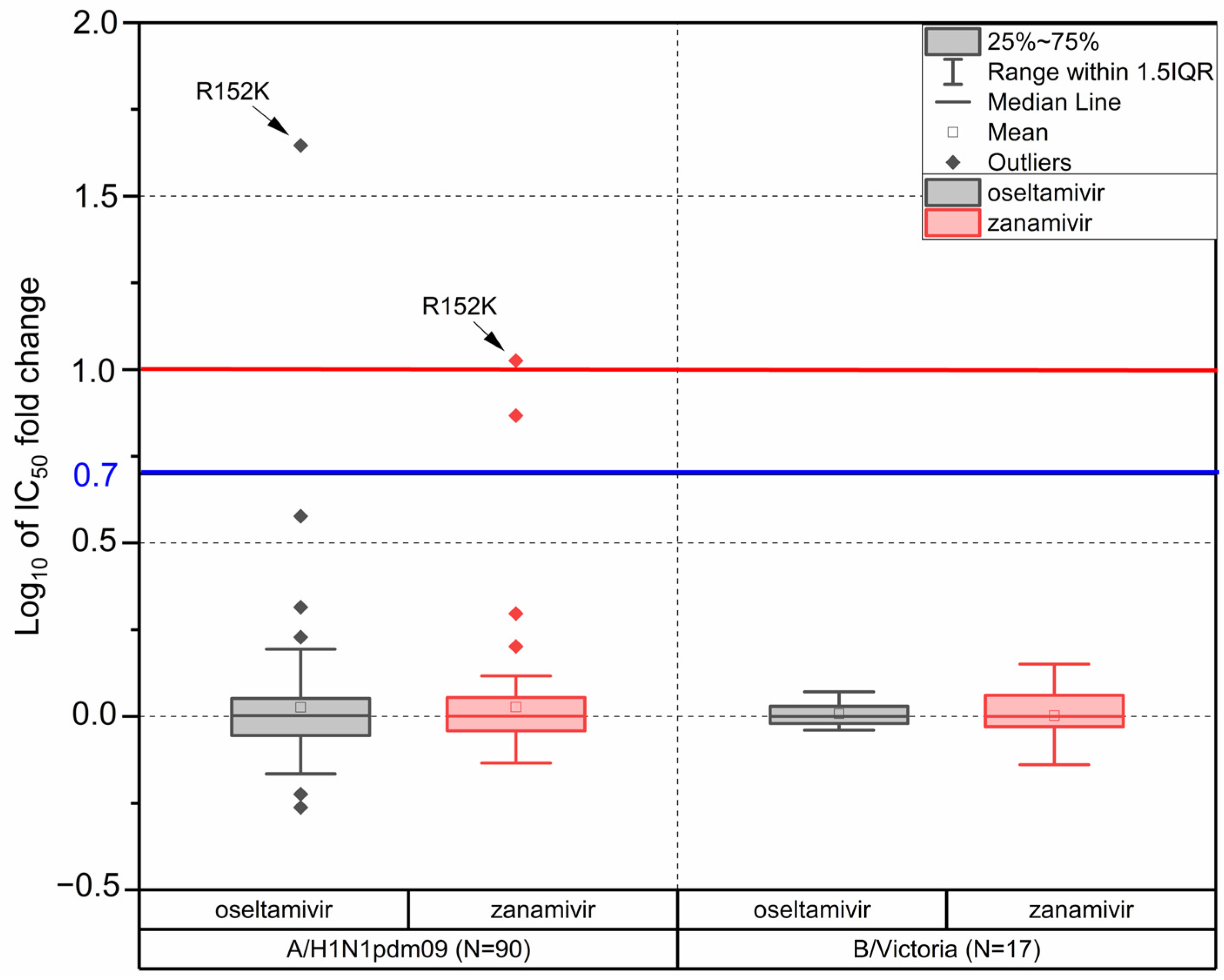
3.6. Drug Susceptibility
4. Discussion
4.1. Re-Emergence of a Widespread Circulation of Influenza A(H1N1pdm09) and B/Victoria Viruses in Russia in 2022–2023
4.2. Genetic and Antigenic Characterization of Influenza Viruses
4.2.1. Influenza A(H1N1)pdm09 Viruses
4.2.2. Influenza A(H3N2) Viruses
4.2.3. Influenza B Viruses
4.3. Drug Susceptibility of Influenza Viruses
4.4. Evaluation of HA-D222G/N Polymorphism in Influenza A(H1N1)pdm09 Viruses
4.4.1. Detection of HA-D222G/N Polymorphism in A(H1N1)pdm09 Viruses from Fatal Cases
4.4.2. Limited Detection of HA-D222G/N Substitutions in Influenza A(H1N1)pdm09 Viruses in Broad Circulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uyeki, T.M. High-risk Groups for Influenza Complications. JAMA 2020, 324, 2334. [Google Scholar] [CrossRef]
- Adlhoch, C.; Mook, P.; Lamb, F.; Ferland, L.; Melidou, A.; Amato-Gauci, A.J.; Pebody, R.; Network, T.E.I.S. Decreased Influenza Activity During the COVID-19 Pandemic—United States, Australia, Chile, and South Africa, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1305–1309. [Google Scholar] [CrossRef]
- Adlhoch, C.; Mook, P.; Lamb, F.; Ferland, L.; Melidou, A.; Amato-Gauci, A.J.; Pebody, R. European Influenza Surveillance Network.Very little influenza in the WHO European Region during the 2020/21 season, weeks 40 2020 to 8 2021. Euro Surveill. 2021, 26, 2100221. [Google Scholar] [CrossRef] [PubMed]
- Bolton, M.J.; Ort, J.T.; McBride, R.; Swanson, N.J.; Wilson, J.; Awofolaju, M.; Furey, C.; Greenplate, A.R.; Drapeau, E.M.; Pekosz, A.; et al. Antigenic and virological properties of an H3N2 variant that continues to dominate the 2021–22 Northern Hemisphere influenza season. Cell Rep. 2022, 39, 110897. [Google Scholar] [CrossRef] [PubMed]
- Influenza Virus Characterization: Summary Report, Europe, February 2023; WHO Regional Office for Europe and European Centre for Disease Prevention and Control: Copenhagen, Denmark; Stockholm, Sweden, 2023; Available online: https://www.ecdc.europa.eu/en/publications-data/influenza-virus-characterization-summary-europe-february-2023 (accessed on 1 October 2023).
- World Health Organization. Recommended Composition of Influenza Virus Vaccines for Use in the 2023–2024 Northern Hemisphere Influenza Season—Composition Recommandée des Vaccins Antigrippaux pour la Saison Grippale 2023–2024 dans L’hémisphère Nord. Wkly Epidemiol. Rec. 2023, 98, 99–110. Available online: https://iris.who.int/handle/10665/366491 (accessed on 1 October 2023).
- World Health Organization. Recommended Composition of Influenza Virus Vaccines for Use in the 2024 Southern Hemisphere Influenza Season—Composition Recommandée des Vaccins Antigrippaux pour la Saison Grippale 2024 dans L’hémisphère Sud Wkly Epidemiol. Rec. 2023, 98, 521–554. Available online: https://iris.who.int/handle/10665/373646 (accessed on 1 October 2023).
- Sominina, A.; Danilenko, D.; Komissarov, A.B.B.; Pisareva, M.; Fadeev, A.V.; Konovalova, N.; Eropkin, M.; Petrova, P.; Zheltukhina, A.; Musaeva, T.D.; et al. Assessing the Intense Influenza A(H1N1)pdm09 Epidemic and Vaccine Effectiveness in the Post-COVID Season in the Russian Federation. Viruses 2023, 15, 1780. [Google Scholar] [CrossRef]
- Goka, E.A.; Vallely, P.J.; Mutton, K.J.; Klapper, P.E. Mutations associated with severity of the pandemic influenza A(H1N1)pdm09 in humans: A systematic review and meta-analysis of epidemiological evidence. Arch. Virol. 2014, 159, 3167–3183. [Google Scholar] [CrossRef]
- Danilenko, A.V.; Kolosova, N.P.; Shvalov, A.N.; Ilyicheva, T.N.; Svyatchenko, S.V.; Durymanov, A.G.; Bulanovich, J.A.; Goncharova, N.I.; Susloparov, I.M.; Marchenko, V.Y.; et al. Evaluation of HA-D222G/N polymorphism using targeted NGS analysis in A(H1N1)pdm09 influenza virus in Russia in 2018–2019. PLoS ONE 2021, 16, e0251019. [Google Scholar] [CrossRef]
- Kolosova, N.P.; Ilyicheva, T.N.; Danilenko, A.V.; Svyatchenko, S.V.; Goncharova, N.I.; Bulanovich, J.A.; Torzhkova, P.Y.; Durymanov, A.G.; Gudymo, A.S.; Shvalov, A.N.; et al. Severe cases of seasonal influenza and detection of seasonal A(H1N2) in Russia in 2018–2019. Arch. Virol. 2020, 165, 2045–2051. [Google Scholar] [CrossRef]
- Kolosova, N.P.; Ilyicheva, T.N.; Danilenko, A.V.; Bulanovich, J.A.; Svyatchenko, S.V.; Durymanov, A.G.; Goncharova, N.I.; Gudymo, A.S.; Shvalov, A.N.; Susloparov, I.M.; et al. Severe cases of seasonal influenza in Russia in 2017-2018. PLoS ONE 2019, 14, e0220401. [Google Scholar] [CrossRef] [PubMed]
- Ilyicheva, T.; Durymanov, A.; Susloparov, I.; Kolosova, N.; Goncharova, N.; Svyatchenko, S.; Petrova, O.; Bondar, A.; Mikheev, V.; Ryzhikov, A. Fatal Cases of Seasonal Influenza in Russia in 2015–2016. PLoS ONE 2016, 11, e0165332. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.H.; Janczewski, T.A.; Lourens, R.M.; Elliot, A.J.; Daniels, R.S.; Berry, C.L.; Oxford, J.S.; Taubenberger, J.K. 1918 influenza pandemic caused by highly conserved viruses with two receptor-binding variants. Emerg. Infect. Dis. 2003, 9, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Preliminary Review of D222G Amino Acid Substitution in the Haemagglutinin of Pandemic Influenza A (H1N1) 2009 Viruses. [Internet]. Vol. 85, Weekly Epidemiological Record = Relevé Épidémiologique Hebdomadaire. 2010, pp. 21–22. Available online: https://apps.who.int/iris/handle/10665/241505 (accessed on 1 October 2023).
- Zheng, B.; Chan, K.-H.; Zhang, A.J.X.; Zhou, J.; Chan, C.C.S.; Poon, V.K.M.; Zhang, K.; Leung, V.H.C.; Jin, D.-Y.; Woo, P.C.Y.; et al. D225G mutation in hemagglutinin of pandemic influenza H1N1 (2009) virus enhances virulence in mice. Exp. Biol. Med. 2010, 235, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Bao, L.; Lv, Q.; Deng, W.; Ma, Y.; Li, F.; Zhan, L.; Zhu, H.; Ma, C.; Qin, C. A single-amino-acid substitution in the HA protein changes the replication and pathogenicity of the 2009 pandemic A (H1N1) influenza viruses in vitro and in vivo. Virol. J. 2010, 7, 325. [Google Scholar] [CrossRef]
- Chan, K.-H.; Zhang, A.J.X.; To, K.K.W.; Chan, C.C.S.; Poon, V.K.M.; Guo, K.; Ng, F.; Zhang, Q.-W.; Leung, V.H.C.; Cheung, A.N.Y.; et al. Wild type and mutant 2009 pandemic influenza a (H1N1) viruses cause more severe disease and higher mortality in pregnant balb/c mice. PLoS ONE 2010, 5, e13757. [Google Scholar] [CrossRef]
- Belser, J.A.; Jayaraman, A.; Raman, R.; Pappas, C.; Zeng, H.; Cox, N.J.; Katz, J.M.; Sasisekharan, R.; Tumpey, T.M. Effect of D222G mutation in the hemagglutinin protein on receptor binding, pathogenesis and transmissibility of the 2009 pandemic H1N1 influenza virus. PLoS ONE 2011, 6, e25091. [Google Scholar] [CrossRef]
- World Health Organization. Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza; WHO Press: Geneva, Switzerland, 2011. [Google Scholar]
- St George, K. Diagnosis of Influenza Virus. In Influenza Virus Methods and Protocols; Humana: Totowa, NJ, USA, 2012; pp. 53–71. [Google Scholar]
- Deng, Y.-M.; Spirason, N.; Iannello, P.; Jelley, L.; Lau, H.; Barr, I.G. A simplified Sanger sequencing method for full genome sequencing of multiple subtypes of human influenza A viruses. J. Clin. Virol. 2015, 68, 43–48. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Leang, S.-K.; Hurt, A.C. Fluorescence-based Neuraminidase Inhibition Assay to Assess the Susceptibility of Influenza Viruses to The Neuraminidase Inhibitor Class of Antivirals. J. Vis. Exp. 2017, e55570. [Google Scholar] [CrossRef]
- Zhou, L.; Feng, Z.; Liu, J.; Chen, Y.; Yang, L.; Liu, S.; Li, X.; Gao, R.; Zhu, W.; Wang, D.; et al. A single N342D substitution in Influenza B Virus NA protein determines viral pathogenicity in mice. Emerg. Microbes Infect. 2020, 9, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Summary of Neuraminidase (NA) Amino Acid Substitutions Associated with Reduced Inhibition by Neuraminidase Inhibitors (NAIs). Available online: https://www.who.int/publications/m/item/summary-of-neuraminidase-(na)-amino-acid-substitutions-associated-with-reduced-inhibition-by-neuraminidase-inhibitors-(nais) (accessed on 27 October 2023).
- Summary of Polymerase Acidic (PA) Protein Amino Acid Substitutions Analysed for Their Effects On Baloxavir Susceptibility. Available online: https://www.who.int/publications/m/item/summary-of-polymerase-acidic-(pa)-protein-amino-acid-substitutions-analysed-for-their-effects-on-baloxavir-susceptibility (accessed on 27 October 2023).
- Meetings of the WHO working group on surveillance of influenza antiviral susceptibility—Geneva, November 2011 and June 2012. Wkly. Epidemiol. Rec. 2012, 87, 369–374.
- Jones, R.P.; Ponomarenko, A. Roles for Pathogen Interference in Influenza Vaccination, with Implications to Vaccine Effectiveness (VE) and Attribution of Influenza Deaths. Infect. Dis. Rep. 2022, 14, 710–758. [Google Scholar] [CrossRef] [PubMed]
- Lytras, T.; Andreopoulou, A.; Gkolfinopoulou, K.; Mouratidou, E.; Tsiodras, S. Association between type-specific influenza circulation and incidence of severe laboratory-confirmed cases; which subtype is the most virulent? Clin. Microbiol. Infect. 2019, 26, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E. Mortality Associated with Different Influenza Subtypes in France between 2015–2019. medRxiv 2022. Available online: http://medrxiv.org/content/early/2022/11/22/2022.11.21.22282612.abstract (accessed on 29 November 2023).
- Sumner, K.M.; Masalovich, S.; O’Halloran, A.; Holstein, R.; Reingold, A.; Kirley, P.D.; Alden, N.B.; Herlihy, R.K.; Meek, J.; Yousey-Hindes, K.; et al. Severity of influenza-associated hospitalisations by influenza virus type and subtype in the USA, 2010–2019: A repeated cross-sectional study. Lancet Microbe 2023, 4, e903–e912. [Google Scholar] [CrossRef]
- Pediatric Flu Deaths during 2019–2020 Reach New High. Available online: https://www.cdc.gov/flu/spotlights/2020-2021/pediatric-flu-deaths-reach-new-high.htm (accessed on 27 October 2023).
- Özkaya, P.Y.; Turanli, E.E.; Metïn, H.; Uysal, A.A.; Çïçek, C.; Karapinar, B. Severe influenza virus infection in children admitted to the PICU: Comparison of influenza A and influenza B virus infection. J. Med. Virol. 2021, 94, 575–581. [Google Scholar] [CrossRef]
- World Health Organization. Recommended Composition of Influenza Virus Vaccines for Use in the 2021 Southern Hemisphere Influenza Season—Composition Recommandée des Vaccins Antigrippaux pour la Saison GRIPPALE 2021 dans l’hémisphère Sud. Wkly. Epidemiol. Rec. 2020, 95, 497–508. Available online: https://iris.who.int/handle/10665/336145 (accessed on 1 October 2023).
- Worldwide Influenza Centre; WHO CC for Reference and Research on Influenza; The Francis Crick Institute. Report Prepared for the WHO Annual Consultation on the Composition of Influenza Vaccines for the Northern Hemisphere 2023–2024. Available online: https://www.crick.ac.uk/sites/default/files/2023-03/F2023-VCM-seasonal_web.pdf (accessed on 27 October 2023).
- Chinese Weekly Influenza Surveillance Report. September 25 to October 1, 2023 (Week 39). Available online: https://ivdc.chinacdc.cn/cnic/en/Surveillance/WeeklyReport/202310/P020231007282742073293.pdf (accessed on 27 October 2023).
- Influenza Activity in the United States during the 2022–23 Season and Composition of the 2023–24 Influenza Vaccine. Available online: https://www.cdc.gov/flu/spotlights/2023-2024/22-23-summary-technical-report.htm (accessed on 27 October 2023).
- Sominina, A.; Danilenko, D.; Komissarov, A.; Karpova, L.; Pisareva, M.; Fadeev, A.; Konovalova, N.; Eropkin, M.; Stolyarov, K.; Shtro, A.; et al. Resurgence of Influenza Circulation in the Russian Federation during the Delta and Omicron COVID-19 Era. Viruses 2022, 14, 1909. [Google Scholar] [CrossRef]
- Influenza Virus Characterization: Summary Report, Europe, August 2023; WHO Regional Office for Europe: Copenhagen, Denmark; European Centre for Disease Prevention and Control: Stockholm, Sweden, 2023; Available online: https://www.ecdc.europa.eu/en/publications-data/influenza-virus-characterization-summary-europe-august-2023 (accessed on 1 October 2023).
- Govorkova, E.A.; Takashita, E.; Daniels, R.S.; Fujisaki, S.; Presser, L.D.; Patel, M.C.; Huang, W.; Lackenby, A.; Nguyen, H.T.; Pereyaslov, D.; et al. Global update on the susceptibilities of human influenza viruses to neuraminidase inhibitors and the cap-dependent endonuclease inhibitor baloxavir, 2018–2020. Antivir. Res. 2022, 200, 105281. [Google Scholar] [CrossRef] [PubMed]
- L’vov, D.K.; Burtseva, E.I.; Prilipov, A.G.; Bogdanova, V.S.; Shchelkanov, M.I.; Bovin, N.V.; Samokhvalov, E.I.; Fediakina, I.T.; Deriabin, P.G.; Kolobukhina, L.V.; et al. A possible association of fatal pneumonia with mutations of pandemic influenza A/H1N1 sw1 virus in the receptor-binding site of the HA1 subunit. Vopr. Virusol. 2010, 55, 4–9. [Google Scholar] [PubMed]
- Chutinimitkul, S.; Herfst, S.; Steel, J.; Lowen, A.C.; Ye, J.; van Riel, D.; Schrauwen, E.J.A.; Bestebroer, T.M.; Koel, B.; Burke, D.F.; et al. Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J. Virol. 2010, 84, 11802–11813. [Google Scholar] [CrossRef] [PubMed]
- Krasnoslobodtsev, K.G.; Lvov, D.K.; Alkhovsky, S.V.; Burtseva, E.I.; Fedyakina, I.T.; Kolobukhina, L.V.; Kirillova, E.S.; Trushakova, S.V.; Oskerko, T.A.; Shchelkanov, M.Y.; et al. Amino acid polymorphism at residue 222 of the receptor-binding site of the hemagglutinin of the pandemic influenza A(H1N1)pdm09 from patients 166 with lethal virus pneumonia in 2012-2014. Probl. Virol. 2016, 61, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Wedde, M.; Wählisch, S.; Wolff, T.; Schweiger, B. Predominance of HA-222D/G Polymorphism in influenza A(H1N1)pdm09 viruses associated with fatal and severe outcomes recently circulating in germany. PLoS ONE 2013, 8, e57059. [Google Scholar] [CrossRef] [PubMed]
- Piralla, A.; Rovida, F.; Girello, A.; Premoli, M.; Mojoli, F.; Belliato, M.; Braschi, A.; Iotti, G.; Pariani, E.; Bubba, L.; et al. Frequency of respiratory virus infections and next-generation analysis of influenza A/H1N1pdm09 dynamics in the lower respiratory tract of patients admitted to the ICU. PLoS ONE 2017, 12, e0178926. [Google Scholar] [CrossRef]
- Selleri, M.; Piralla, A.; Rozera, G.; Giombini, E.; Bartolini, B.; Abbate, I.; Campanini, G.; Rovida, F.; Dossena, L.; Capobianchi, M.; et al. Detection of haemagglutinin D222 polymorphisms in influenza A(H1N1)pdm09-infected patients by ultra-deep pyrosequencing. Clin. Microbiol. Infect. 2013, 19, 668–673. [Google Scholar] [CrossRef]
- Stevens, J.; Blixt, O.; Glaser, L.; Taubenberger, J.K.; Palese, P.; Paulson, J.C.; Wilson, I.A. Glycan Microarray Analysis of the Hemagglutinins from Modern and Pandemic Influenza Viruses Reveals Different Receptor Specificities. J. Mol. Biol. 2006, 355, 1143–1155. [Google Scholar] [CrossRef]
- Takayama, I.; Nguyen, B.G.; Dao, C.X.; Pham, T.T.; Dang, T.Q.; Truong, P.T.; Van Do, T.; Pham, T.T.P.; Fujisaki, S.; Odagiri, T.; et al. Next-Generation Sequencing Analysis of the Within-Host Genetic Diversity of Influenza A(H1N1)pdm09 Viruses in the Upper and Lower Respiratory Tracts of Patients with Severe Influenza. mSphere 2021, 6, e01043-20. [Google Scholar] [CrossRef]
- Abed, Y.; Pizzorno, A.; Hamelin, M.-E.; Leung, A.; Joubert, P.; Couture, C.; Kobasa, D.; Boivin, G. The 2009 Pandemic H1N1 D222G Hemagglutinin Mutation Alters Receptor Specificity and Increases Virulence in Mice but Not in Ferrets. J. Infect. Dis. 2011, 204, 1008–1016. [Google Scholar] [CrossRef][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data on Cases | Total | A(H1N1)pdm09 | A(H3N2) | Influenza B |
|---|---|---|---|---|
| Number of laboratory-confirmed cases/proportion | 780 | 563 (72.2%) | 22 (2.8%) | 195 (25%) |
| Number of cases with fatal outcomes/proportion | 178 | 156 (87.6%) | 0 | 22 (12.4%) |
| Age group, years old | ||||
| Proportion of patients in different age groups among cases with fatal outcomes | 0–17 | 15.4% | - | 45.4% |
| 18–64 | 49.4% | - | 36.4% | |
| 65 and over | 35.2% | - | 18.2% | |
| Proportion of patients in risk groups for influenza among cases with fatal outcomes | 71% | - | 68% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolosova, N.P.; Boldyrev, N.D.; Svyatchenko, S.V.; Danilenko, A.V.; Goncharova, N.I.; Shadrinova, K.N.; Danilenko, E.I.; Onkhonova, G.S.; Kosenko, M.N.; Antonets, M.E.; et al. An Investigation of Severe Influenza Cases in Russia during the 2022–2023 Epidemic Season and an Analysis of HA-D222G/N Polymorphism in Newly Emerged and Dominant Clade 6B.1A.5a.2a A(H1N1)pdm09 Viruses. Pathogens 2024, 13, 1. https://doi.org/10.3390/pathogens13010001
Kolosova NP, Boldyrev ND, Svyatchenko SV, Danilenko AV, Goncharova NI, Shadrinova KN, Danilenko EI, Onkhonova GS, Kosenko MN, Antonets ME, et al. An Investigation of Severe Influenza Cases in Russia during the 2022–2023 Epidemic Season and an Analysis of HA-D222G/N Polymorphism in Newly Emerged and Dominant Clade 6B.1A.5a.2a A(H1N1)pdm09 Viruses. Pathogens. 2024; 13(1):1. https://doi.org/10.3390/pathogens13010001
Chicago/Turabian StyleKolosova, Natalia P., Nikita D. Boldyrev, Svetlana V. Svyatchenko, Alexey V. Danilenko, Natalia I. Goncharova, Kyunnei N. Shadrinova, Elena I. Danilenko, Galina S. Onkhonova, Maksim N. Kosenko, Maria E. Antonets, and et al. 2024. "An Investigation of Severe Influenza Cases in Russia during the 2022–2023 Epidemic Season and an Analysis of HA-D222G/N Polymorphism in Newly Emerged and Dominant Clade 6B.1A.5a.2a A(H1N1)pdm09 Viruses" Pathogens 13, no. 1: 1. https://doi.org/10.3390/pathogens13010001
APA StyleKolosova, N. P., Boldyrev, N. D., Svyatchenko, S. V., Danilenko, A. V., Goncharova, N. I., Shadrinova, K. N., Danilenko, E. I., Onkhonova, G. S., Kosenko, M. N., Antonets, M. E., Susloparov, I. M., Ilyicheva, T. N., Marchenko, V. Y., & Ryzhikov, A. B. (2024). An Investigation of Severe Influenza Cases in Russia during the 2022–2023 Epidemic Season and an Analysis of HA-D222G/N Polymorphism in Newly Emerged and Dominant Clade 6B.1A.5a.2a A(H1N1)pdm09 Viruses. Pathogens, 13(1), 1. https://doi.org/10.3390/pathogens13010001