
Comparison of the Whole-Genome Sequence of the African Swine Fever Virus from a Mongolian Wild Boar with Genotype II Viruses from Asia and Europe
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Rapid ASFV Detection
2.2. DNA Extraction, ASFV Detection, and Genotyping
2.3. ASFV Whole-Genome Sequence and Assembly
2.4. Comparative Analysis of ASFV Genomes and Phylogenetic Analysis
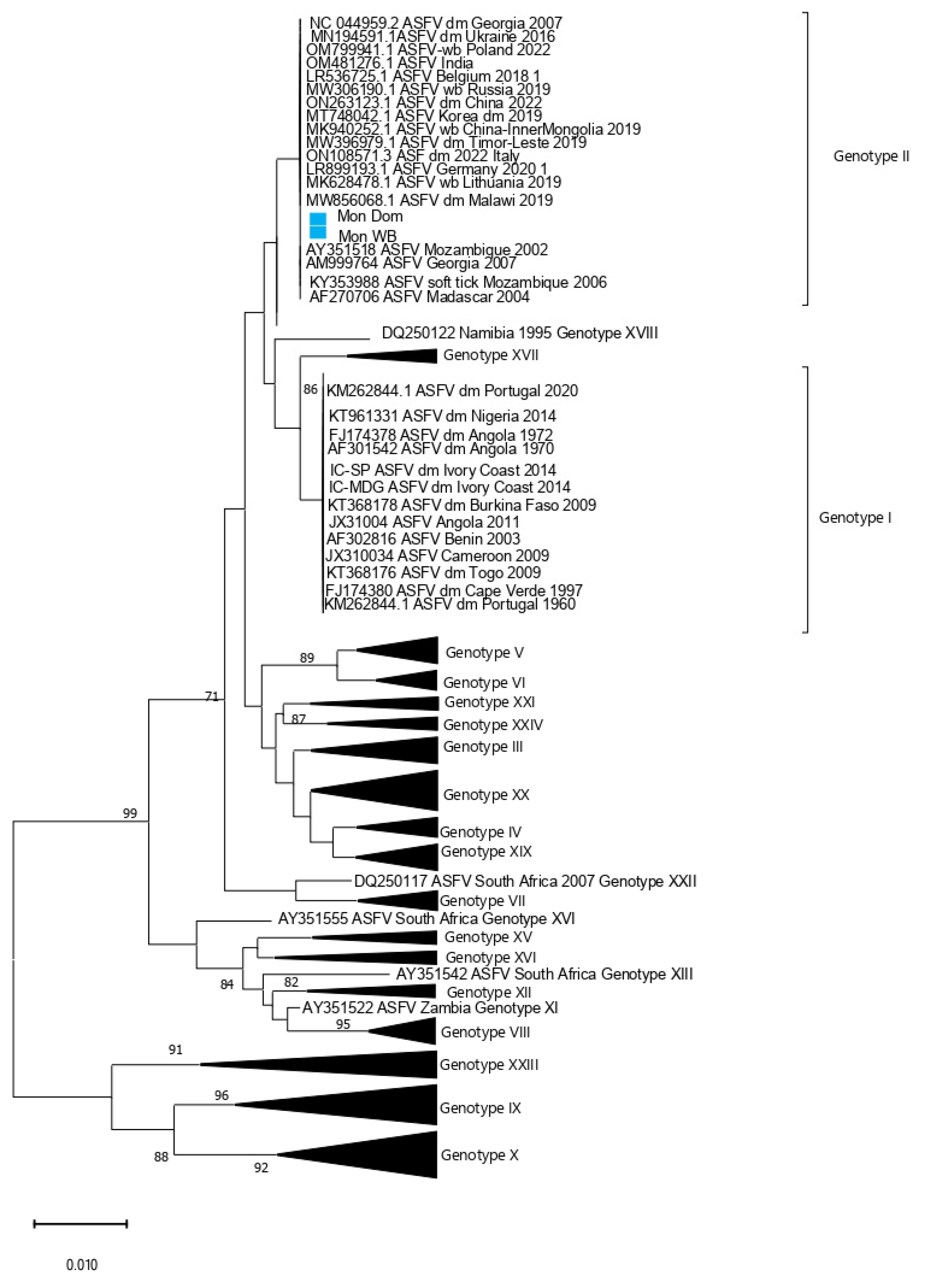
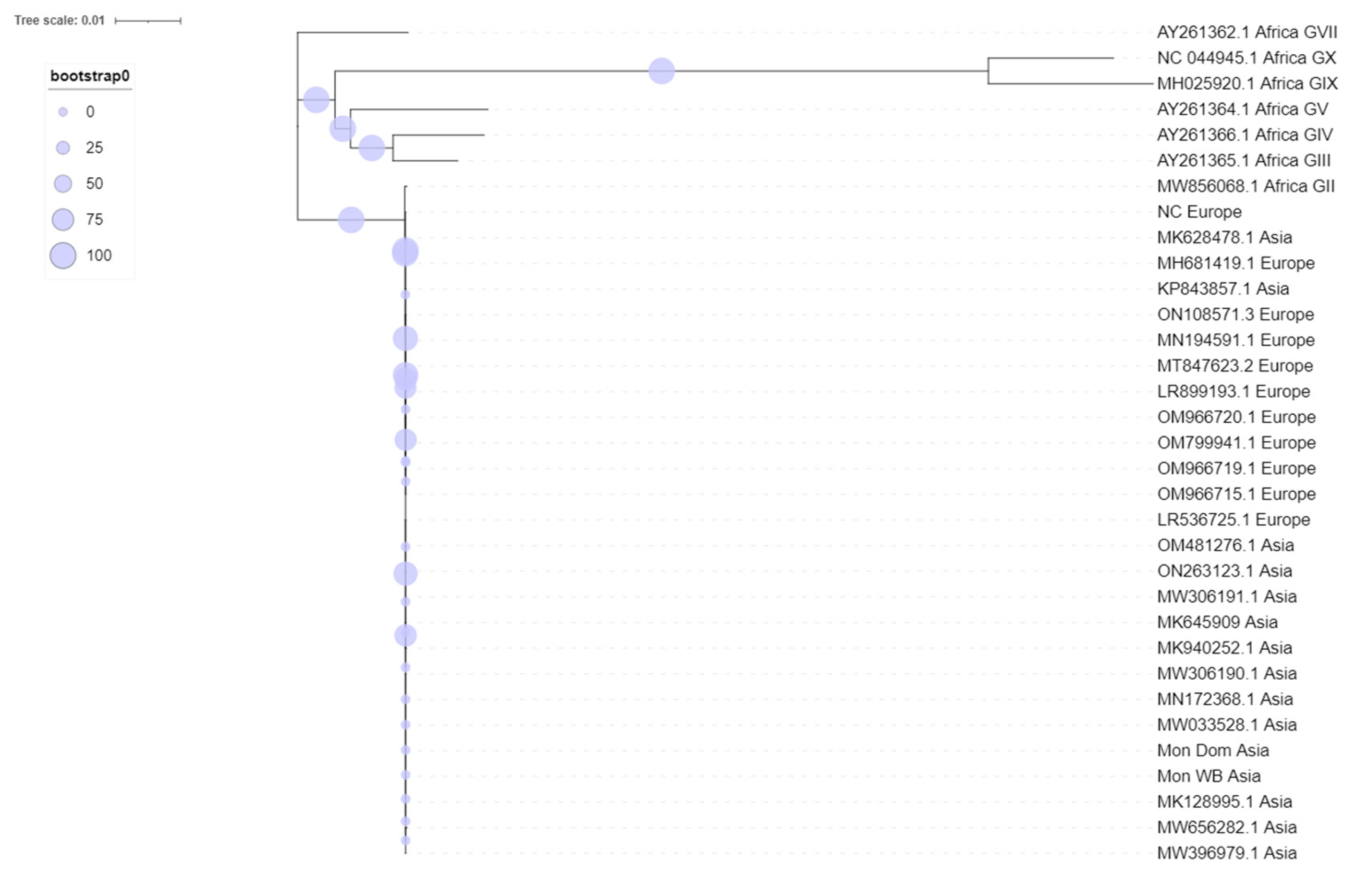
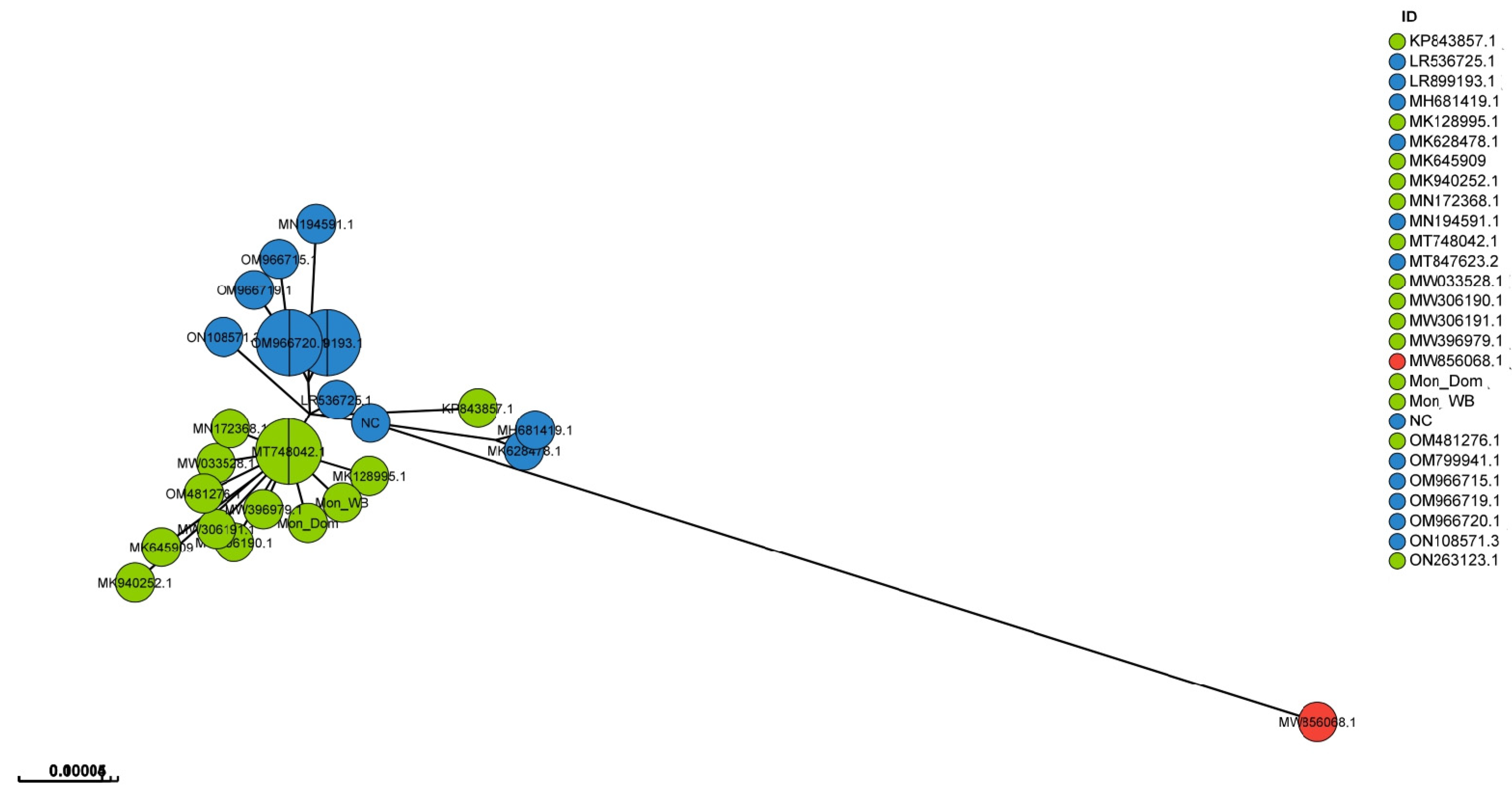
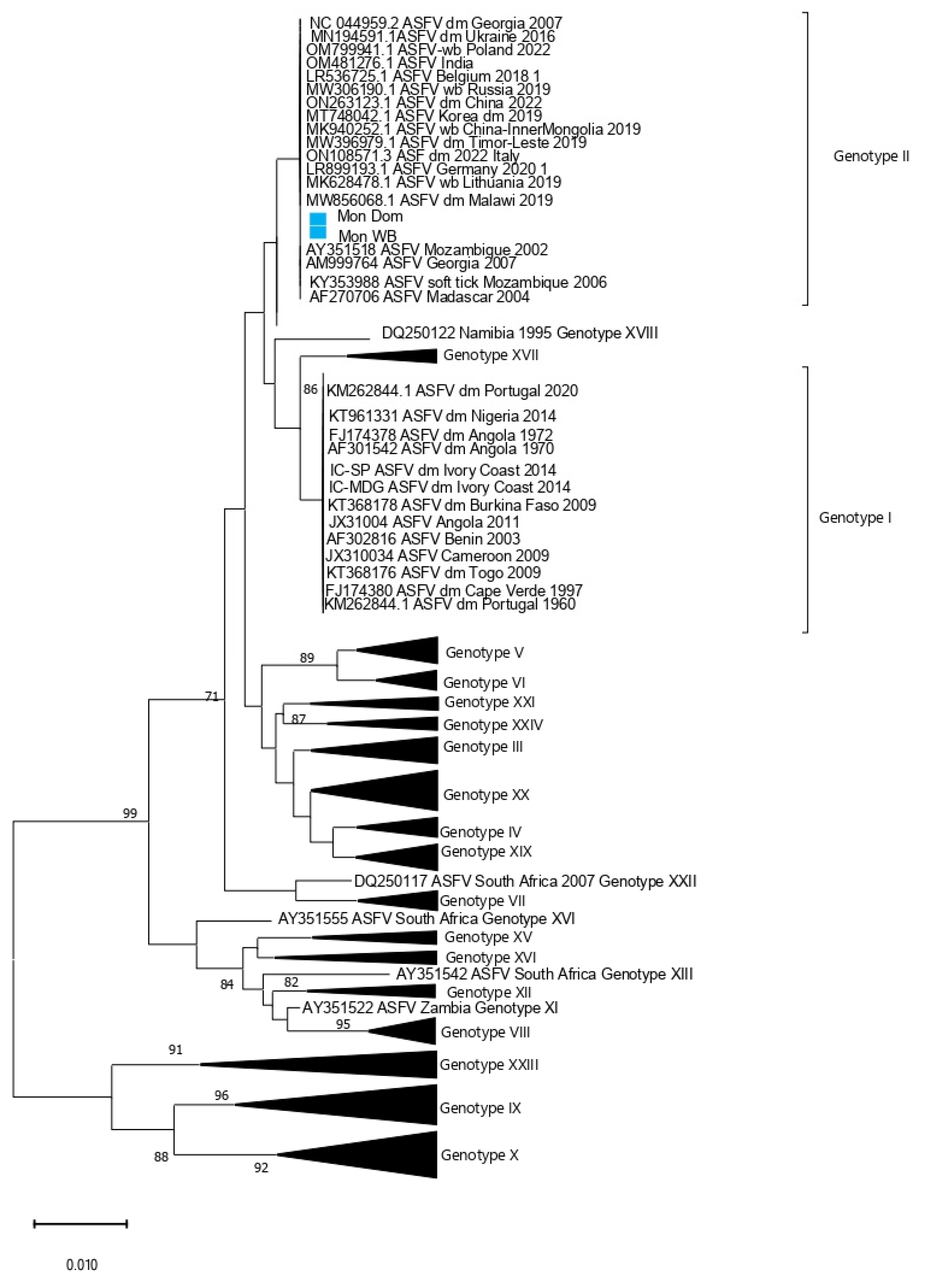
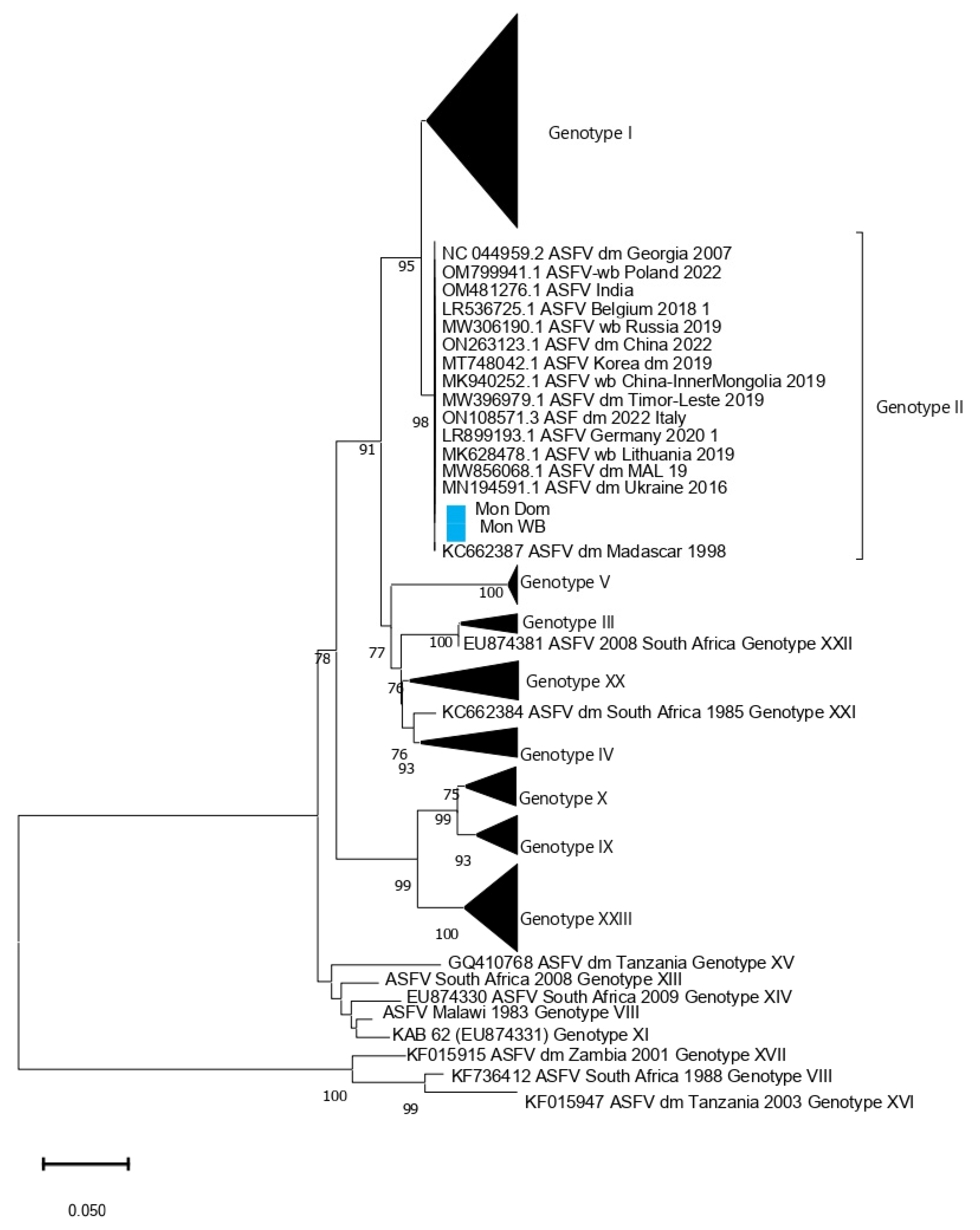
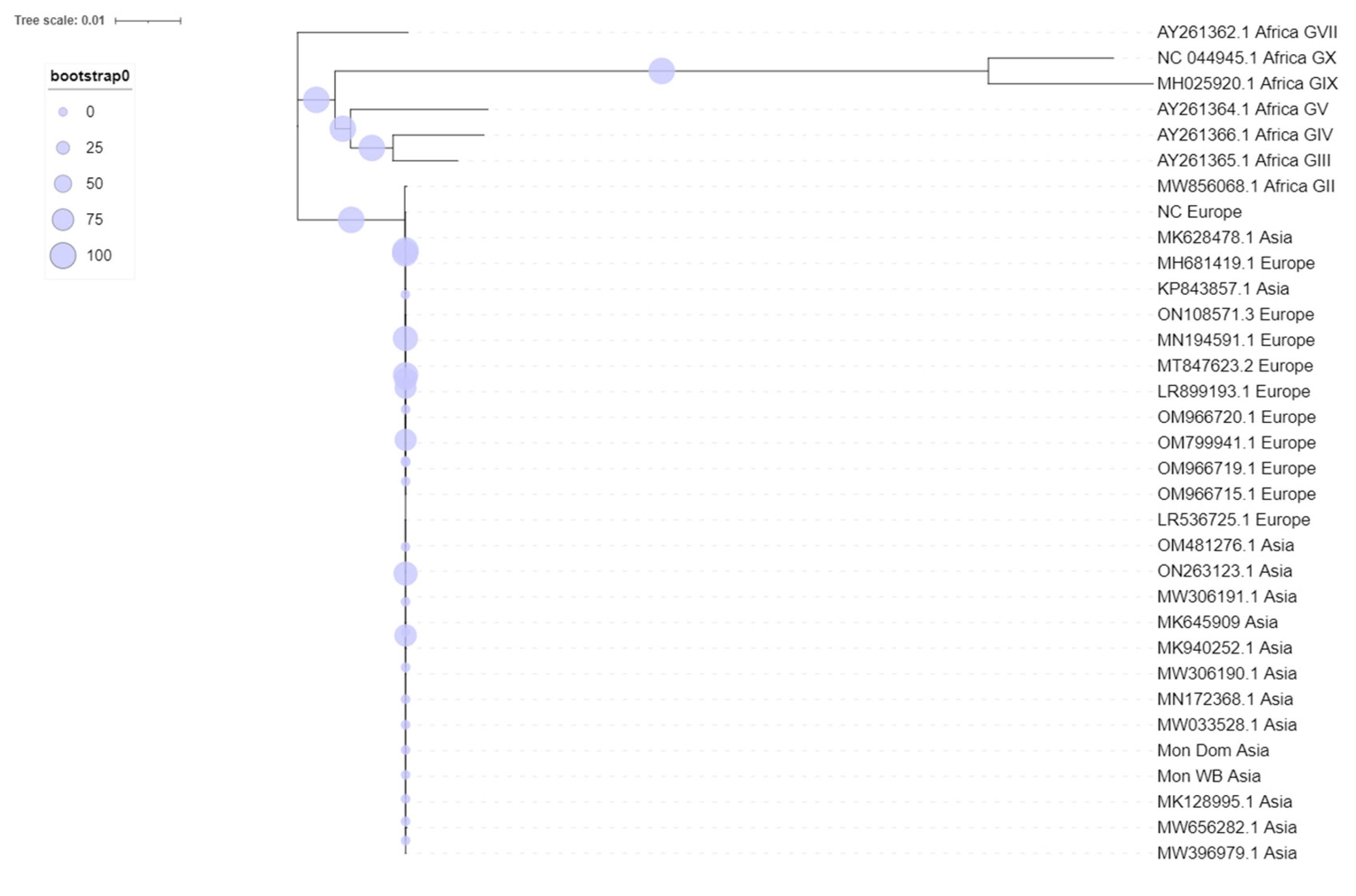
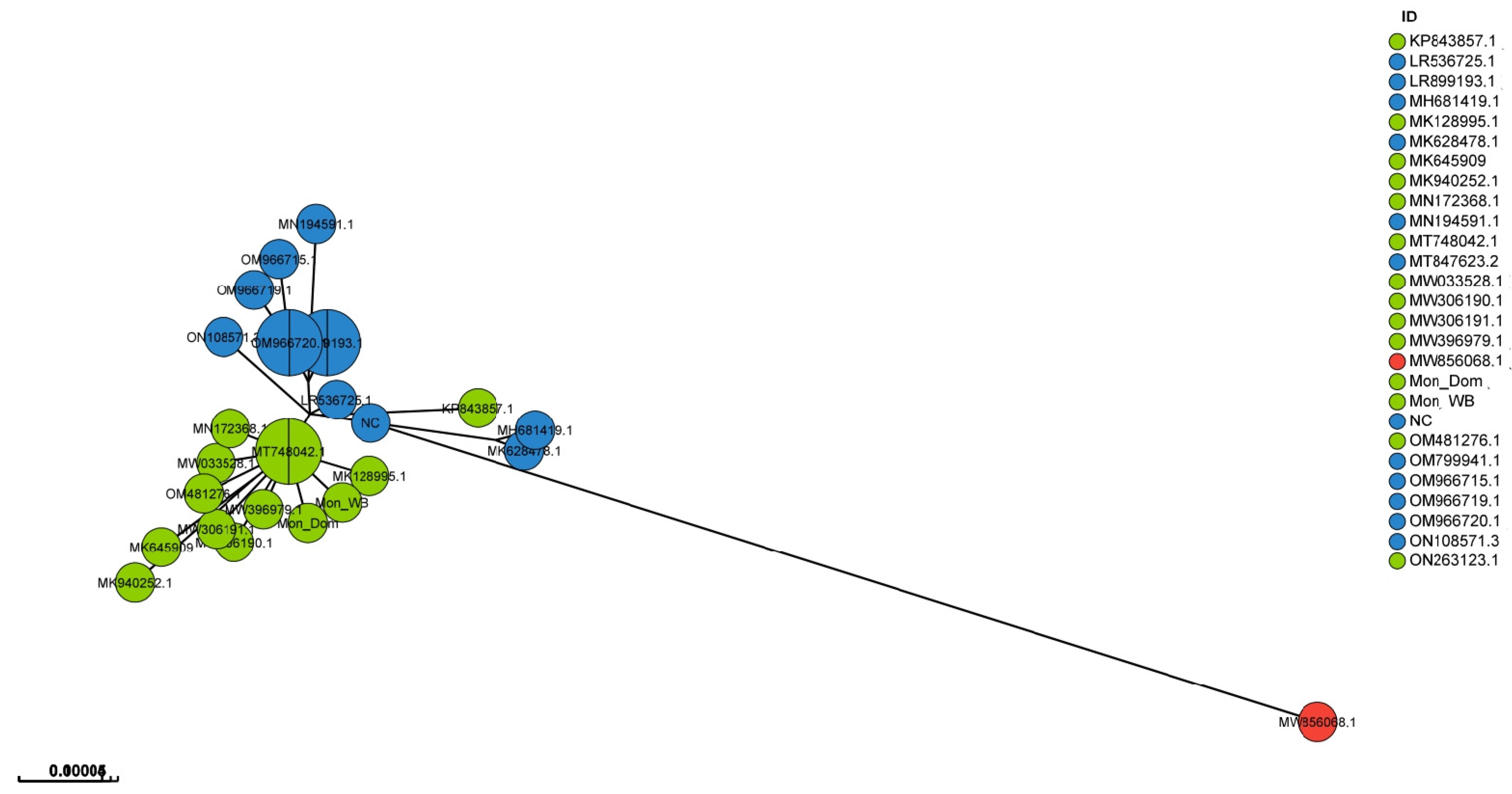
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Bao, J.; Zhang, Y.; Shi, C.; Wang, Q.; Wang, S.; Wu, X.; Cao, S.; Xu, F.; Wang, Z. Genome-Wide Diversity Analysis of African Swine Fever Virus Based on a Curated Dataset. Animals 2022, 12, 2446. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.; Boklund, A. The Epidemiology of African Swine Fever, Its Complexity and the Requirement for Multiple Solution Approaches. Animals 2020, 10, 1900. [Google Scholar] [CrossRef] [PubMed]
- Njau, E.P.; Domelevo Entfellner, J.B.; Machuka, E.M.; Bochere, E.N.; Cleaveland, S.; Shirima, G.M.; Kusiluka, L.J.; Upton, C.; Bishop, R.P.; Pelle, R.; et al. The first genotype II African swine fever virus isolated in Africa provides insight into the current Eurasian pandemic. Sci. Rep. 2021, 11, 13081. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.E. On a form of swine fever occurring in British east-Africa (Kenya Colony). J. Comp. Pathol. Ther. 1921, 34, 159–191. [Google Scholar] [CrossRef]
- Giammarioli, M.; Alessandro, D.; Cammà, C.; Masoero, L.; Torresi, C.; Marcacci, M.; Zoppi, S.; Curini, V.; Rinaldi, A.; Rossi, E.; et al. Molecular Characterization of the First African Swine Fever Virus Genotype II Strains Identified from Mainland Italy, 2022. Pathogens 2023, 12, 372. [Google Scholar] [CrossRef] [PubMed]
- Portugal, R.; Coelho, J.; Hoeper, D.; Little, N.; Smithson, C.; Upton, C.; Martins, C.; Leitão, A.; Keil, G. Related strains of African swine fever virus with different virulence: Genome comparison and analysis. J. Gen. Virol. 2014, 96, 408–419. [Google Scholar] [CrossRef]
- Fiori, M.S.; Sanna, D.; Scarpa, F.; Floris, M.; Di Nardo, A.; Ferretti, L.; Loi, F.; Cappai, S.; Sechi, A.M.; Angioi, P.P.; et al. A Deeper Insight into Evolutionary Patterns and Phylogenetic History of ASFV Epidemics in Sardinia (Italy) through Extensive Genomic Sequencing. Viruses 2021, 13, 1994. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-Alcrudo, D.; Lubroth, J.; Depner, K.; De La Rocque, S. African swine fever in the Caucasus. FAO Empres. Watch. 2008, 1, 1–8. [Google Scholar]
- Zhou, X.; Li, N.; Luo, Y.; Liu, Y.; Miao, F.; Chen, T.; Zhang, S.; Cao, P.; Li, X.; Tian, K.; et al. Emergence of African Swine Fever in China, 2018. Transbound. Emerg. Dis. 2018, 65, 1482–1484. [Google Scholar] [CrossRef]
- Ankhanbaatar, U.; Sainnokhoi, T.; Khanui, B.; Ulziibat, G.; Jargalsaikhan, T.; Dulam, P.; Settypalli, B.K.; Flannery, J.; Dundon, W.; Basan, G.; et al. African Swine Fever Virus Genotype II in Mongolia, 2019. Transbound. Emerg. Dis. 2021, 68, 2787–2794. [Google Scholar] [CrossRef]
- Afonso, C.L.; Piccone, M.E.; Zaffuto, K.M.; Neilan, J.; Kutish, G.F.; Lu, Z.; Balinsky, C.A.; Gibb, T.R.; Bean, T.J.; Zsak, L.; et al. African swine fever virus multigene family 360 and 530 genes affect host interferon response. J. Virol. 2004, 78, 1858–1864. [Google Scholar] [CrossRef] [PubMed]
- King, D.P.; Reid, S.M.; Hutchings, G.H.; Grierson, S.S.; Wilkinson, P.J.; Dixon, L.K.; Bastos, A.D.; Drew, T.W. Development of a TaqMan PCR assay with internal amplification control for the detection of African swine fever virus. J. Virol. Methods 2003, 107, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Mwaengo, D.M.; Macharia, J.M.; Arias, M.; Taracha, E.A.; Soler, A.; Okoth, E.; Martín, E.; Kasiti, J.; Bishop, R.P. Enhanced discrimination of African swine fever virus isolates through nucleotide sequencing of the p54, p72, and pB602L (CVR) genes. Virus Genes 2009, 38, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2018, 35, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.E.; Handsaker, R.; Lunter, G.; Marth, G.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R. Rstudio; PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 1 September 2022).
- NCBI-National Center for Biotechnology Information (NCBI) [Internet]. National Library of Medicine (US). National Center for Biotechnology Information: Bethesda, MD, USA. 1988. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 6 March 2023).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms Molecular Biology and Evolution; Oxford University Press: Oxford, UK, 2018; Volume 35, p. 1547. [Google Scholar]
- Zhang, Y.; Ke, J.; Zhang, J.; Yang, J.; Yue, H.; Zhou, X.; Qi, Y.; Zhu, R.; Miao, F.; Li, Q.; et al. African Swine Fever Virus Bearing an I226R Gene Deletion Elicits Robust Immunity in Pigs to African Swine Fever. J. Virol. 2021, 95, e0119921. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Alikhan, N.F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Olesen, A.S.; Kodama, M.; Lohse, L.; Accensi, F.; Rasmussen, T.B.; Lazov, C.M.; Limborg, M.T.; Gilbert, M.T.P.; Bøtner, A.; Belsham, G.J. Identification of African Swine Fever Virus Transcription within Peripheral Blood Mononuclear Cells of Acutely Infected Pigs. Viruses 2021, 13, 2333. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Núñez, D.; Castillo-Rosa, E.; Vigara-Astillero, G.; García-Belmonte, R.; Gallardo, C.; Revilla, Y. Identification and Isolation of Two Different Subpopulations within African Swine Fever Virus Arm/07 Stock. Vaccines 2020, 8, 625. [Google Scholar] [CrossRef] [PubMed]
- Rolesu, S.; Mandas, D.; Loi, F.; Oggiano, A.; Dei Giudici, S.; Franzoni, G.; Guberti, V.; Cappai, S. African swine fever in smallholder Sardinian farms: Last ten years of network transmission reconstruction and analysis. Front. Vet. Med. 2021, 8, 692448. [Google Scholar] [CrossRef]
- Kim, G.; Park, J.-E.; Kim, S.-J.; Kim, Y.; Kim, W.; Kim, Y.-K.; Jheong, W. Complete genome analysis of the African swine fever virus isolated from a wild boar responsible for the first viral outbreak in Korea, 2019. Front. Vet. Sci. 2023, 9, 1080397. [Google Scholar] [CrossRef]
- Hakizimana, J.N.; Ntirandekura, J.B.; Yona, C.; Nyabongo, L.; Kamwendo, G.; Chulu, J.L.C.; Ntakirutimana, D.; Kamana, O.; Nauwynck, H.; Misinzo, G. Complete genome analysis of African swine fever virus responsible for outbreaks in domestic pigs in 2018 in Burundi and 2019 in Malawi. Trop. Anim. Health Prod. 2021, 53, 438. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Wozniakowski, G.; Niemczuk, K. The first complete genomic sequences of african swine fever virus isolated in Poland. Sci. Rep. 2019, 9, 4556. [Google Scholar] [CrossRef] [PubMed]
- Forth, J.H.; Forth, L.F.; Blome, S.; Höper, D.; Beer, M. African swine fever whole-genome sequencing—Quantity wanted but quality needed. PLoS Pathog. 2020, 16, e1008779. [Google Scholar] [CrossRef]
- Guinat, C.; Gogin, A.; Blome, S.; Keil, G.; Pollin, R.; Pfeiffer, D.U.; Dixon, L. Transmission routes of African swine fever virus to domestic pigs: Current knowledge and future research directions. Vet. Rec. 2016, 178, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Cadenas-Fernández, E.; Ito, S.; Aguilar-Vega, C.; Sánchez-Vizcaíno, J.M.; Bosch, J. The Role of the Wild Boar Spreading African Swine Fever Virus in Asia: Another Underestimated Problem. Front. Vet. Sci. 2022, 9, 844209. [Google Scholar] [CrossRef] [PubMed]
- Pautienius, A.; Grigas, J.; Pileviciene, S.; Zagrabskaite, R.; Buitkuviene, J.; Pridotkas, G.; Stankevicius, R.; Streimikyte, Z.; Salomskas, A.; Zienius, D.; et al. Prevalence and spatiotemporal distribution of African swine fever in Lithuania, 2014–2017. Virol. J. 2018, 15, 177. [Google Scholar] [CrossRef] [PubMed]
- Cwynar, P.; Stojkov, J.; Wlazlak, K. African Swine Fever Status in Europe. Viruses 2019, 11, 310. [Google Scholar] [CrossRef]
- Guberti, V.; Khomenko, S.; Masiulis, M.; Kerba, S. African Swine Fever in Wild Boar—Ecology and Biosecurity, 2nd ed.; FAO Animal Production and Health Manual No. 28; FAO; World Organisation for Animal Health and European Commission: Rome, Italy, 2022. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Provinces | Area | Hunted/Found Dead | Date | Sample Type |
|---|---|---|---|---|---|
| 1 | Selenge | Minjiin Khangai | Hunted | March 2021 | Blood, lung, heart, liver, spleen, kidney, tonsil, blood, marrow |
| 2 | Selenge | Minjiin Khangai | Found dead | December 2020 | Spleen |
| 3 | Tuv | Eluur river | Found dead | January 2021 | Kidney, spleen, heart, liver, lung |
| No. | NCBI Reference | Geographical Region | Country | Year Collected | Host Species | Genotype |
|---|---|---|---|---|---|---|
| 1 | NC044959 | Europe | Georgia | 2007 | dm | II |
| 2 | MN194591 | Europe | Ukraine | 2016 | dm | |
| 3 | OM966715 | Europe | Poland | 2018 | wb | |
| 4 | MH681419 | Europe | Poland | 2015 | dm | |
| 5 | MK628478 | Europe | Lithuania | 2014 | wb | |
| 6 | OM799941 | Europe | Poland | 2022 | wb | |
| 7 | OM966720 | Europe | Poland | 2018 | wb | |
| 8 | OM966719 | Europe | Poland | 2018 | wb | |
| 9 | MT847623 | Europe | Poland | 2019 | wb | |
| 10 | LR899193 | Europe | Germany | 2020 | NA | |
| 11 | ON108571 | Europe | Italy | 2022 | dm | |
| 12 | LR536725 | Europe | Belgium | 2018 | NA | |
| 13 | KP843857 | Asia | Russia | 2014 | wb | |
| 14 | OM481276 | Asia | India | 2022 | NA | |
| 15 | MW306190 | Asia | Russia | 2019 | wb | |
| 16 | MW306191 | Asia | China | 2019 | wb | |
| 17 | ON263123 | Asia | China | 2022 | dm | |
| 18 | MK128995 | Asia | China | 2018 | dm | |
| 19 | Mon_Dom | Asia | Mongolia | 2019 | dm | |
| 20 | Mon_WB | Asia | Mongolia | 2021 | wb | II (This study) |
| 21 | MN172368 | Asia | China | 2019 | dm | II |
| 22 | MW033528 | Asia | China | 2019 | wb | |
| 23 | MK940252 | Asia | China | 2019 | wb | |
| 24 | MW396979 | Asia | Timor-Leste | 2019 | dm | |
| 25 | MK645909 | Asia | China | 2019 | wb | |
| 26 | MT748042 | Asia | Korea | 2019 | dm | |
| 27 | MW856068 | Africa | Malawi | 2019 | dm | |
| 28 | AY261365 | Africa | South Africa | 2004 | tick | III |
| 29 | AY261366 | Africa | Namibia | 2004 | warthog | IV |
| 30 | AY261364 | Africa | Malawi | 2004 | dm | V |
| 31 | AY261362 | Africa | South Africa | 1979 | tick | VII |
| 32 | MH025920 | Africa | Uganda | 2018 | dm | IX |
| 33 | NC_044945 | Africa | Kenya | 2020 | tick | X |
| No. | NCBI Reference | Region | Nucleotide at Position | ||
|---|---|---|---|---|---|
| (1) 9114 | (2) 22996 | (3) 99933 | |||
| 1 | NC044959 | Europe | a | t | gap |
| 2 | MN194591 | Europe | a | t | gap |
| 3 | OM966715 | Europe | a | t | gap |
| 4 | MH681419 | Europe | gap | t | gap |
| 5 | MK628478 | Europe | gap | t | gap |
| 6 | OM799941 | Europe | a | t | gap |
| 7 | OM966720 | Europe | a | t | gap |
| 8 | OM966719 | Europe | a | t | gap |
| 9 | MT847623 | Europe | a | t | gap |
| 10 | LR899193 | Europe | a | t | gap |
| 11 | ON108571 | Europe | a | t | gap |
| 12 | LR536725 | Europe | a | t | gap |
| 13 | KP843857 | Asia | gap | t | gap |
| 14 | OM481276 | Asia | a | c | gap |
| 15 | MW306190 | Asia | gap | c | g |
| 16 | MW306191 | Asia | gap | c | g |
| 17 | ON263123 | Asia | gap | c | g |
| 18 | MK128995 | Asia | gap | c | g |
| 19 | Mon_Dom | Asia | gap | c | g |
| 20 | Mon_WB | Asia | gap | c | g |
| 21 | MN172368 | Asia | gap | c | g |
| 22 | MW033528 | Asia | gap | c | g |
| 23 | MK940252 | Asia | gap | c | g |
| 24 | MW396979 | Asia | gap | c | g |
| 25 | MK645909 | Asia | gap | c | g |
| 26 | MT748042 | Asia | gap | c | g |
| 27 | MW856068 | Africa | a | t | gap |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© FAO, 2023. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY NC SA) license (https://creativecommons.org/licenses/by-nc-sa/3.0/igo/).
Share and Cite
Ankhanbaatar, U.; Auer, A.; Ulziibat, G.; Settypalli, T.B.K.; Gombo-Ochir, D.; Basan, G.; Takemura, T.; Tseren-Ochir, E.-O.; Ouled Ahmed, H.; Meki, I.K.; et al. Comparison of the Whole-Genome Sequence of the African Swine Fever Virus from a Mongolian Wild Boar with Genotype II Viruses from Asia and Europe. Pathogens 2023, 12, 1143. https://doi.org/10.3390/pathogens12091143
Ankhanbaatar U, Auer A, Ulziibat G, Settypalli TBK, Gombo-Ochir D, Basan G, Takemura T, Tseren-Ochir E-O, Ouled Ahmed H, Meki IK, et al. Comparison of the Whole-Genome Sequence of the African Swine Fever Virus from a Mongolian Wild Boar with Genotype II Viruses from Asia and Europe. Pathogens. 2023; 12(9):1143. https://doi.org/10.3390/pathogens12091143
Chicago/Turabian StyleAnkhanbaatar, Ulaankhuu, Agathe Auer, Gerelmaa Ulziibat, Tirumala B. K. Settypalli, Delgerzul Gombo-Ochir, Ganzorig Basan, Taichiro Takemura, Erdene-Ochir Tseren-Ochir, Hatem Ouled Ahmed, Irene Kasindi Meki, and et al. 2023. "Comparison of the Whole-Genome Sequence of the African Swine Fever Virus from a Mongolian Wild Boar with Genotype II Viruses from Asia and Europe" Pathogens 12, no. 9: 1143. https://doi.org/10.3390/pathogens12091143
APA StyleAnkhanbaatar, U., Auer, A., Ulziibat, G., Settypalli, T. B. K., Gombo-Ochir, D., Basan, G., Takemura, T., Tseren-Ochir, E.-O., Ouled Ahmed, H., Meki, I. K., Datta, S., Soumare, B., Metlin, A., Cattoli, G., & Lamien, C. E. (2023). Comparison of the Whole-Genome Sequence of the African Swine Fever Virus from a Mongolian Wild Boar with Genotype II Viruses from Asia and Europe. Pathogens, 12(9), 1143. https://doi.org/10.3390/pathogens12091143