
Genomic Characterization of Listeria innocua Isolates Recovered from Cattle Farms, Beef Abattoirs, and Retail Outlets in Gauteng Province, South Africa

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Source of L. innocua Used in the Current Study
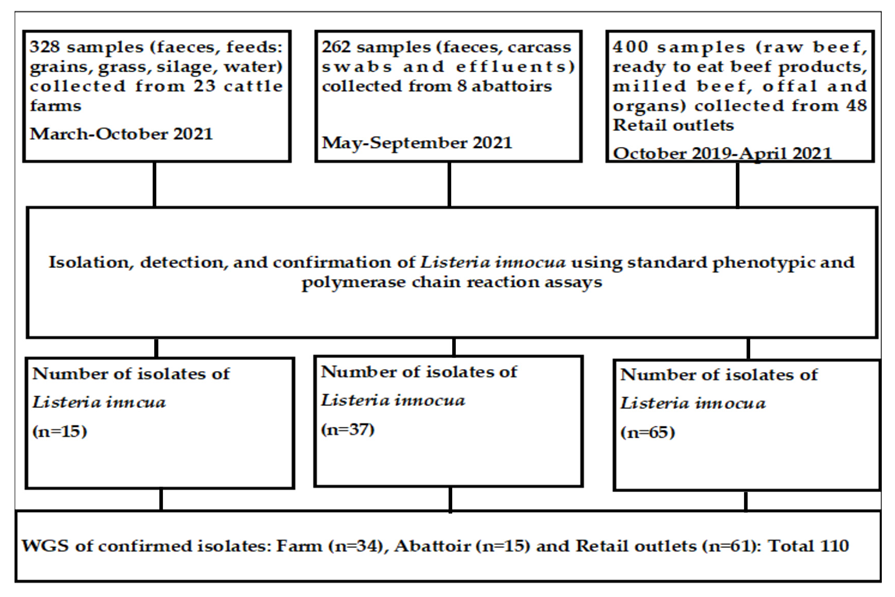
2.2. Study Design and Sources of Samples
2.3. Variables of Beef Industries Investigated in the Cross-Sectional Studies
2.3.1. Cattle Farms
- i.
- Communal farms: South Africa has about 18 million hectares of communal land. This is owned by the government but managed by tribal authorities. Livestock owned by several owners graze collectively in the community and are taken to vaccination and ectoparasite control centers in the area as a large herd (owned by many farmers) for vector control. Ten communal farms were sampled for the current study.
- ii.
- Cow-calf operations: These operations refer to farms that breed and raise cattle to sell them. These farmers are focused on raising quality cattle that are suitable for the specific industry they sell them to, such as dairy cattle or beef cattle. A total of 10 cow-calf operations were included in the current study.
- iii.
- Feedlots: Feedlots purchase cattle and prepare them for the final stage of the beef production process. Feedlots are focused on feeding mature cattle and ensuring that they have the right medical clearance to continue the beef production process. The input of private veterinary services is optimal at the feedlots, and the majority have their own abattoirs. Samples for the current study were collected from three feedlots. In South Africa, feedlots contribute 60–65% of the cattle slaughtered.
2.3.2. Abattoirs
- i.
- Butcheries: These are small operations by individuals where cattle are slaughtered primarily from small farms (communal and cow-calf operations) and the beef is sold fresh on-site to the consumers. The animals slaughtered at these facilities are not inspected by the veterinary public health (VPH) personnel, either pre-slaughter or post-slaughter. The samples originating from these slaughter facilities are mostly sold directly to consumers.
- ii.
- Low-throughput (LT): The facilities are so classified by the veterinary public health section of the Department of Agriculture, Forestry, and Fisheries (DAFF). In the country, all abattoirs are privately owned, but all animals slaughtered at these facilities are legally expected to be inspected before and after slaughter by VPH personnel. LT abattoirs slaughter livestock, including red-meat livestock such as sheep, pigs, and goats. Game and poultry are slaughtered at different abattoirs in the country. LT abattoirs are classified based on the maximum daily throughput of livestock, ranging from 20 units for cattle, 30 for pigs, and 40 for sheep and goats if only one species is slaughtered (Red Meat Regulation R1072 from Meat Safety Act 40 of 2000) [39]. Cattle slaughtered at LT abattoirs primarily originate from communal farms and cow-calf operations. For our study, eight LT abattoirs were randomly selected for sampling where cattle were slaughtered.
- iii.
- High-throughput (HT): The activities that take place at the HT abattoirs are similar to those at the LT abattoirs, except for the fact that they have a higher maximum daily throughput that is determined by the provincial executive officer on the grounds of lairage capacity and hourly throughput potential relative to available equipment and infrastructure, as stated by the Red Meat Regulation R1072 from Meat Safety Act 40 of 2000 [39]. HT abattoirs are classified based on units of daily slaughter exceeding those stated for LT abattoirs, as stated above. Most feedlots have their own individual HT abattoirs. A total of six HT abattoirs were sampled in the current study.
2.3.3. Retail Outlets
- i.
- Chain retail outlets: These are retail outlets with two or more outlets distributed across the province. Samples were collected from 30 chain retail outlets.
- ii.
- Large retail outlets: Outlets with six or more cashiers, from which 10 were recruited for sampling.
- iii.
- Medium retail outlets: These are outlets with 3–5 cashiers with sampling from 6 outlets.
- iv.
- Small retail outlets: Outlets with 1–2 cashiers, from which two outlets were sampled.
2.4. Variables of Sample Types Investigated in the Cross-Sectional Studies and Investigating the Potential Effect of the Source of Samples on the Distribution and the Characteristics of L. innocua
2.5. Isolation, Identification of L. innocua, and Determination of AMR
2.6. Selection of Antimicrobial Agents Used and Determination of the Resistance of Listeria Isolates to Antimicrobial Agents
2.7. Whole-Genome Sequencing, Genomic Analysis, Assembly, and Annotation
2.8. In Silico MLST
2.9. Resistance and Virulence Profiles
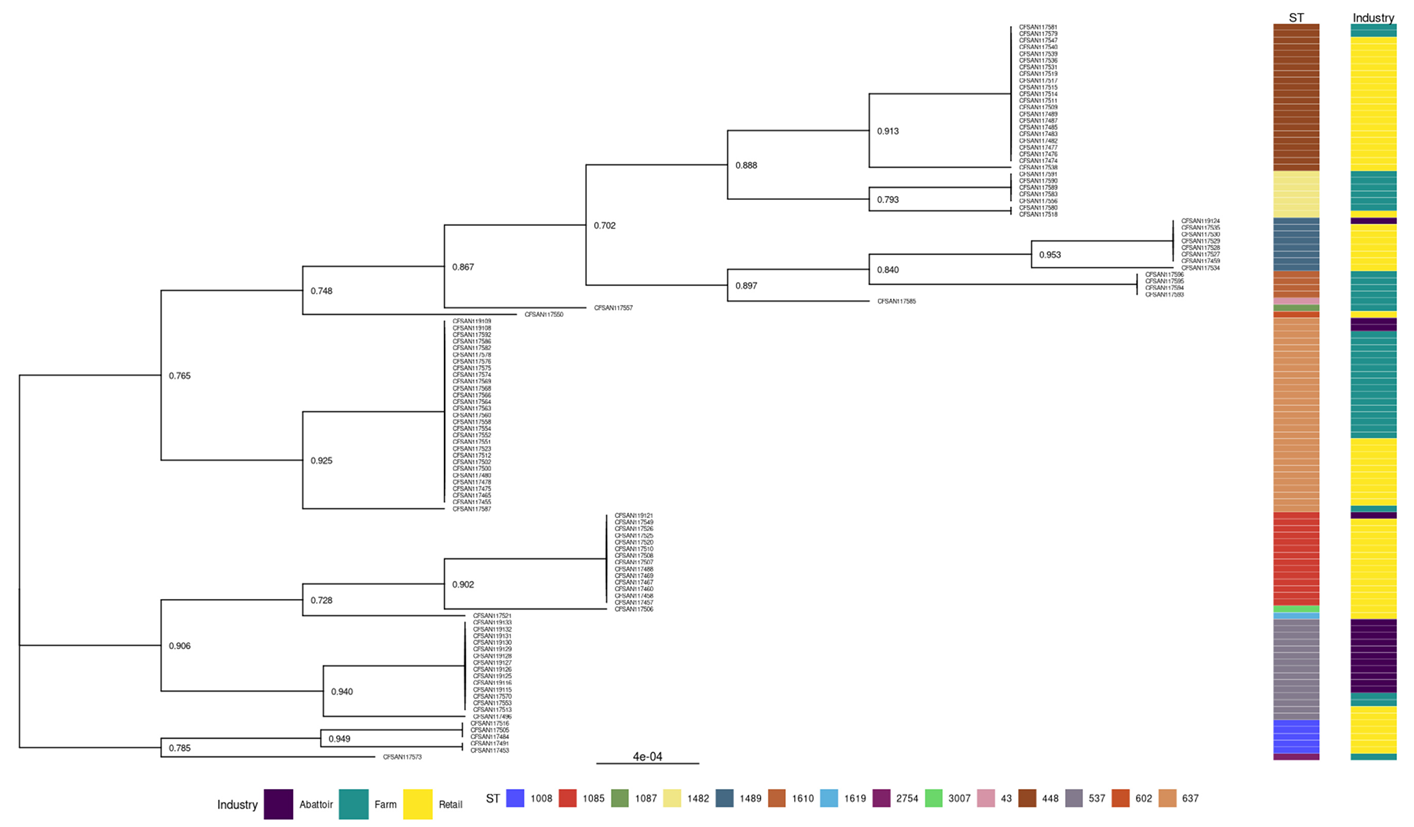
2.10. Construction of the Phylogenetic Tree for L. innocua Isolates and Correlation with Source and Type of Samples
2.11. Data Analysis
3. Results
3.1. Effect of the Three Beef Industries (Cattle Farms, Abattoirs, and Retail) on the Frequency of Detection of STs, AMR Genes, and Virulence Genes in L. innocua Isolates
3.2. Frequency of STs in Innocua
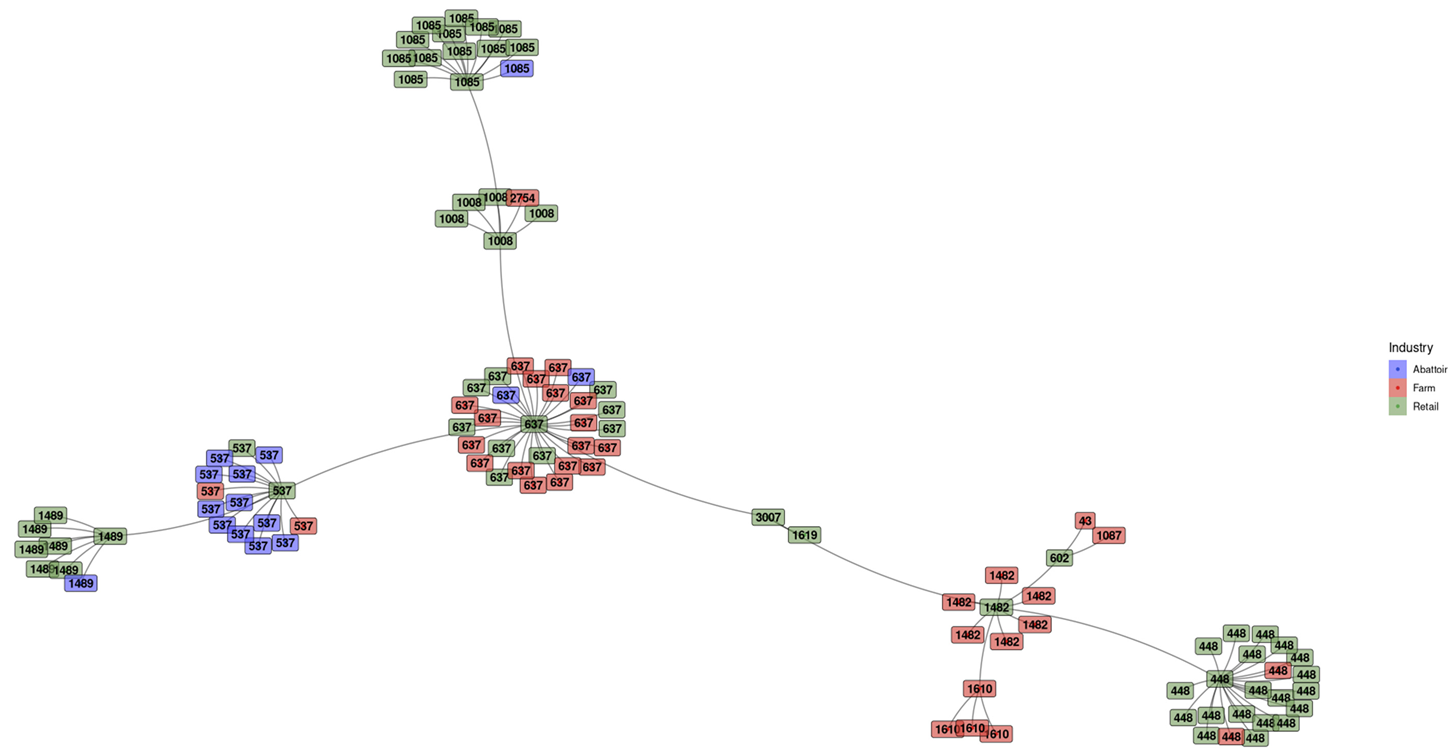
3.2.1. Minimum Spanning Tree (MST) Based on ST Profiles
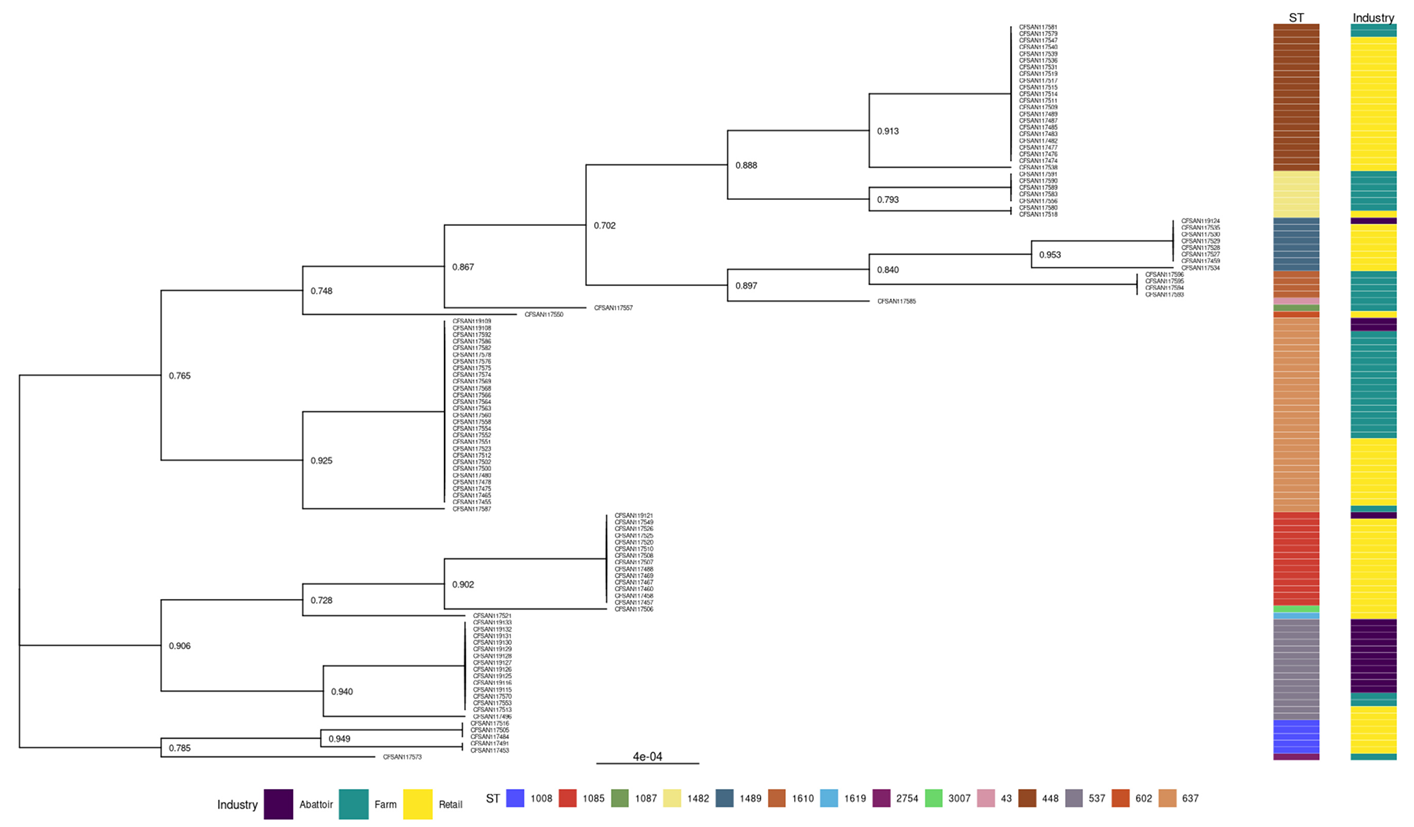
3.2.2. Phylogenies of L. innocua Isolates According to the STs and Industry
3.3. Detection of Antimicrobial Resistance Genes in L. innocua
3.3.1. Patterns of Multiple Antimicrobial Resistance Genes
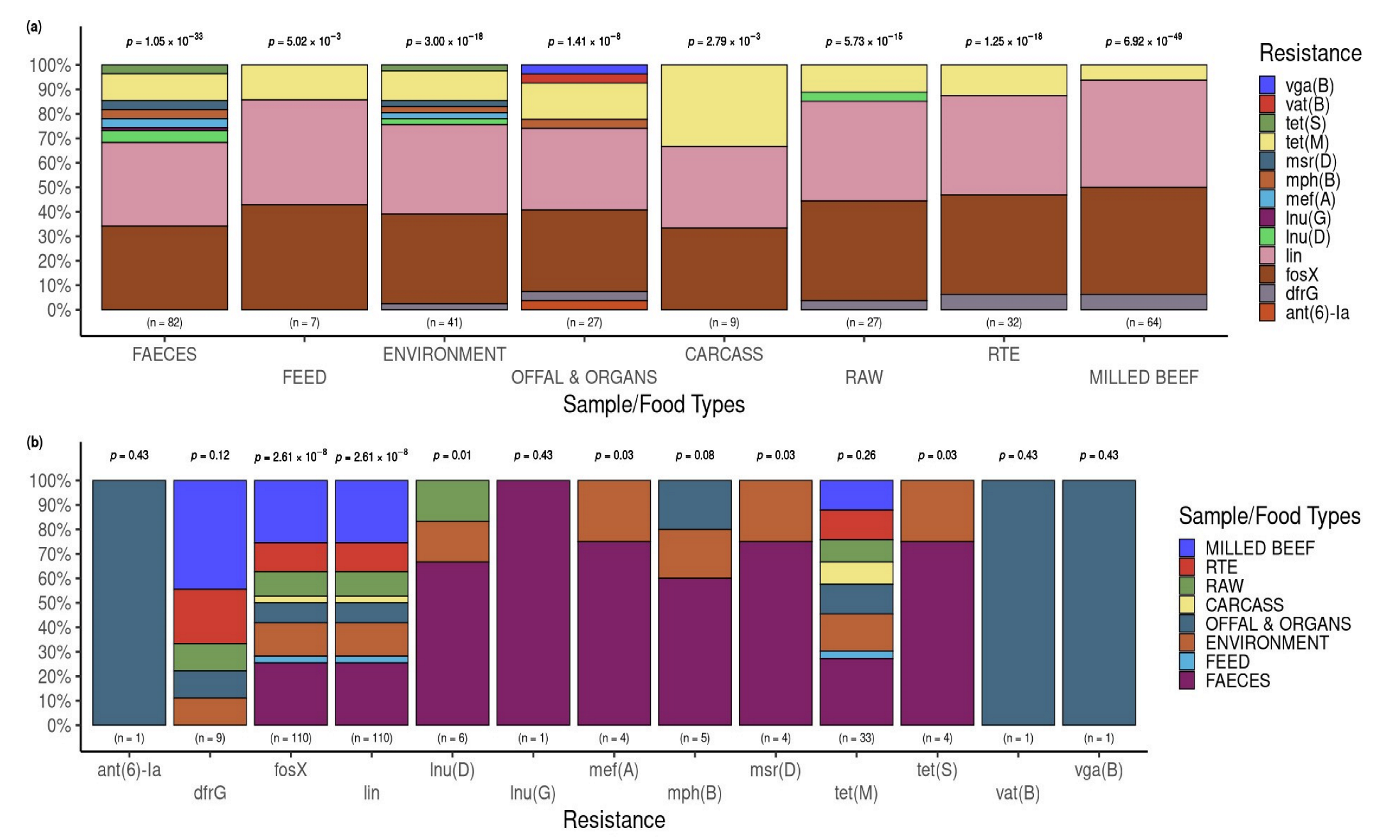
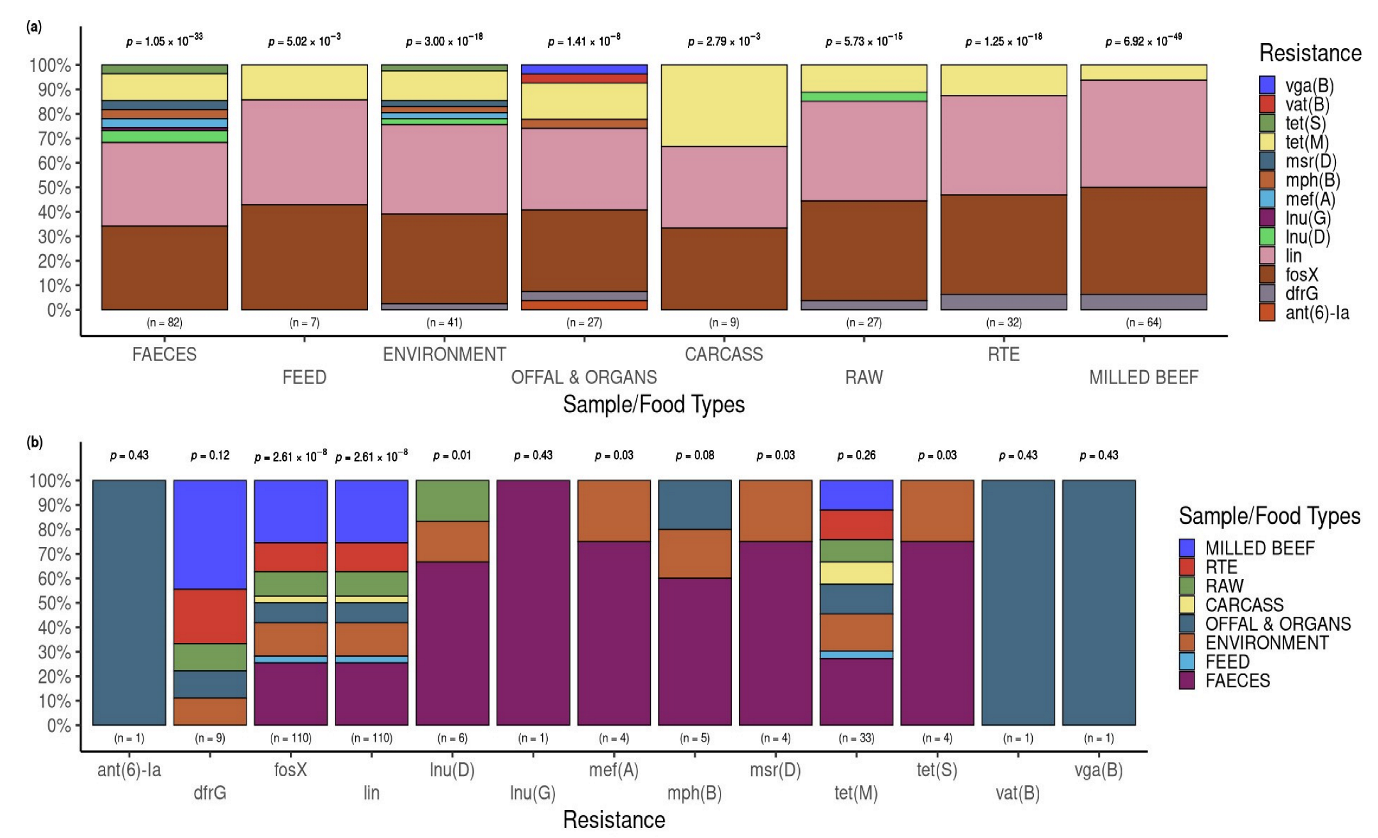
3.3.2. Putative Resistance Phenotypes According to Beef Industries of Origin and the Sample Types
3.3.3. Relationship between Phenotypic AMR Profile and Genomic AMR Gene
3.3.4. Resistance and Virulence Genes across the Industries
3.4. Occurrence of AMR Genes in L. innocua Isolates per Food and Sample Type
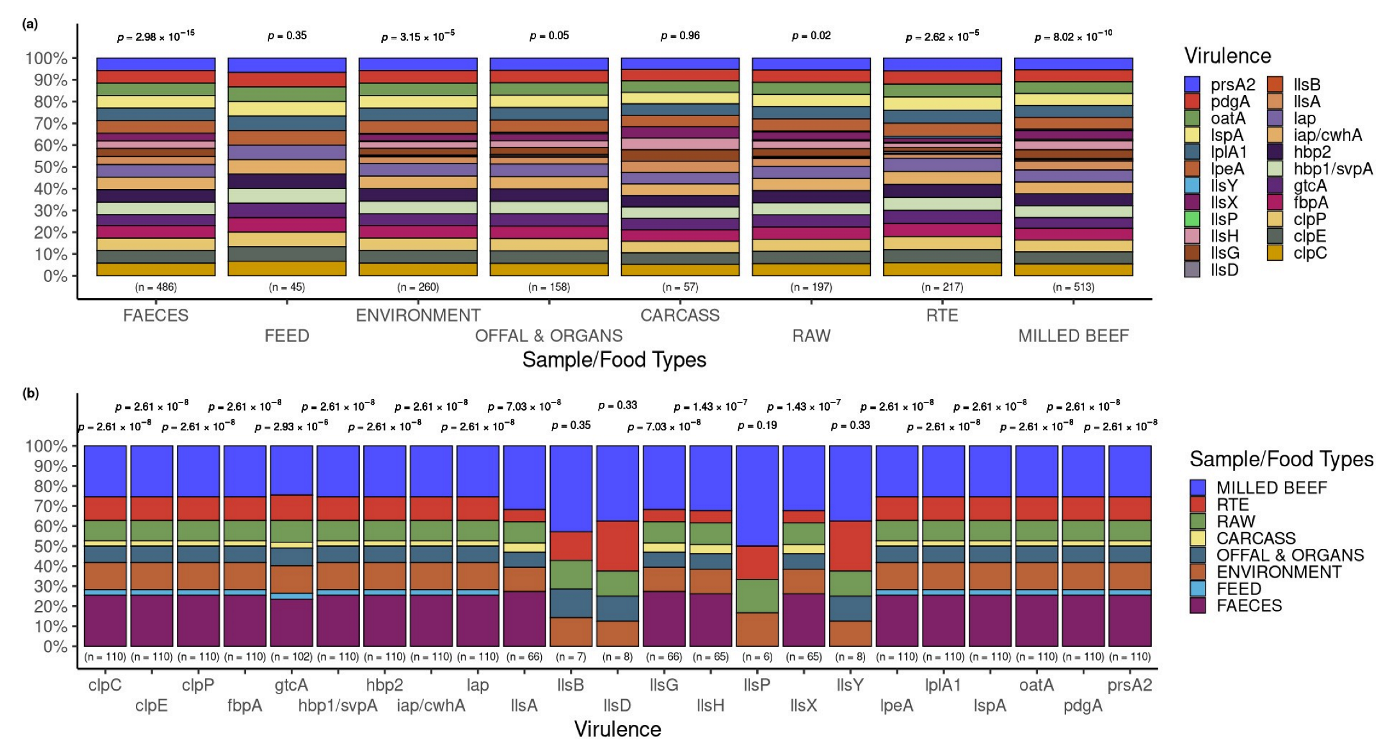
3.5. Occurrence of Virulence Genes in L. innocua Isolates per Food and Sample Type
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gebretsadik, S.; Kassa, T.; Alemayehu, H.; Huruy, K.; Kebede, N. Isolation and characterization of Listeria monocytogenes and other Listeria species in foods of animal origin in Addis Ababa, Ethiopia. J. Infect. Public Health 2011, 4, 22–29. [Google Scholar] [CrossRef]
- Carlin, C.R.; Liao, J.; Weller, D.; Guo, X.; Orsi, R.; Wiedmann, M. Listeria cossartiae sp. nov., Listeria immobilis sp. nov., Listeria portnoyi sp. nov. and Listeria rustica sp. nov., isolated from agricultural water and natural environments. Int. J. Syst. Evol. Microbiol. 2021, 71, 004795. [Google Scholar] [CrossRef] [PubMed]
- Castro, H.; Jaakkonen, A.; Hakkinen, M.; Korkeala, H.; Lindström, M. Occurrence, persistence, and contamination routes of Listeria monocytogenes genotypes on three Finnish dairy cattle farms: A longitudinal study. Appl. Environ. Microbiol. 2018, 84, e02000-17. [Google Scholar] [CrossRef]
- Murray, E.G.D.; Webb, R.A.; Swann, M.B.R. A disease of rabbits characterised by a large mononuclear leucocytosis, caused by a hitherto undescribed bacillus Bacterium monocytogenes (n. sp.). J. Pathol. Bacteriol. 1926, 29, 407–439. [Google Scholar] [CrossRef]
- Molla, B.; Yilma, R.; Alemayehu, D. Listeria monocytogenes and other Listeria species in retail meat and milk products in Addis Ababa, Ethiopia. Ethiopia J. Health Dev. 2004, 18, 208–212. [Google Scholar] [CrossRef]
- Zoellner, C.; Wiedmann, M.; Ivanek, R. An Assessment of Listeriosis Risk Associated with a Contaminated Production Lot of Frozen Vegetables Consumed under Alternative Consumer Handling Scenarios. J. Food Prot. 2019, 82, 2174–2193. [Google Scholar] [CrossRef] [PubMed]
- Acciari, V.; Iannetti, L.; Schirone, M.; Neri, D.; Visciano, P.; Acciari, V.A.; Centorotola, G.; Mangieri, M.S.; Torresi, M.; Santarelli, G.A.; et al. Listeria monocytogenes in poultry: Detection and strain characterization along an integrated production chain in Italy. Food Microbiol. 2020, 91, 103533. [Google Scholar]
- Kureljušić, J.; Rokvić, N.; Jezdimirović, N.; Kureljušić, B.; Pisinov, B.; Karabasil, N. Isolation and detection of Listeria monocytogenes in poultry meat by standard culture methods and PCR. In IOP Conference Series: Earth and Environmental Science; 2017; Volume 85, p. 012069. Available online: https://iopscience.iop.org/article/10.1088/1755-1315/85/1/012069/pdf (accessed on 7 July 2023).
- Favaro, M.; Sarmati, L.; Sancesario, G.; Fontana, C. First case of Listeria innocua meningitis in a patient on steroids and eternecept. JMM Case Rep. 2014, 1, e003103. [Google Scholar] [CrossRef]
- Gradovska, S.; Šteingolde, Ž.; Ķibilds, J.; Meistere, I.; Avsejenko, J.; Streikiša, M.; Alksne, L.; Terentjeva, M.; Bērziņš, A. Genetic diversity and known virulence genes in Listeria innocua strains isolated from cattle abortions and farm environment. Vet. Anim. Sci. 2023, 19, 100276. [Google Scholar] [CrossRef]
- Seçil, A.B.A.Y.; Aydin, F.; Sumerkan, A.B. Molecular typing of Listeria spp. isolated from different sources. Ankara Üniversitesi Veteriner Fakültesi Dergisi. 2012, 59, 183–190. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Q.; Jiang, L.; Cheng, C.; Bai, F.; Wang, J.; Mo, F.; Fang, W. Internalin profiling and multilocus sequence typing suggest four Listeria innocua subgroups with different evolutionary distances from Listeria monocytogenes. BMC Microbiol. 2010, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Disson, O.; Moura, A.; Lecuit, M. Making sense of the biodiversity and virulence of Listeria monocytogenes. Trends Microbiol. 2021, 29, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Clayton, E.M.; Daly, K.M.; Guinane, C.M.; Hill, C.; Cotter, P.D.; Ross, P.R. Atypical Listeria innocua strains possess an intact LIPI-3. BMC Microbiol. 2014, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Moura, A.; Disson, O.; Lavina, M.; Thouvenot, P.; Huang, L.; Leclercq, A.; Fredriksson-Ahomaa, M.; Eshwar, A.K.; Stephan, R.; Lecuit, M. Atypical hemolytic Listeria innocua isolates are virulent, albeit less than Listeria monocytogenes. Infect. Immun. 2019, 87, e00758-18. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Jinneman, K.; Stelma, G.; Smith, B.G.; Lye, D.; Messer, J.; Ulaszek, J.; Evsen, L.; Gendel, S.; Bennett, R.W.; et al. Natural atypical Listeria innocua strains with Listeria monocytogenes pathogenicity island 1 Genes. Appl. Environ. Microbiol. 2004, 70, 4256–4266. [Google Scholar] [CrossRef]
- Chen, Y.; Gonzalez-Escalona, N.; Hammack, T.S.; Allard, M.W.; Strain, E.A.; Brown, E.W. Core genome multilocus sequence typing for identification of globally distributed clonal groups and differentiation of outbreak strains of Listeria monocytogenes. Appl. Environ. Microbiol. 2016, 82, 6258–6272. [Google Scholar] [CrossRef]
- Kaszoni-Rückerl, I.; Mustedanagic, A.; Muri-Klinger, S.; Brugger, K.; Wagner, K.H.; Wagner, M.; Stessl, B. Predominance of distinct Listeria innocua and Listeria monocytogenes in recurrent contamination events at dairy processing facilities. Microorganisms. 2020, 10, 234. [Google Scholar] [CrossRef]
- Oswaldi, V.; Lüth, S.; Dzierzon, J.; Meemken, D.; Schwarz, S.; Feßler, A.T.; Félix, B.; Langforth, S. Distribution and characteristics of Listeria spp. in pigs and pork production chains in Germany. Microorganisms 2022, 26, 512. [Google Scholar] [CrossRef]
- Tomáštíková, Z.; Gelbíčová, T.; Karpíšková, R. Population structure of Listeria monocytogenes isolated from human listeriosis cases and from ready-to-eat foods in the Czech Republic. J. Food Nutr. Res. 2019, 1, 58. [Google Scholar]
- Olanya, O.M.; Hoshide, A.K.; Ijabadeniyi, O.A.; Ukuku, D.O.; Mukhopadhyay, S.; Niemira, B.A.; Ayeni, O. Cost estimation of listeriosis (Listeria monocytogenes) occurrence in South Africa in 2017 and its food safety implications. Food Contr. 2019, 1, 231–239. [Google Scholar] [CrossRef]
- Hyden, P.; Pietzka, A.; Lennkh, A.; Murer, A.; Springer, B.; Blaschitz, M.; Indra, A.; Huhulescu, S.; Allerberger, F.; Ruppitsch, W.; et al. Whole genome sequence-based serogrouping of Listeria monocytogenes isolates. J. Biotechnol. 2016, 235, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.C.; McCallum, N.; Sintchenko, V.; Howden, B.P. Whole genome sequencing in clinical and public health microbiology. Pathology 2015, 47, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Segerman, B. The most frequently used sequencing technologies and assembly methods in different time segments of the bacterial surveillance and RefSeq genome databases. Front. Cell. Infect. Microbial. 2020, 10, 527102. [Google Scholar] [CrossRef] [PubMed]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.S.; Call, D.R.; Broschat, S.L. PARGT: A software tool for predicting antimicrobial resistance in bacteria. Sci. Rep. 2020, 1, 11033. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, K.; Matsuyama, T.; Kündig, T.M.; Wakeham, A.; Kishihara, K.; Shahinian, A.; Wiegmann, K.; Ohashi, P.S.; Krönke, M.; Mak, T.W. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to Listeria. monocytogenes infection. Cell 1993, 7, 457–467. [Google Scholar] [CrossRef]
- Shamloo, E.; Hosseini, H.; Moghadam, Z.A.; Larsen, M.H.; Haslberger, A.; Alebouyeh, M. Importance of Listeria monocytogenes in food safety: A review of its prevalence, detection, and antibiotic resistance. Iran J. Vet. Res. 2019, 20, 241. [Google Scholar]
- Wu, L.; Bao, H.; Yang, Z.; He, T.; Tian, Y.; Zhou, Y.; Pang, M.; Wang, R.; Zhang, H. Antimicrobial susceptibility, multilocus sequence typing, and virulence of Listeria isolated from a slaughterhouse in Jiangsu, China. BMC Microbiol. 2021, 21, 327. [Google Scholar] [CrossRef]
- Hosain, M.Z.; Kabir, S.L.; Kamal, M.M. Antimicrobial uses for livestock production in developing countries. Vet. World 2021, 14, 210. [Google Scholar] [CrossRef]
- Allam, M.; Tau, N.; Smouse, S.L.; Mtshali, P.S.; Mnyameni, F.; Khumalo, Z.T.; Ismail, A.; Govender, N.; Thomas, J.; Smith, A.M. Whole-genome sequences of Listeria monocytogenes sequence type 6 isolates associated with a large foodborne outbreak in South Africa, 2017 to 2018. Genome Annonc. 2018, 6, e00538-18. [Google Scholar] [CrossRef]
- Matle, I.; Mbatha, K.R.; Lentsoane, O.; Magwedere, K.; Morey, L.; Madoroba, E. Occurrence, serotypes, and characteristics of Listeria monocytogenes in meat and meat products in South Africa between 2014 and 2016. J. Food Saf. 2019, 39, e12629. [Google Scholar] [CrossRef]
- Gana, J.; Gcebe, N.; Pierneef, R.; Moerane, R.; Adesiyun, A.A. Multiple-Locus Variable-Number Tandem Repeat Analysis Genotypes of Listeria monocytogenes Isolated from Farms, Abattoirs, and Retail in Gauteng Province, South Africa. J. Food Prot. 2022, 85, 1249–1257. [Google Scholar] [CrossRef]
- Manqele, A.; Gcebe, N.; Pierneef, R.E.; Moerane, R.; Adesiyun, A.A. Identification of Listeria species and Multilocus Variable-Number Tandem Repeat Analysis (MLVA) Typing of Listeria innocua and Listeria monocytogenes Isolates from Cattle Farms and Beef and Beef-Based Products from Retail Outlets in Mpumalanga and North West Provinces, South Africa. Pathogens 2023, 12, 147. [Google Scholar] [PubMed]
- Mafuna, T.; Matle, I.; Magwedere, K.; Pierneef, R.E.; Reva, O.N. Comparative Genomics of Listeria Species Recovered from Meat and Food Processing Facilities. Microbiol. Spect. 2022, 10, e01189-22. [Google Scholar] [CrossRef]
- ElZowalaty, M.E.; Hickman, R.A.; Moura, A.; Lecuit, M.; Zishiri, O.T.; Noyes, N.; Järhult, J.D. Genome sequence of Listeria innocua strain MEZLIS26, isolated from a goat in South Africa. Microbiol. Resour. Announc. 2019, 8, e00991-19. [Google Scholar]
- Gana, J. Prevalence. Risk Factors and Molecular Characterization of Listeria Species from Cattle Farms, Beef Abattoir and Retail Outlets in Gauteng, South Africa. Ph.D. Thesis, University of Pretoria: Pretoria, South Africa, 2022. [Google Scholar]
- Thrusfield, M. Sample size determination. Vet. Epidemiol. 2007, 3, 185–189. [Google Scholar]
- Department of Agriculture. Meat Safety Act. 2000 (Act 40 of 2000). Red Meat Regulations; Department of Agriculture: Washington, DC, USA, 2004; No.1072. [Google Scholar]
- Soumet, C.; Ermel, G.; Fach, P.; Colin, P. Evaluation of different DNA extraction procedures for the detection of Salmonella from chicken products by polymerase chain reaction. Lett. Appl. Microbiol. 1994, 19, 294–298. [Google Scholar] [CrossRef]
- Jamali, H.; Chai, L.C.; Thong, K.L. Detection and isolation of Listeria spp. and Listeria monocytogenes in ready-to-eat foods with various selective culture media. Food Contr. 2013, 32, 19–24. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing, 23rd ed.; informational supplement (M100-S23); CLSI: Wayne, PA, USA, 2013. [Google Scholar]
- Conter, M.; Paludi, D.; Zanardi, E.; Ghidini, S.; Vergara, A.; Ianieri, A. Characterization of antimicrobial resistance of foodborne Listeria monocytogenes. Int. J. Food Microbial. 2009, 128, 497–500. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. mlst Github. Available online: https://github.com/tseemann/mlst (accessed on 7 July 2023).
- Moura, A.; Criscuolo, A.; Pouseele, H.; Maury, M.M.; Leclercq, A.; Tarr, C.; Björkman, J.T.; Dallman, T.; Reimer, A.; Enouf, V.; et al. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol. 2016, 2, 16185. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Abricate, Github. Available online: https://github.com/tseemann/abricate (accessed on 28 July 2023).
- Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- RStudio Team. RStudio: Integrated Development Environment for R. RStudio; PBC: Boston, MA, USA, 2022; Available online: http://www.rstudio (accessed on 7 July 2023).
- Maechler, M. Finding groups in data: Cluster analysis extended Rousseeuw et al. R Package Version 2019, 2, 242–248. [Google Scholar]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Inter. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Tyner, R.C.; Briatte, F.; Hofmann, H. Network Visualization with ggplot2. R J. 2017, 9, 26–59. [Google Scholar]
- Patil, I. Visualizations with statistical details: The ‘ggstatsplot’ approach. J. Open. Source Soft. 2021, 61, 3167. [Google Scholar] [CrossRef]
- Xiao, N. ggsci: Scientific Journal and Sci-Fi Themed Color Palettes for ‘ggplot2’. In R Package Version; R Core Team: Vienna, Austria, 2018; Volume 2, p. 9. [Google Scholar]
- Kassambara, A.; Kassambara, M.A. Package ‘ggpubr’. In R Package Version 0.1; R Core Team: Vienna, Austria, 2020; Volume 6. [Google Scholar]
- Kaptchouang Tchatchouang, C.D.; Fri, J.; De Santi, M.; Brandi, G.; Schiavano, G.F.; Amagliani, G.; Ateba, C.N. Listeriosis outbreak in South Africa: A comparative analysis with previously reported cases worldwide. Microorganisms 2020, 8, 135. [Google Scholar] [CrossRef]
- Escolar, C.; Gómez, D.; del Carmen Rota García, M.; Conchello, P.; Herrera, A. Antimicrobial resistance profiles of Listeria monocytogenes and Listeria innocua isolated from ready-to-eat products of animal origin in Spain. Foodborne Path. Dis. 2017, 14, 357–363. [Google Scholar] [CrossRef]
- Hanes, R.M.; Huang, Z. Investigation of Antimicrobial Resistance Genes in Listeria monocytogenes from 2010 through to 2021. Int. J. Environ. Res. Public. Health 2022, 19, 5506. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, J.; Bland, R.; Waite-Cusic, J.; Kovacevic, J. Diversity and antimicrobial resistance of Listeria spp. and Listeria monocytogenes clones from produce handling and processing facilities in the Pacific Northwest. Food Contr. 2021, 123, 107665. [Google Scholar] [CrossRef]
- Palaiodimou, L.; Fanning, S.; Fox, E.M. Genomic insights into persistence of Listeria species in the food processing environment. J. Appl. Microbiol. 2021, 131, 2082–2094. [Google Scholar] [CrossRef] [PubMed]
- Mpondo, L.; Ebomah, K.E.; Okoh, A.I. Multidrug-resistant Listeria species shows abundance in environmental waters of a key district municipality in South Africa. Int. J. Environ. Res. Public. Health. 2021, 18, 481. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, H.; Al-Ashmaw, M.; Soliman, A.M.; Elbediwi, M.; Sabeq, I.; Yousef, M.; Algammal, A.A.; Hiott, L.M.; Berrang, M.E.; Frye, J.G.; et al. Whole-genome sequencing of Listeria innocua recovered from retail milk and dairy products in Egypt. Front. Microbiol. 2023, 14, 1160244. [Google Scholar] [CrossRef] [PubMed]
- Scortti, M.; Han, L.; Alvarez, S.; Leclercq, A.; Moura, A.; Lecuit, M.; Vazquez-Boland, J. Epistatic control of intrinsic resistance by virulence genes in Listeria. PLoS Genet. 2018, 14, e1007525. [Google Scholar] [CrossRef]
- Parra-Flores, J.; Holý, O.; Bustamante, F.; Lepuschitz, S.; Pietzka, A.; Contreras-Fernández, A.; Castillo, C.; Ovalle, C.; Alarcón-Lavín, M.P.; Cruz-Córdova, A.; et al. Virulence and Antibiotic Resistance Genes in Listeria monocytogenes Strains Isolated From Ready-to-Eat Foods in Chile. Front. Microbiol. 2022, 12, 796040. [Google Scholar] [CrossRef]
- Mupfunya, C.R.; Qekwana, D.N.; Naidoo, V. Antimicrobial use practices and resistance in indicator bacteria in communal cattle in the Mnisi community, Mpumalanga, South Africa. Vet. Med. Sci. 2021, 7, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Adesiyun, A.A.; Nkuna, C.; Mokgoatlheng Mamogobo, M.; Malepe, K.; Simanda, L. Food safety risk posed to consumers of table eggs from layer farms in Gauteng Province, South Africa: Prevalence of Salmonella species and Escherichia coli, antimicrobial residues, and antimicrobial resistant bacteria. J. Food Saf. 2020, 40, e12783. [Google Scholar] [CrossRef]
- Van, T.T.H.; Yidana, Z.; Smooker, P.M.; Coloe, P.J. Antibiotic use in food animals worldwide, with a focus on Africa: Pluses and minuses. J. Glob. Antimicrob. Resist. 2020, 20, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Gray, J.; Chandry, P.S.; Fox, E.M. Phenotypic and genotypic analysis of antimicrobial resistance among Listeria monocytogenes isolated from Australian food production chains. Genes 2018, 9, 80. [Google Scholar] [CrossRef]
- Salyers, A.A.; Amabile-Cuevas, C.F. Why are antibiotic resistance genes so resistant to elimination? Antimicrob. Age Chem. 1997, 41, 2321–2325. [Google Scholar] [CrossRef]
- Hummel, A.S.; Hertel, C.; Holzapfel, W.H.; Franz, C.M. Antibiotic resistances of starter and probiotic strains of lactic acid bacteria. Appl. Environ. Microbiol. 2007, 73, 730–739. [Google Scholar] [CrossRef]
- Olaimat, A.N.; Al-Holy, M.A.; Shahbaz, H.M.; Al-Nabulsi, A.A.; Abu Ghoush, M.H.; Osaili, T.M.; Ayyash, M.M.; Holley, R.A. Emergence of antibiotic resistance in Listeria monocytogenes isolated from food products: A comprehensive review. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1277–1292. [Google Scholar] [CrossRef]
- Maury, M.M.; Chenal-Francisque, V.; Bracq-Dieye, H.; Han, L.; Leclercq, A.; Vales, G.; Moura, A.; Gouin, E.; Scortti, M.; Disson, O.; et al. Spontaneous loss of virulence in natural populations of Listeria monocytogenes. Infect. Immun. 2017, 85, e00541-17. [Google Scholar] [CrossRef]
- Liu, D.; Lawrence, M.L.; Austin, F.W.; Ainsworth, A.J. A multiplex PCR for species-and virulence-specific determination of Listeria monocytogenes. J. Microbiol. Methods 2007, 71, 133–140. [Google Scholar] [CrossRef]
- Rabinovich, L.; Sigal, N.; Borovok, I.; Nir-Paz, R.; Herskovits, A.A. Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 2012, 150, 792–802. [Google Scholar] [CrossRef]
- Gilmour, M.W.; Graham, M.; Van Domselaar, G.; Tyler, S.; Kent, H.; Trout-Yakel, K.M.; Larios, O.; Allen, V.; Lee, B.; Nadon, C. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genom. 2010, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Wurtzel, O.; Sesto, N.; Mellin, J.R.; Karunker, I.; Edelheit, S.; Bécavin, C.; Archambaud, C.; Cossart, P.; Sorek, R. Comparative transcriptomics of pathogenic and non-pathogenic Listeria species. Mol. Syst. Biol. 2012, 8, 583. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, E.M.; Björkman, J.T.; Kiil, K.; Grant, K.; Dallman, T.; Painset, A.; Amar, C.; Roussel, S.; Guillier, L.; Félix, B.; et al. Closing gaps for performing a risk assessment on Listeria monocytogenes in ready-to-eat (RTE) foods: Activity 3, the comparison of isolates from different compartments along the food chain and from humans using whole genome sequencing (WGS) analysis. EFSA Support. Publ. 2017, 14, 1151E. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Isolates of L. innocua | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of Samples | No. (%) Positive | No. of Isolates | No. (%) of Isolates That Belong to ST a: | ||||||||||||||
| Industry | Tested | For L. innocua | T\ested | ST637 | ST448 | ST537 | ST1085 | ST1087 | ST1489 | ST1482 | ST1008 | ST602 | ST43 | ST 1619 | ST 1610 | ST3007 | ST 2754 |
| Cattle farms b | 328 | 37 (11.3) | 34 | 17 (50.0) | 2 (5.9) | 2 (5.9) | 1 (2.9) | 1 (2.9) | 0 (0.0) | 6 (17.6) | 0 (0.0) | 0 (0.0) | 1 (2.9) | 0 (0.0) | 4 (11.8) | 0 (0.0) | 1 (2.9) |
| Abattoirs c | 262 | 15 (5.7) | 15 | 2 (13.3) | 0 (0.0) | 11 (73.3) | 0 (0.0) | 0 (0.0) | 1 (6.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Retail outlets d | 400 | 65 (16.3) | 61 | 10 (16.4) | 20 (32.8) | 2 (3.3) | 13 (21.3) | 0 (0.0) | 7 (11.5) | 1 (1.6) | 5 (8.2) | 1 (1.6) | 0 (0.0) | 1 (1.6) | 0 (0.0) | 1 (1.6) | 0 (0.0) |
| Total | 990 | 117 (11.8) | 110 | 29 (26.4) | 22 (20.0) | 15 (13.6) | 14 (12.7) | 1 (0.9) | 8 (7.3) | 7 (6.4) | 5 (4.5) | 1 (0.9) | 1 (0.9) | 1 (0.9) | 4 (3.6) | 1 (0.9) | 1 (0.9) |
| No. of samples | No. (%) positive | No. of isolates | No (%) positive for AMR gene e | ||||||||||||||
| Industry | tested | for L. innocua | tested | fosX | Lin | tet (M) | dfrG7 | imuD | ImuG | mefA | mph(B) | mrs(D) | tet(S) | vatB | vga | ant.6.1a | |
| Cattle farms b | 328 | 37 (11.3) | 34 | 34 (100.0) | 34 (100.0) | 7 (20.6) | 0 (0.0) | 5 (14.7) | 1 (2.9) | 4 (11.8) | 4 (11.8) | 4 (11.8) | 4 (11.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Abattoirs c | 262 | 15 (5.7) | 15 | 15 (100.0) | 15 (100.0) | 11 (73.3) | 1 (6.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Retail outlets d | 400 | 65 (16.3) | 61 | 61 (100.0) | 61 (100.0) | 15 (24.6) | 8 (13.1) | 1 (1.6) | 0 (0.0) | 0 (0.0) | 1 (1.6) | 0 (0.0) | 0 (0.0) | 1 (1.6) | 1 (1.6) | 1 (1.6) | |
| Total | 990 | 117 (11.8) | 110 | 110 (100.0) | 110 (100.0) | 33 (30.0) | 9 (8.2) | 6 (5.5) | 1 (0.9) | 4 (3.6) | 5 (4.5) | 4 (3.6) | 4 (3.6) | 1 (0.9) | 1 (0.9) | 1 (0.9) | |
| Number of Resistance | No. (%) of L. innocua Isolates with Resistance Genes by Source of Samples | Total (No., %), | ||||
|---|---|---|---|---|---|---|
| Genes a (No. of Patterns) | Resistance Gene Pattern | Cattle Farm (n = 34) c | Beef Abattoirs (n = 15) d | Retail Outlets (n = 61) e | p-Value | n = 110 |
| 2 (1) | b fosX-lin | 23 (67.6) | 3 (20.0) | 38 (62.3) | 0.0048 | 64 (58.2) |
| 3 (2) | fosX-lin-tet(M) | 5 (14.7) | 11 (73.3) | 11 (18.0) | 0.00001 | 27 (24.5) |
| dfrG-fosX-lin | 0 (0.0) | 1 (6.7) | 7 (11.4) | 0.0474 | 8 (7.3) | |
| 4 (4) | fosX-lin-Inu(D)-tet(M) | 1 (2.9) | 0 (0.0) | 1 (1.6) | 1 | 2 (1.8) |
| fosX-lin-Inu(G)-tet(M) | 1 (2.9) | 0 (0.0) | 0 (0.0) | 0.7539 | 1 (0.9) | |
| fosX-lin-tet(M)-vat(B) | 0 (0.0) | 0 (0.0) | 1 (1.6) | 1 | 1 (0.9) | |
| dfrG-fosX-lin-tet(M) | 0 (0.0) | 0 (0.0) | 1 (1.6) | 1 | 1 (0.9) | |
| 5 (2) | ant(6)-Ia-fosX-lin-mph(B)-tet(M) | 0 (0.0) | 0 (0.0) | 1 (1.6) | 1 | 1 (0.9) |
| fosX-lin-tet(M)-vat(B)-vga(B) | 0 (0.0) | 0 (0.0) | 1 (1.6) | 1 | 1 (0.9) | |
| 7 (1) | fosX-lin-Inu(D)-mef(A)-mph(B)-msr(D), tet(S) | 4 (11.8) | 0 (0.0) | 0 (0.0) | 0.146 | 4 (3.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gana, J.; Gcebe, N.; Pierneef, R.E.; Chen, Y.; Moerane, R.; Adesiyun, A.A. Genomic Characterization of Listeria innocua Isolates Recovered from Cattle Farms, Beef Abattoirs, and Retail Outlets in Gauteng Province, South Africa. Pathogens 2023, 12, 1062. https://doi.org/10.3390/pathogens12081062
Gana J, Gcebe N, Pierneef RE, Chen Y, Moerane R, Adesiyun AA. Genomic Characterization of Listeria innocua Isolates Recovered from Cattle Farms, Beef Abattoirs, and Retail Outlets in Gauteng Province, South Africa. Pathogens. 2023; 12(8):1062. https://doi.org/10.3390/pathogens12081062
Chicago/Turabian StyleGana, James, Nomakorinte Gcebe, Rian Ewald Pierneef, Yi Chen, Rebone Moerane, and Abiodun Adewale Adesiyun. 2023. "Genomic Characterization of Listeria innocua Isolates Recovered from Cattle Farms, Beef Abattoirs, and Retail Outlets in Gauteng Province, South Africa" Pathogens 12, no. 8: 1062. https://doi.org/10.3390/pathogens12081062
APA StyleGana, J., Gcebe, N., Pierneef, R. E., Chen, Y., Moerane, R., & Adesiyun, A. A. (2023). Genomic Characterization of Listeria innocua Isolates Recovered from Cattle Farms, Beef Abattoirs, and Retail Outlets in Gauteng Province, South Africa. Pathogens, 12(8), 1062. https://doi.org/10.3390/pathogens12081062