
Analysis of Lung Microbiome in COVID-19 Patients during Time of Hospitalization

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Sample Collection
2.2. Microbial DNA Extraction
2.3. Nanopore-Targeted Sequencing and Analysis
2.3.1. Amplification
2.3.2. Library Construction
2.3.3. Sequencing and Analyzing
2.4. Statistical Analysis
3. Results
3.1. Clinical Characteristics of the Study Population
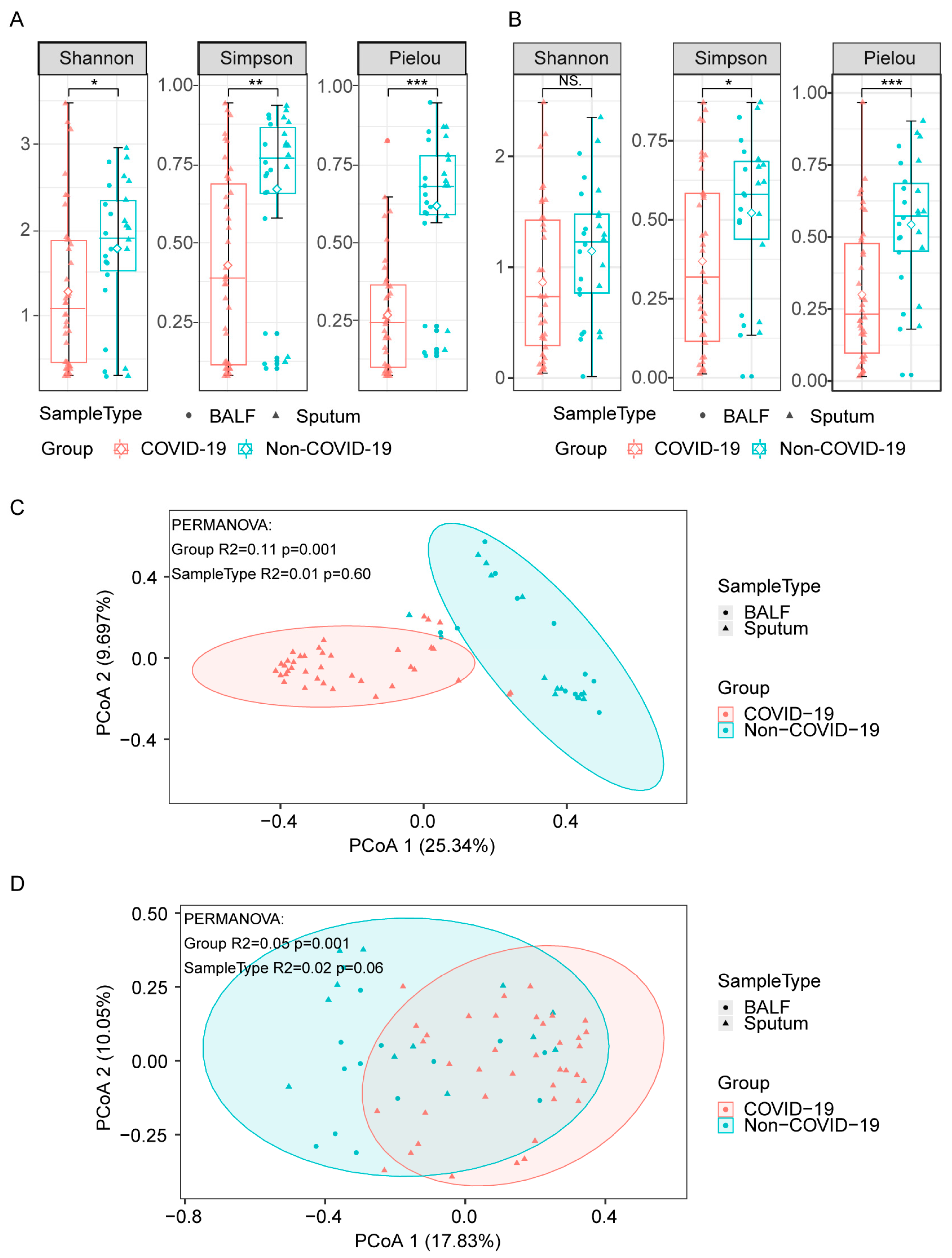
3.2. Lung Microbiome Diversity in COVID-19 and Non-COVID-19 Patients
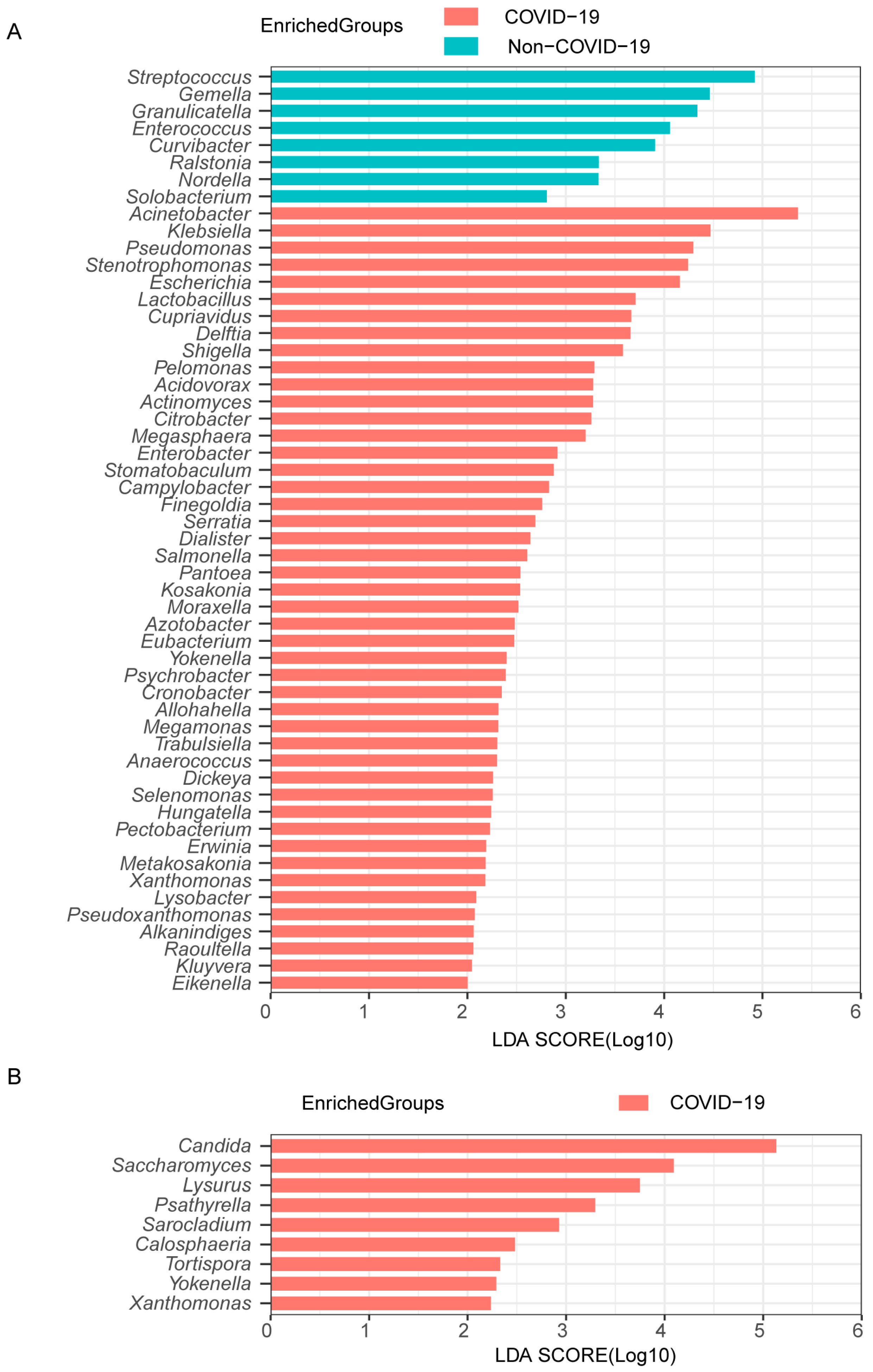
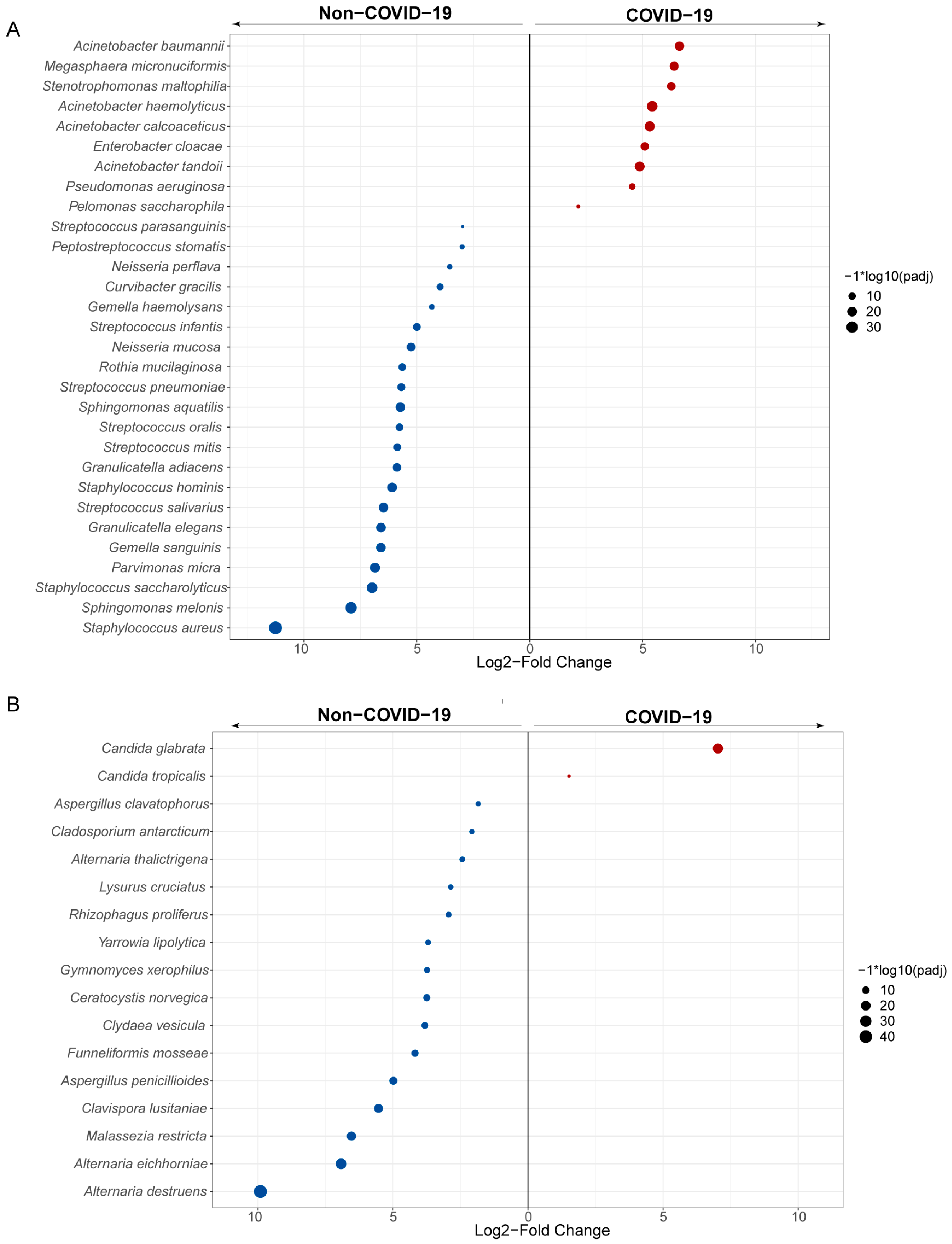
3.3. Lung Microbiome Composition in COVID-19 and Non-COVID-19 Patients
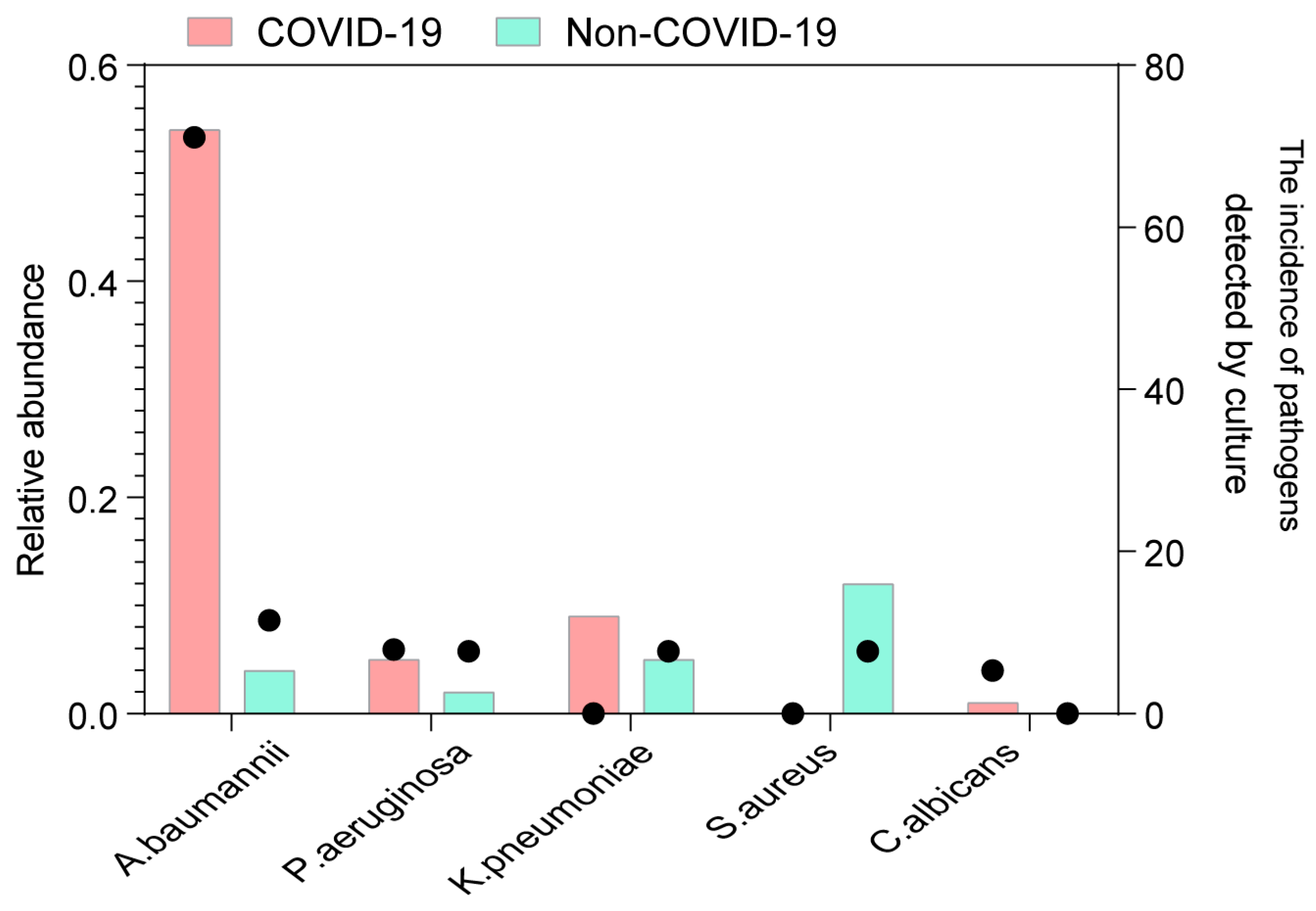
3.4. Codetection of Pathogens in the Lung of COVID-19 Patients
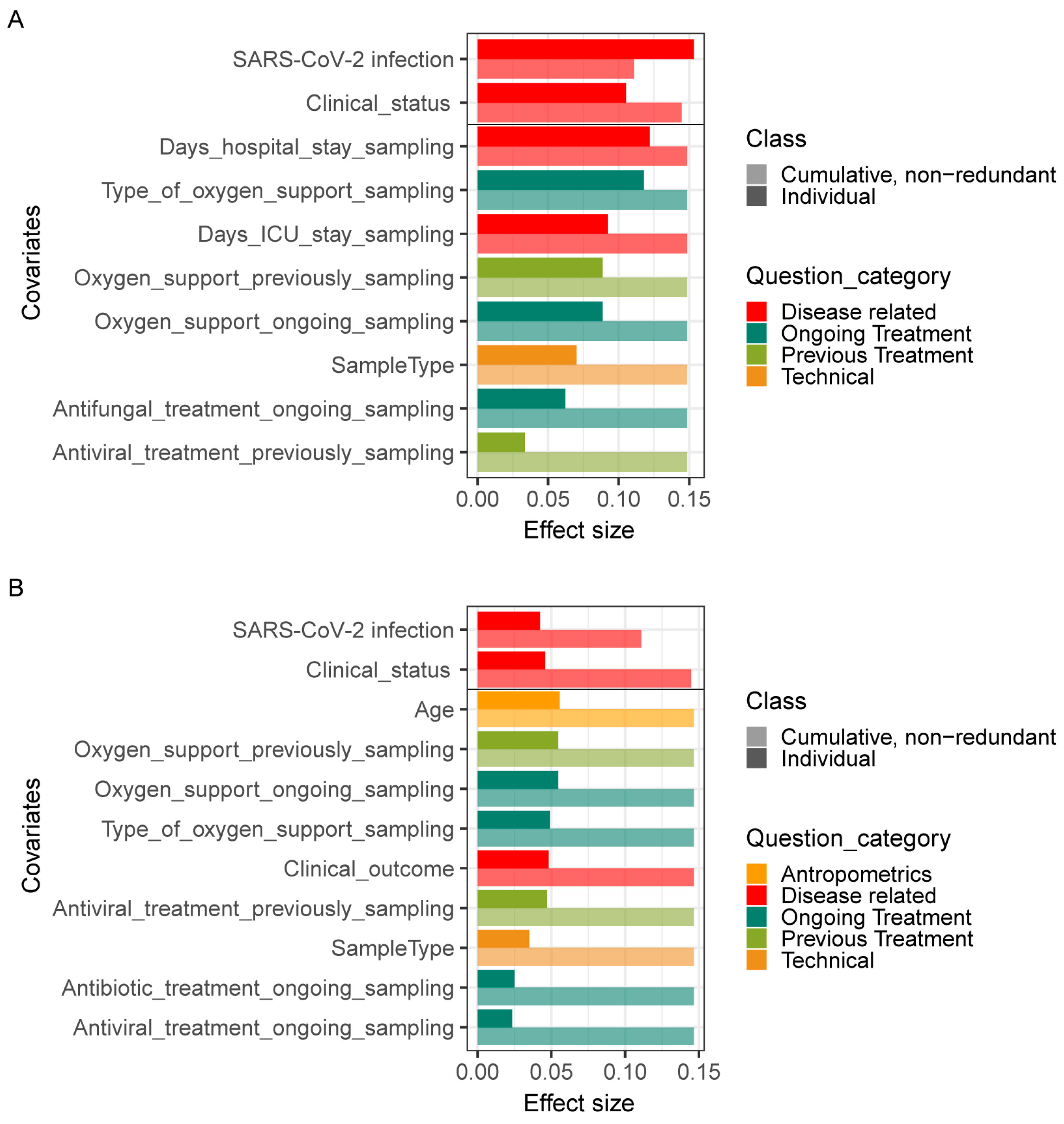
3.5. Associations between Lung Microbiome Composition and Clinical Practices in COVID-19 and Non-COVID-19 Patients
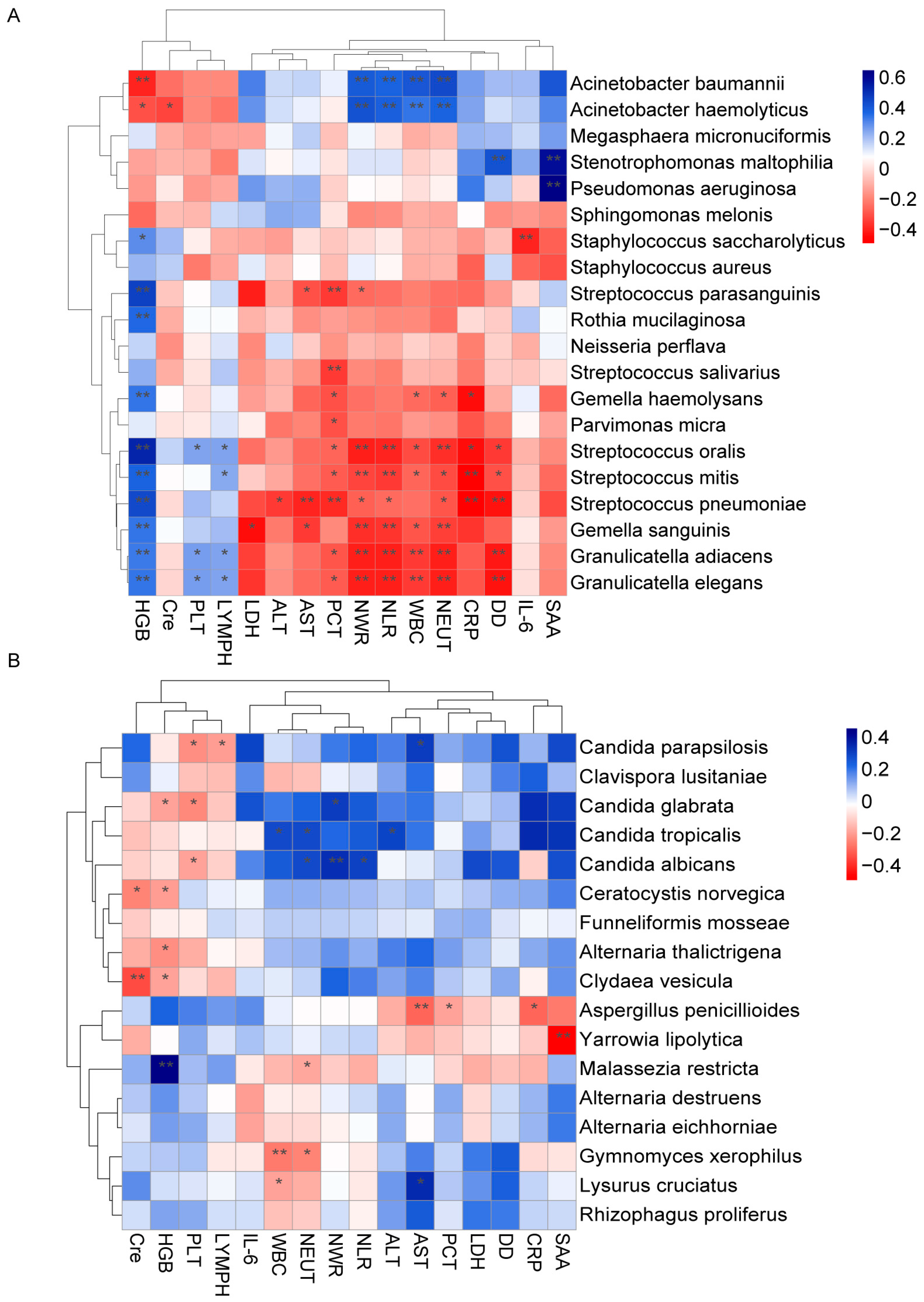
3.6. Correlation between the Lung Microbiota and Clinical Indicators during Time of Hospitalization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castagnoli, R.; Votto, M.; Licari, A.; Brambilla, I.; Bruno, R.; Perlini, S.; Rovida, F.; Baldanti, F.; Marseglia, G.L. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection in Children and Adolescents: A Systematic Review. JAMA Pediatr. 2020, 174, 882–889. [Google Scholar] [CrossRef]
- Ni, L.; Ye, F.; Cheng, M.L.; Feng, Y.; Deng, Y.Q.; Zhao, H.; Wei, P.; Ge, J.; Gou, M.; Li, X.; et al. Detection of SARS-CoV-2-Specific Humoral and Cellular Immunity in COVID-19 Convalescent Individuals. Immunity 2020, 52, 971–977.e973. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Wang, C.Y.; Hsueh, P.R. Co-infections among patients with COVID-19: The need for combination therapy with non-anti-SARS-CoV-2 agents? J. Microbiol. Immunol. Infect. = Wei Mian Yu Gan Ran Za Zhi 2020, 53, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Tufan, A.; Avanoglu Guler, A.; Matucci-Cerinic, M. COVID-19, immune system response, hyperinflammation and repurposing antirheumatic drugs. Turk. J. Med. Sci. 2020, 50, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Beovic, B.; Dousak, M.; Ferreira-Coimbra, J.; Nadrah, K.; Rubulotta, F.; Belliato, M.; Berger-Estilita, J.; Ayoade, F.; Rello, J.; Erdem, H. Antibiotic use in patients with COVID-19: A ‘snapshot’ Infectious Diseases International Research Initiative (ID-IRI) survey. J. Antimicrob. Chemother. 2020, 75, 3386–3390. [Google Scholar] [CrossRef]
- Rawson, T.M.; Moore, L.S.P.; Zhu, N.; Ranganathan, N.; Skolimowska, K.; Gilchrist, M.; Satta, G.; Cooke, G.; Holmes, A. Bacterial and Fungal Coinfection in Individuals With Coronavirus: A Rapid Review To Support COVID-19 Antimicrobial Prescribing. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 2459–2468. [Google Scholar] [CrossRef]
- Man, W.H.; de Steenhuijsen Piters, W.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef]
- Gao, Z.; Kang, Y.; Yu, J.; Ren, L. Human pharyngeal microbiome may play a protective role in respiratory tract infections. Genom. Proteom. Bioinform. 2014, 12, 144–150. [Google Scholar] [CrossRef]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef]
- Unger, S.A.; Bogaert, D. The respiratory microbiome and respiratory infections. J. Infect. 2017, 74 (Suppl. S1), S84–S88. [Google Scholar] [CrossRef] [PubMed]
- Bakaletz, L.O. Viral-bacterial co-infections in the respiratory tract. Curr. Opin. Microbiol. 2017, 35, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef] [PubMed]
- Siegel, S.J.; Roche, A.M.; Weiser, J.N. Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source. Cell Host Microbe 2014, 16, 55–67. [Google Scholar] [CrossRef]
- Tsang, T.K.; Lee, K.H.; Foxman, B.; Balmaseda, A.; Gresh, L.; Sanchez, N.; Ojeda, S.; Lopez, R.; Yang, Y.; Kuan, G.; et al. Association Between the Respiratory Microbiome and Susceptibility to Influenza Virus Infection. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 1195–1203. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, F.; Zhou, F.; Li, H.; Ge, W.; Gan, R.; Nie, H.; Li, B.; Wang, Y.; Wu, M.; et al. Metagenomic analysis reveals oropharyngeal microbiota alterations in patients with COVID-19. Signal Transduct. Target. Ther. 2021, 6, 191. [Google Scholar] [CrossRef]
- Merenstein, C.; Liang, G.; Whiteside, S.A.; Cobian-Guemes, A.G.; Merlino, M.S.; Taylor, L.J.; Glascock, A.; Bittinger, K.; Tanes, C.; Graham-Wooten, J.; et al. Signatures of COVID-19 Severity and Immune Response in the Respiratory Tract Microbiome. mBio 2021, 12, e0177721. [Google Scholar] [CrossRef]
- Iebba, V.; Zanotta, N.; Campisciano, G.; Zerbato, V.; Di Bella, S.; Cason, C.; Luzzati, R.; Confalonieri, M.; Palamara, A.T.; Comar, M. Profiling of Oral Microbiota and Cytokines in COVID-19 Patients. Front. Microbiol. 2021, 12, 671813. [Google Scholar] [CrossRef]
- Hernandez-Teran, A.; Mejia-Nepomuceno, F.; Herrera, M.T.; Barreto, O.; Garcia, E.; Castillejos, M.; Boukadida, C.; Matias-Florentino, M.; Rincon-Rubio, A.; Avila-Rios, S.; et al. Dysbiosis and structural disruption of the respiratory microbiota in COVID-19 patients with severe and fatal outcomes. Sci. Rep. 2021, 11, 21297. [Google Scholar] [CrossRef]
- Ren, Z.; Wang, H.; Cui, G.; Lu, H.; Wang, L.; Luo, H.; Chen, X.; Ren, H.; Sun, R.; Liu, W.; et al. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut 2021, 70, 1253–1265. [Google Scholar] [CrossRef]
- de Castilhos, J.; Zamir, E.; Hippchen, T.; Rohrbach, R.; Schmidt, S.; Hengler, S.; Schumacher, H.; Neubauer, M.; Kunz, S.; Muller-Esch, T.; et al. Severe dysbiosis and specific Haemophilus and Neisseria signatures as hallmarks of the oropharyngeal microbiome in critically ill COVID-19 patients. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2021, 75, e1063–e1071. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Rico, V.; Gregory, A.C.; Van Weyenbergh, J.; Jansen, S.; Van Buyten, T.; Qian, J.; Braz, M.; Menezes, S.M.; Van Mol, P.; Vanderbeke, L.; et al. Clinical practices underlie COVID-19 patient respiratory microbiome composition and its interactions with the host. Nat. Commun. 2021, 12, 6243. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Liu, L.; Zhang, M.; Hu, Y.; Yang, Q.; Guo, J.; Guo, Y.; Dai, Y.; Xu, Y.; Cai, Y.; et al. Co-infections of SARS-CoV-2 with multiple common respiratory pathogens in infected patients. Sci. China Life Sci. 2020, 63, 606–609. [Google Scholar] [CrossRef]
- Massey, B.W.; Jayathilake, K.; Meltzer, H.Y. Respiratory Microbial Co-infection with SARS-CoV-2. Front. Microbiol. 2020, 11, 2079. [Google Scholar] [CrossRef]
- Chen, X.; Liao, B.; Cheng, L.; Peng, X.; Xu, X.; Li, Y.; Hu, T.; Li, J.; Zhou, X.; Ren, B. The microbial coinfection in COVID-19. Appl. Microbiol. Biotechnol. 2020, 104, 7777–7785. [Google Scholar] [CrossRef] [PubMed]
- Langford, B.J.; So, M.; Raybardhan, S.; Leung, V.; Westwood, D.; MacFadden, D.R.; Soucy, J.-P.R.; Daneman, N. Bacterial co-infection and secondary infection in patients with COVID-19: A living rapid review and meta-analysis. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2020, 26, 1622–1629. [Google Scholar] [CrossRef]
- Soltani, S.; Zakeri, A.; Zandi, M.; Kesheh, M.M.; Tabibzadeh, A.; Dastranj, M.; Faramarzi, S.; Didehdar, M.; Hafezi, H.; Hosseini, P.; et al. The Role of Bacterial and Fungal Human Respiratory Microbiota in COVID-19 Patients. BioMed Res. Int. 2021, 2021, 6670798. [Google Scholar] [CrossRef]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic Diversity of Severe Acute Respiratory Syndrome-Coronavirus 2 in Patients With Coronavirus Disease 2019. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 713–720. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the Respiratory Tract. Annu. Rev. Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef]
- Richardson, M.; Bowyer, P.; Sabino, R. The human lung and Aspergillus: You are what you breathe in? Med. Mycol. 2019, 57, S145–S154. [Google Scholar] [CrossRef]
- Calus, S.T.; Ijaz, U.Z.; Pinto, A.J. NanoAmpli-Seq: A workflow for amplicon sequencing for mixed microbial communities on the nanopore sequencing platform. Gigascience 2018, 7, giy140. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.I.; Senda, Y.; Nakaguchi, S.; Hashimoto, T. Multiplex PCR using internal transcribed spacer 1 and 2 regions for rapid detection and identification of yeast strains. J. Clin. Microbiol. 2001, 39, 3617–3622. [Google Scholar] [CrossRef]
- Wang, M.; Fu, A.; Hu, B.; Shen, G.; Liu, R.; Zhao, W.; Jiang, S.; Cai, X.; Li, C.; Li, J.; et al. Same-Day Simultaneous Diagnosis of Bacterial and Fungal Infections in Clinical Practice by Nanopore Targeted Sequencing. medRxiv 2020. [Google Scholar] [CrossRef]
- Pan, Y.; Ye, G.; Zeng, X.; Liu, G.; Zeng, X.; Jiang, X.; Zhao, J.; Chen, L.; Guo, S.; Deng, Q.; et al. Can routine laboratory tests discriminate SARS-CoV-2-infected pneumonia from other causes of community-acquired pneumonia? Clin. Transl. Med. 2020, 10, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, X.; Gao, Y.; Zhou, J.; Wang, S.; Huang, B.; Wu, J.; Cao, Q.; Chen, Y.; Wang, Z.; et al. The lung tissue microbiota features of 20 deceased patients with COVID-19. J. Infect. 2020, 81, e64–e67. [Google Scholar] [CrossRef]
- Joly-Guillou, M.L. Clinical impact and pathogenicity of Acinetobacter. Clin. Microbiol. Infect. 2005, 11, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Dizbay, M.; Tunccan, O.G.; Sezer, B.E.; Hizel, K. Nosocomial imipenem-resistant Acinetobacter baumannii infections: Epidemiology and risk factors. Scand. J. Infect. Dis. 2010, 42, 741–746. [Google Scholar] [CrossRef]
- Sileem, A.E.; Said, A.M.; Meleha, M.S. Acinetobacter baumannii in ICU patients: A prospective study highlighting their incidence, antibiotic sensitivity pattern and impact on ICU stay and mortality. Egypt. J. Chest Dis. Tuberc. 2017, 66, 6. [Google Scholar] [CrossRef]
- Xiao, D.; Wang, L.; Zhang, D.; Xiang, D.; Liu, Q.; Xing, X. Prognosis of patients with Acinetobacter baumannii infection in the intensive care unit: A retrospective analysis. Exp. Ther. Med. 2017, 13, 1630–1633. [Google Scholar] [CrossRef]
- Falagas, M.E.; Rafailidis, P.I. Attributable mortality of Acinetobacter baumannii: No longer a controversial issue. Crit. Care 2007, 11, 134. [Google Scholar] [CrossRef] [PubMed]
- Sharifipour, E.; Shams, S.; Esmkhani, M.; Khodadadi, J.; Fotouhi-Ardakani, R.; Koohpaei, A.; Doosti, Z.; Ej Golzari, S. Evaluation of bacterial co-infections of the respiratory tract in COVID-19 patients admitted to ICU. BMC Infect. Dis. 2020, 20, 646. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.; Innes, G.K.; Walters, M.S.; Mehr, J.; Arias, J.; Greeley, R.; Chew, D. Increase in Hospital-Acquired Carbapenem-Resistant Acinetobacter baumannii Infection and Colonization in an Acute Care Hospital During a Surge in COVID-19 Admissions—New Jersey, February–July 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1827–1831. [Google Scholar] [CrossRef]
- Dickson, R.P.; Huffnagle, G.B. The Lung Microbiome: New Principles for Respiratory Bacteriology in Health and Disease. PLoS Pathog. 2015, 11, e1004923. [Google Scholar] [CrossRef] [PubMed]
- Viciani, E.; Gaibani, P.; Castagnetti, A.; Liberatore, A.; Bartoletti, M.; Viale, P.; Lazzarotto, T.; Ambretti, S.; Lewis, R.; Cricca, M. Critically ill patients with COVID-19 show lung fungal dysbiosis with reduced microbial diversity in patients colonized with Candida spp. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2022, 117, 233–240. [Google Scholar] [CrossRef]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Xiang, P.; Pu, L.; Xiong, H.; Li, C.; Zhang, M.; Tan, J.; Xu, Y.; Song, R.; et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J. Transl. Med. 2020, 18, 206. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef]
- Rozaliyani, A.; Antariksa, B.; Nurwidya, F.; Zaini, J.; Setianingrum, F.; Hasan, F.; Nugrahapraja, H.; Yusva, H.; Wibowo, H.; Bowolaksono, A.; et al. The Fungal and Bacterial Interface in the Respiratory Mycobiome with a Focus on Aspergillus spp. Life 2023, 13, 1017. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COVID-19 Cases (n = 38) | Non-COVID-19 Cases (n = 26) | p-Value | |
|---|---|---|---|
| Sex [no. (%)] | |||
| Female | 9 (23.7) | 7 (26.9) | 1.000 |
| Male | 21 (76.3) | 19 (73.1) | |
| Age, y | |||
| Mean (Range) | 61.2 (31–95) | 57.0 (24–89) | 0.360 |
| Chronic medical illness [no. (%)] | 29 (76.3) | 20 (76.9) | 1.000 |
| Hypertension | 9 (23.7) | 10 (38.5) | 0.321 |
| Diabetes | 8 (21.1) | 5 (19.2) | 1.000 |
| Hyperlipidemia | 2 (5.3) | 0 (0.0) | 0.648 |
| Coronary artery disease | 12 (31.6) | 2 (7.7) | 0.050 |
| Chronic lung disease | 0 (0.0) | 2 (7.7) | 0.315 |
| Cancer | 3 (7.9) | 4 (15.4) | 0.593 |
| Pulmonary tuberculosis | 5 (13.2) | 2 (7.7) | 0.779 |
| HIV infection | 0 (0.0) | 3 (11.5) | 0.123 |
| HBV infection | 1 (2.6) | 2 (7.7) | 0.735 |
| Treatment [no. (%)] | |||
| Oxygen support | 36 (94.7) | 14 (53.8) | 0.001 |
| Noninvasive ventilation | 25 (65.8) | 7 (26.9) | 0.005 |
| Invasive ventilation | 35 (92.1) | 12 (46.2) | 0.001 |
| ECMO | 13 (34.2) | 1 (3.8) | 0.010 |
| Antibiotic treatment | 38 (100.0) | 25 (100) | 0.847 |
| Antiviral treatment | 26 (68.4) | 3 (11.5) | 0.001 |
| Antifungal treatment | 30 (78.9) | 10 (38.5) | 0.003 |
| Clinical outcome [no. (%)] | |||
| Alive | 26 (68.4) | 20 (76.9) | 0.646 |
| Died | 12 (31.6) | 6 (23.1) | |
| Clinical indicators [mean ± SD] | |||
| WBC, ×109/L | 13.36 ± 8.51 | 10.30 ± 6.99 | 0.134 |
| Neutrophil count, ×109/L | 12.13 ± 8.18 | 8.41 ± 6.73 | 0.060 |
| Lymphocyte count, ×109/L | 0.56 ± 0.33 | 0.98 ± 0.69 | 0.002 |
| Neutrophil/white blood cell ratio (%) | 88.19 ± 9.88 | 76.63 ± 16.37 | 0.001 |
| Neutrophil/lymphocyte ratio (%) | 30.41 ± 28.03 | 18.96 ± 26.06 | 0.104 |
| Hemoglobin level, g/L | 97.31 ± 22.09 | 112.32 ± 24.77 | 0.015 |
| Platelet count, ×109/L | 161.95 ± 104.03 | 183.27 ± 87.72 | 0.395 |
| Alanine aminotransferase, U/L | 58.61 ± 94.12 | 30.71 ± 25.83 | 0.194 |
| Aspartate aminotransferase, U/L | 56.89 ± 73.39 | 43.62 ± 42.94 | 0.464 |
| Lactate dehydrogenase, U/L | 475.00 ± 256.24 | 368.00 ± 320.32 | 0.395 |
| Creatinine, μmol/L | 76.11 ± 38.18 | 92.61 ± 37.55 | 0.127 |
| D-dimer, mg/L | 3698.00 ± 7220.31 | 2534.86 ± 4562.80 | 0.525 |
| PCT, μg/L | 1.47 ± 4.52 | 3.31 ± 6.35 | 0.191 |
| CRP, mg/L | 124.09 ± 148.84 | 54.65 ± 82.24 | 0.161 |
| SAA, mg/L | 134.31 ± 70.31 | 71.65 ± 72.69 | 0.048 |
| IL-6, pg/mL | 435.90 ± 1134.76 | 187.43 ± 303.47 | 0.358 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, L.; Chen, L.; Li, X.; Zhou, J.; Tian, H.; Zhao, J.; Li, Z.; Li, Y. Analysis of Lung Microbiome in COVID-19 Patients during Time of Hospitalization. Pathogens 2023, 12, 944. https://doi.org/10.3390/pathogens12070944
Xie L, Chen L, Li X, Zhou J, Tian H, Zhao J, Li Z, Li Y. Analysis of Lung Microbiome in COVID-19 Patients during Time of Hospitalization. Pathogens. 2023; 12(7):944. https://doi.org/10.3390/pathogens12070944
Chicago/Turabian StyleXie, Linlin, Liangjun Chen, Xinran Li, Junying Zhou, Hongpan Tian, Jin Zhao, Zhiqiang Li, and Yirong Li. 2023. "Analysis of Lung Microbiome in COVID-19 Patients during Time of Hospitalization" Pathogens 12, no. 7: 944. https://doi.org/10.3390/pathogens12070944
APA StyleXie, L., Chen, L., Li, X., Zhou, J., Tian, H., Zhao, J., Li, Z., & Li, Y. (2023). Analysis of Lung Microbiome in COVID-19 Patients during Time of Hospitalization. Pathogens, 12(7), 944. https://doi.org/10.3390/pathogens12070944