
Metavirome Analysis Reveals a High Prevalence of Porcine Hemagglutination Encephalomyelitis Virus in Clinically Healthy Pigs in China



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statements
2.2. Sample Collection
2.3. Sample Preparation and Sequencing
2.4. Viral Genome Discovery
2.5. Phylogenetic Analysis
2.6. Nucleotide Sequence Accession Number
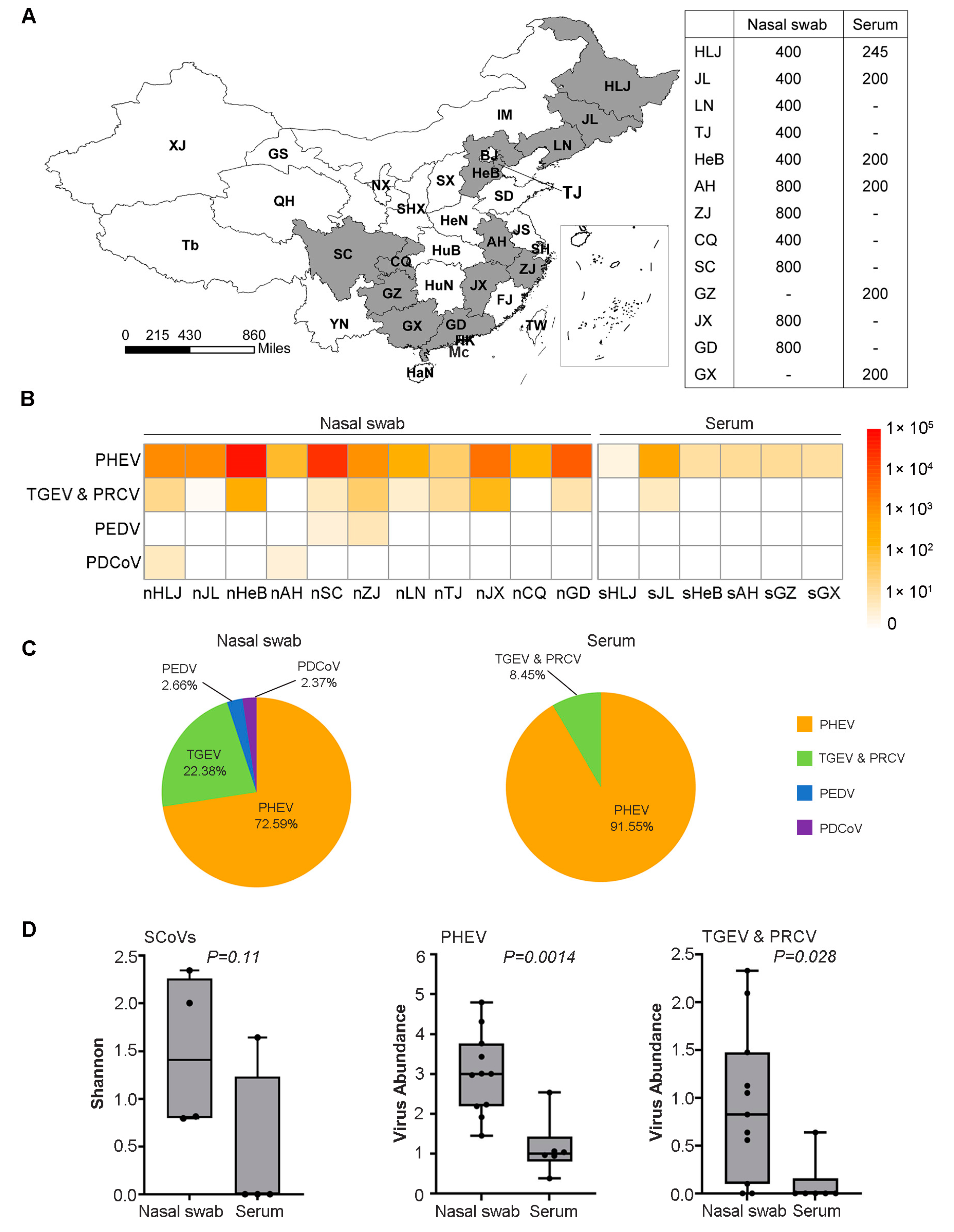
3. Results
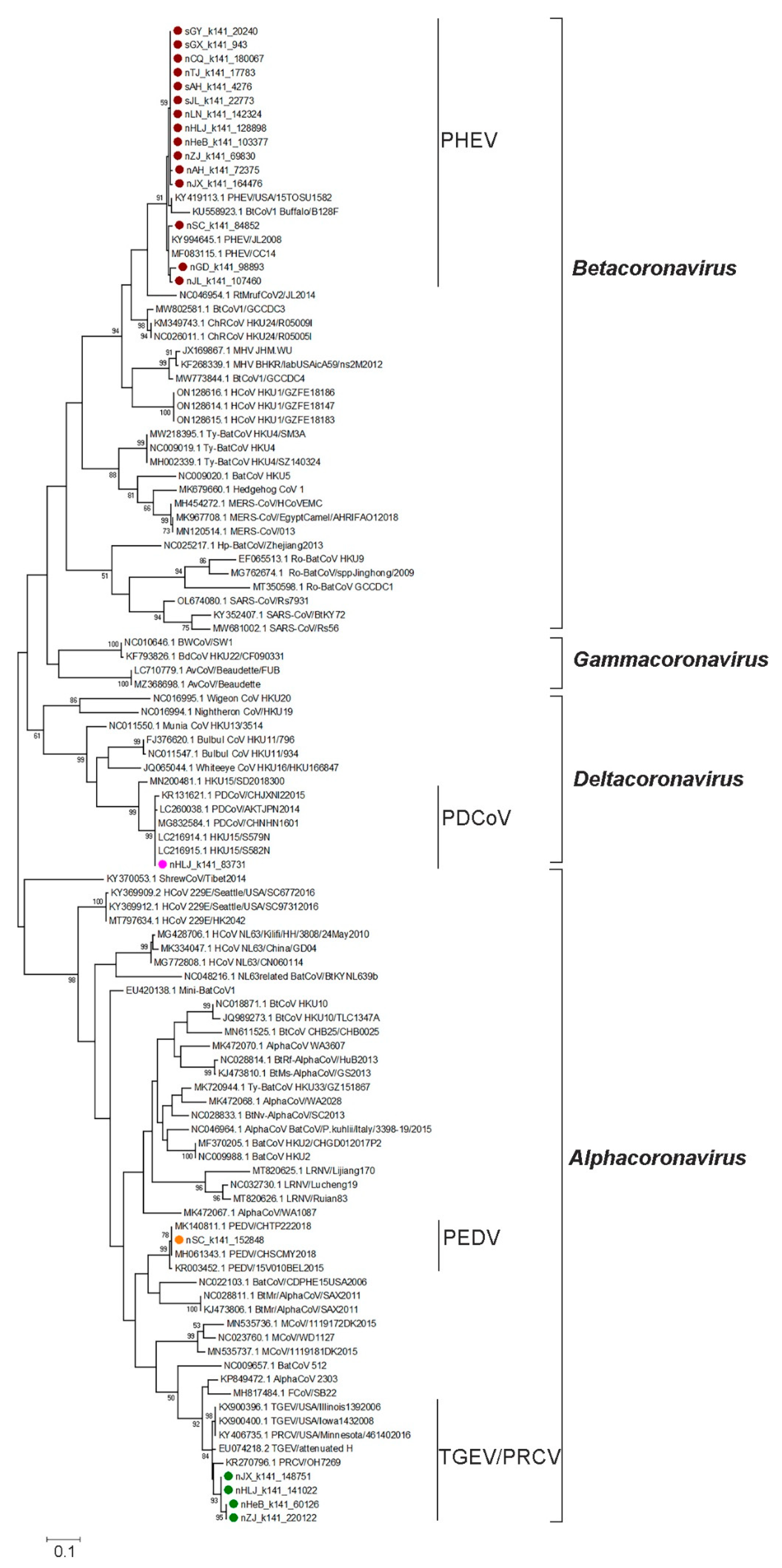
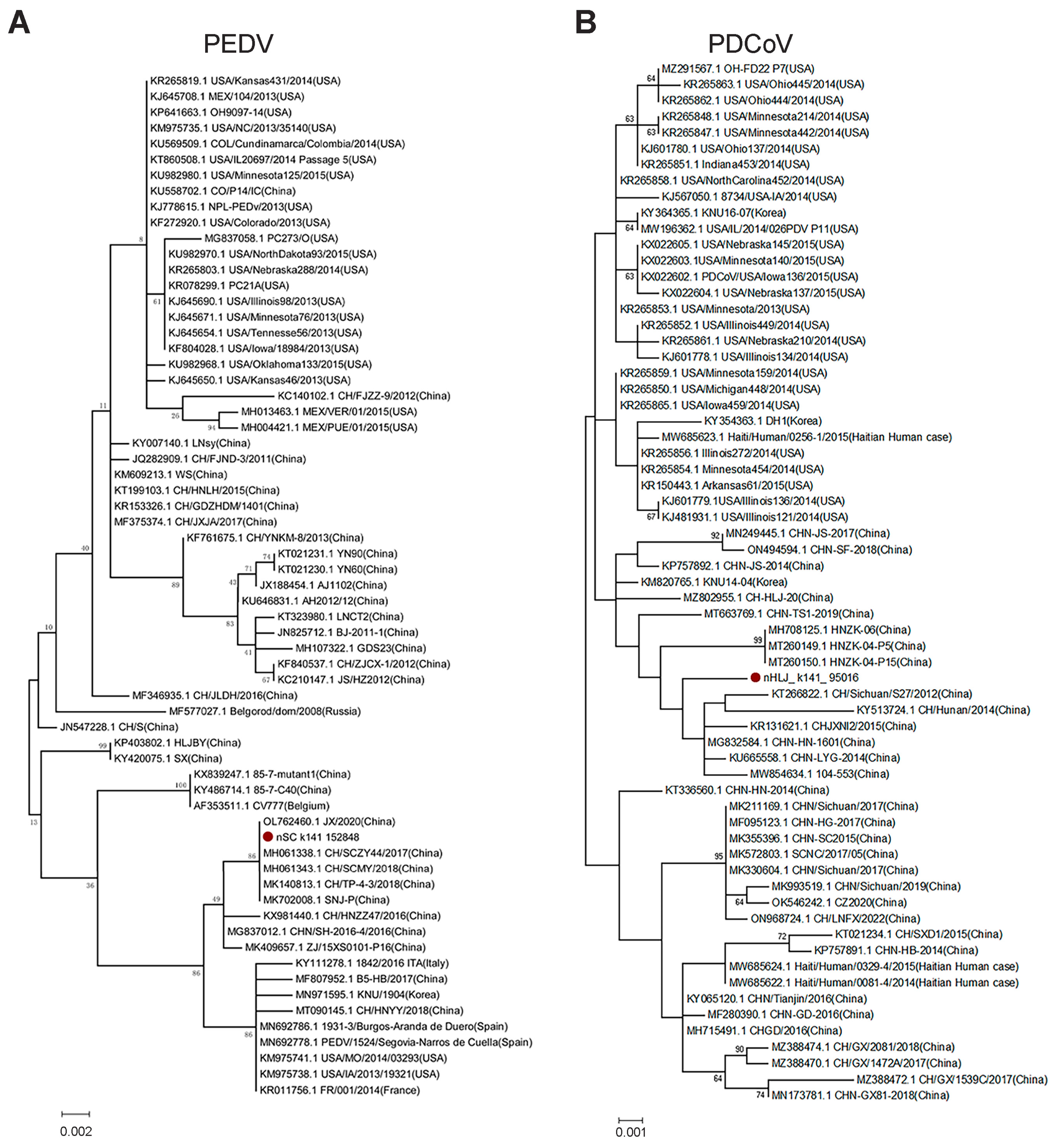
3.1. Diversity and Phylogenetic Evolution of SCoVs
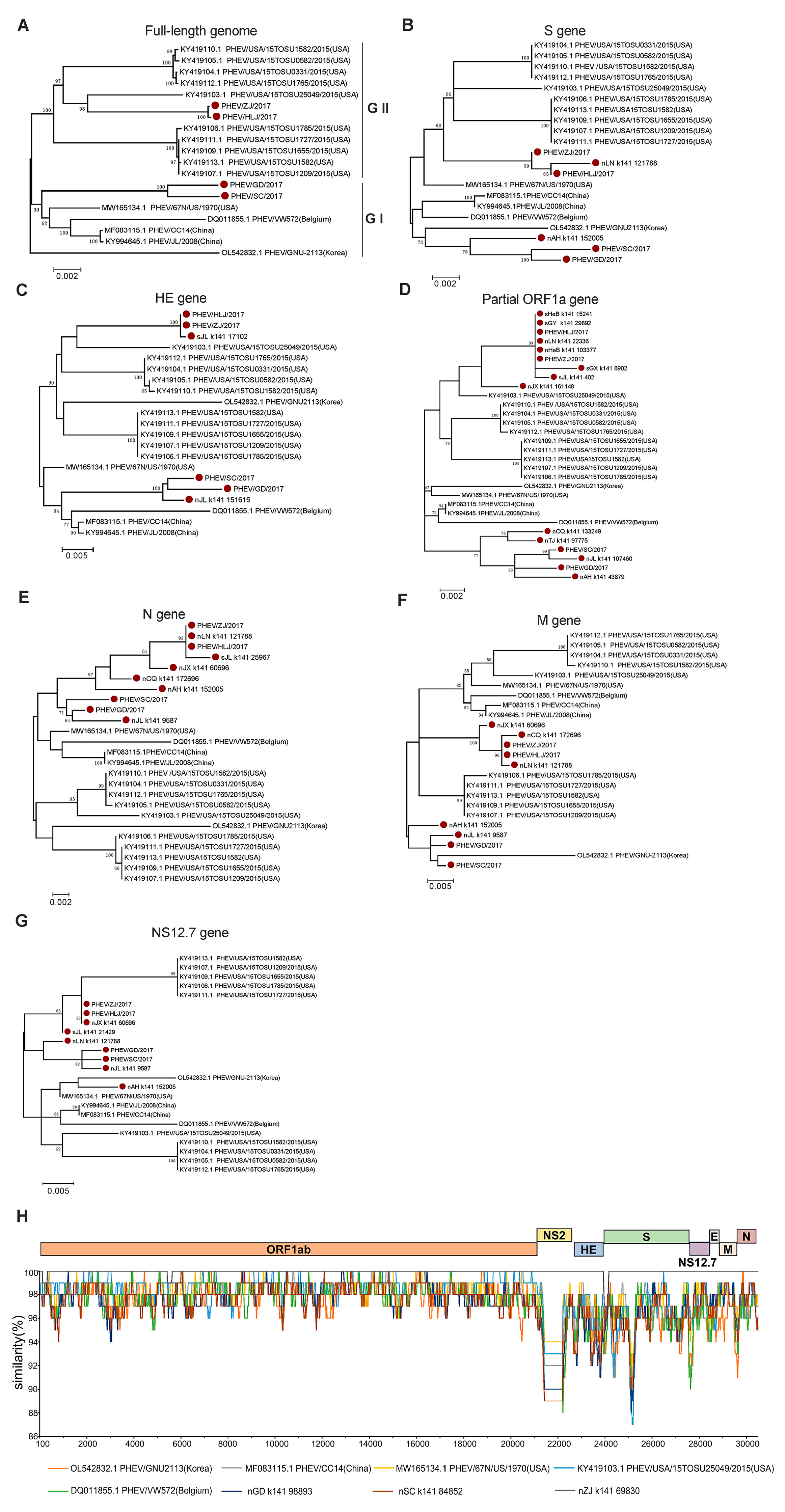
3.2. Genetic Diversity and Evolution of PHEV
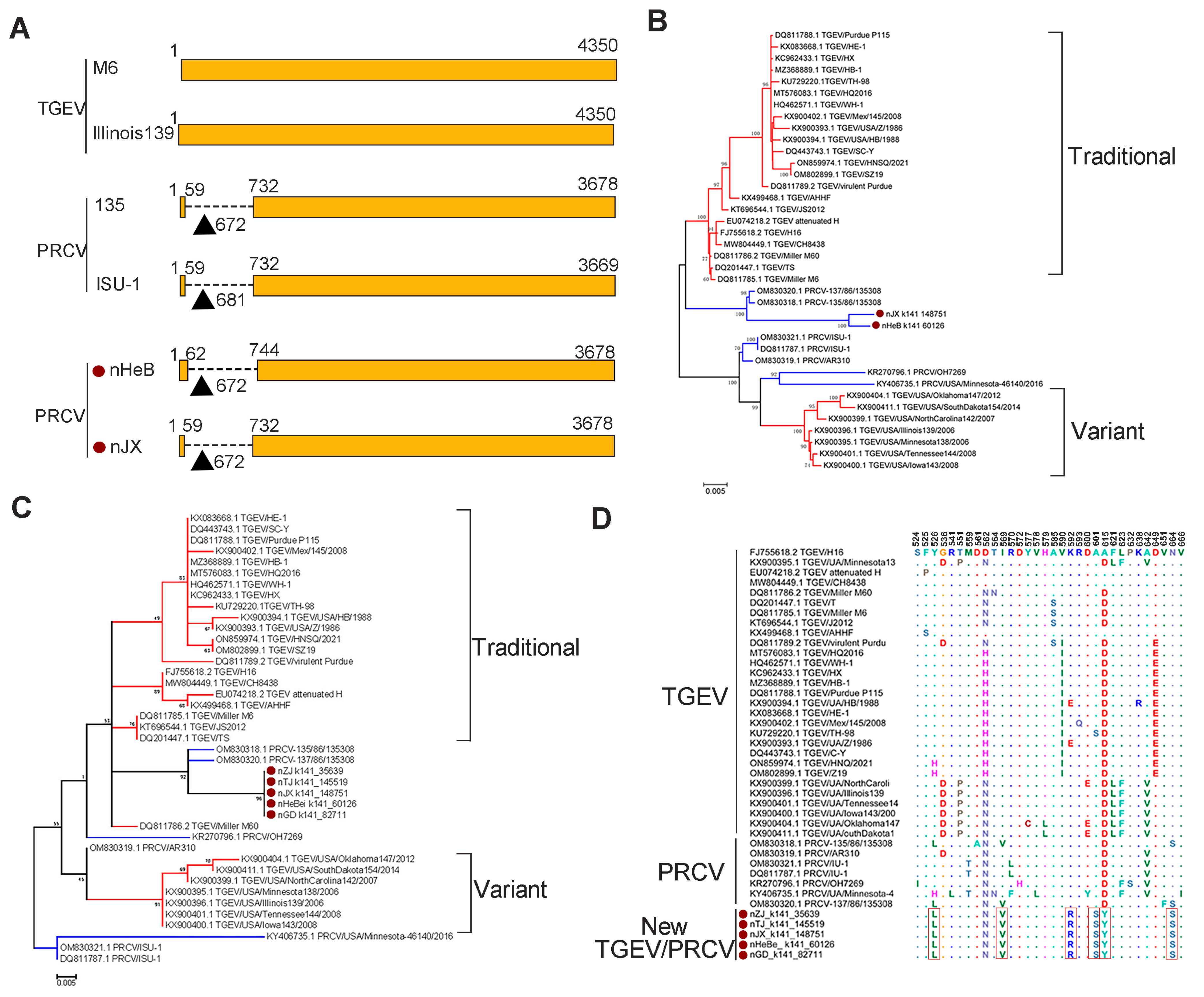
3.3. Genetic Diversity of Other SCoVs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hammerberg, C.; Schurig, G.G.; Ochs, D.L. Immunodeficiency in young pigs. Am. J. Vet. Res. 1989, 50, 868–874. [Google Scholar]
- Woo, P.C.; Lau, S.K.; Yip, C.C.; Huang, Y.; Tsoi, H.W.; Chan, K.H.; Yuen, K.Y. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef]
- Zhong, N.S.; Zheng, B.J.; Li, Y.M.; Poon; Xie, Z.H.; Chan, K.H.; Li, P.H.; Tan, S.Y.; Chang, Q.; Xie, J.P.; et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef]
- Hijawi, B.; Abdallat, M.; Sayaydeh, A.; Alqasrawi, S.; Haddadin, A.; Jaarour, N.; Alsheikh, S.; Alsanouri, T. Novel coronavirus infections in Jordan, April 2012: Epidemiological findings from a retrospective investigation. East Mediterr. Health J. 2013, 19 (Suppl. S1), S12–S18. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Turlewicz-Podbielska, H.; Pomorska-Mol, M. Porcine Coronaviruses: Overview of the State of the Art. Virol. Sin. 2021, 36, 833–851. [Google Scholar] [CrossRef]
- Wang, Q.; Vlasova, A.N.; Kenney, S.P.; Saif, L.J. Emerging and re-emerging coronaviruses in pigs. Curr. Opin. Virol. 2019, 34, 39–49. [Google Scholar] [CrossRef]
- Qian, S.; Jia, X.; Gao, Z.; Zhang, W.; Xu, Q.; Li, Z. Isolation and Identification of Porcine Deltacoronavirus and Alteration of Immunoglobulin Transport Receptors in the Intestinal Mucosa of PDCoV-Infected Piglets. Viruses 2020, 12, 79. [Google Scholar] [CrossRef]
- Jung, K.; Saif, L.J.; Wang, Q. Porcine epidemic diarrhea virus (PEDV): An update on etiology, transmission, pathogenesis, and prevention and control. Virus Res. 2020, 286, 198045. [Google Scholar] [CrossRef]
- Thakor, J.C.; Dinesh, M.; Manikandan, R.; Bindu, S.; Sahoo, M.; Sahoo, D.; Dhawan, M.; Pandey, M.K.; Tiwari, R.; Emran, T.B.; et al. Swine coronaviruses (SCoVs) and their emerging threats to swine population, inter-species transmission, exploring the susceptibility of pigs for SARS-CoV-2 and zoonotic concerns. Vet. Q. 2022, 42, 125–147. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Wang, X.; Zhou, J.; Ma, L.; Li, J.; Yang, L.; Ouyang, H.; Yuan, H.; Pang, D. Transmissible Gastroenteritis Virus: An Update Review and Perspective. Viruses 2023, 15, 359. [Google Scholar] [CrossRef]
- Wood, E.N. An apparently new syndrome of porcine epidemic diarrhoea. Vet. Rec. 1977, 100, 243–244. [Google Scholar] [CrossRef]
- Pensaert, M.B.; de Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef]
- Sun, M.; Ma, J.; Wang, Y.; Wang, M.; Song, W.; Zhang, W.; Lu, C.; Yao, H. Genomic and epidemiological characteristics provide new insights into the phylogeographical and spatiotemporal spread of porcine epidemic diarrhea virus in Asia. J. Clin. Microbiol. 2015, 53, 1484–1492. [Google Scholar] [CrossRef]
- Yu, L.; Liu, Y.; Wang, S.; Zhang, L.; Liang, P.; Wang, L.; Dong, J.; Song, C. Molecular Characteristics and Pathogenicity of Porcine Epidemic Diarrhea Virus Isolated in Some Areas of China in 2015–2018. Front. Vet. Sci. 2020, 7, 607662. [Google Scholar] [CrossRef]
- Li, W.; Li, H.; Liu, Y.; Pan, Y.; Deng, F.; Song, Y.; Tang, X.; He, Q. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg. Infect. Dis. 2012, 18, 1350–1353. [Google Scholar] [CrossRef]
- Zhang, F.; Luo, S.; Gu, J.; Li, Z.; Li, K.; Yuan, W.; Ye, Y.; Li, H.; Ding, Z.; Song, D.; et al. Prevalence and phylogenetic analysis of porcine diarrhea associated viruses in southern China from 2012 to 2018. BMC Vet. Res. 2019, 15, 470. [Google Scholar] [CrossRef]
- Fan, H.; Zhang, J.; Ye, Y.; Tong, T.; Xie, K.; Liao, M. Complete genome sequence of a novel porcine epidemic diarrhea virus in south China. J. Virol. 2012, 86, 10248–10249. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, J.; Deng, X.; Ye, Y.; Liao, M.; Fan, H. Complete genome sequence of a highly prevalent isolate of porcine epidemic diarrhea virus in South China. J. Virol. 2012, 86, 9551. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Marthaler, D.; Wang, Q.; Culhane, M.R.; Rossow, K.D.; Rovira, A.; Collins, J.; Saif, L.J. Distinct characteristics and complex evolution of PEDV strains, North America, May 2013–February 2014. Emerg. Infect. Dis. 2014, 20, 1620–1628. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar]
- Tang, P.; Cui, E.; Song, Y.; Yan, R.; Wang, J. Porcine deltacoronavirus and its prevalence in China: A review of epidemiology, evolution, and vaccine development. Arch. Virol. 2021, 166, 2975–2988. [Google Scholar] [CrossRef]
- Marthaler, D.; Raymond, L.; Jiang, Y.; Collins, J.; Rossow, K.; Rovira, A. Rapid detection, complete genome sequencing, and phylogenetic analysis of porcine deltacoronavirus. Emerg. Infect. Dis. 2014, 20, 1347–1350. [Google Scholar] [CrossRef]
- Wang, L.; Byrum, B.; Zhang, Y. Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerg. Infect. Dis. 2014, 20, 1227–1230. [Google Scholar] [CrossRef]
- Lee, S.; Lee, C. Complete Genome Characterization of Korean Porcine Deltacoronavirus Strain KOR/KNU14-04/2014. Genome Announc. 2014, 2, e01191-14. [Google Scholar] [CrossRef]
- Jung, K.; Hu, H.; Saif, L.J. Porcine deltacoronavirus infection: Etiology, cell culture for virus isolation and propagation, molecular epidemiology and pathogenesis. Virus Res. 2016, 226, 50–59. [Google Scholar] [CrossRef]
- Pan, Y.; Tian, X.; Qin, P.; Wang, B.; Zhao, P.; Yang, Y.L.; Wang, L.; Wang, D.; Song, Y.; Zhang, X.; et al. Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Vet. Microbiol. 2017, 211, 15–21. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Q.N.; Su, J.N.; Chen, G.H.; Wu, Z.X.; Luo, Y.; Wu, R.T.; Sun, Y.; Lan, T.; Ma, J.Y. The re-emerging of SADS-CoV infection in pig herds in Southern China. Transbound. Emerg. Dis. 2019, 66, 2180–2183. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, H.Y. Porcine enteric coronaviruses: An updated overview of the pathogenesis, prevalence, and diagnosis. Vet. Res. Commun. 2021, 45, 75–86. [Google Scholar] [CrossRef]
- Chen, F.; Knutson, T.P.; Rossow, S.; Saif, L.J.; Marthaler, D.G. Decline of transmissible gastroenteritis virus and its complex evolutionary relationship with porcine respiratory coronavirus in the United States. Sci. Rep. 2019, 9, 3953. [Google Scholar] [CrossRef]
- Magtoto, R.; Poonsuk, K.; Baum, D.; Zhang, J.; Chen, Q.; Ji, J.; Pineyro, P.; Zimmerman, J.; Gimenez-Lirola, L.G. Evaluation of the Serologic Cross-Reactivity between Transmissible Gastroenteritis Coronavirus and Porcine Respiratory Coronavirus Using Commercial Blocking Enzyme-Linked Immunosorbent Assay Kits. mSphere 2019, 4, e00017-19. [Google Scholar] [CrossRef]
- Mora-Díaz, J.C.; Piñeyro, P.E.; Houston, E.; Zimmerman, J.; Giménez-Lirola, L.G. Porcine Hemagglutinating Encephalomyelitis Virus: A Review. Front. Vet. Sci. 2019, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Krempl, C.; Schultze, B.; Laude, H.; Herrler, G. Point mutations in the S protein connect the sialic acid binding activity with the enteropathogenicity of transmissible gastroenteritis coronavirus. J. Virol. 1997, 71, 3285–3287. [Google Scholar] [CrossRef]
- Kim, L.; Hayes, J.; Lewis, P.; Parwani, A.V.; Chang, K.O.; Saif, L.J. Molecular characterization and pathogenesis of transmissible gastroenteritis coronavirus (TGEV) and porcine respiratory coronavirus (PRCV) field isolates co-circulating in a swine herd. Arch. Virol. 2000, 145, 1133–1147. [Google Scholar] [CrossRef]
- Mengeling, W.L.; Cutlip, R.C. Pathogenicity of field isolants of hemagglutinating encephalomyelitis virus for neonatal pigs. J. Am. Vet. Med. Assoc. 1976, 168, 236–239. [Google Scholar]
- Quiroga, M.A.; Cappuccio, J.; Pineyro, P.; Basso, W.; More, G.; Kienast, M.; Schonfeld, S.; Cancer, J.L.; Arauz, S.; Pintos, M.E.; et al. Hemagglutinating encephalomyelitis coronavirus infection in pigs, Argentina. Emerg. Infect. Dis. 2008, 14, 484–486. [Google Scholar] [CrossRef]
- Hirano, N.; Ono, K. A serological survey of human coronavirus in pigs of the Tohoku District of Japan. Adv. Exp. Med. Biol. 1998, 440, 491–494. [Google Scholar]
- Kwok, K.T.T.; Nieuwenhuijse, D.F.; Phan, M.V.T.; Koopmans, M.P.G. Virus Metagenomics in Farm Animals: A Systematic Review. Viruses 2020, 12, 107. [Google Scholar] [CrossRef]
- Davila-Ramos, S.; Castelan-Sanchez, H.G.; Martinez-Avila, L.; Sanchez-Carbente, M.D.R.; Peralta, R.; Hernandez-Mendoza, A.; Dobson, A.D.W.; Gonzalez, R.A.; Pastor, N.; Batista-Garcia, R.A. A Review on Viral Metagenomics in Extreme Environments. Front. Microbiol. 2019, 10, 2403. [Google Scholar] [CrossRef]
- Mohsin, H.; Asif, A.; Fatima, M.; Rehman, Y. Potential role of viral metagenomics as a surveillance tool for the early detection of emerging novel pathogens. Arch. Microbiol. 2021, 203, 865–872. [Google Scholar] [CrossRef]
- Guo, J.; Fang, L.; Ye, X.; Chen, J.; Xu, S.; Zhu, X.; Miao, Y.; Wang, D.; Xiao, S. Evolutionary and genotypic analyses of global porcine epidemic diarrhea virus strains. Transbound. Emerg. Dis. 2019, 66, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.S.; Kim, Y.; Lee, E.S.; Lim, J.S.; Hong, S.K.; Lee, I.S.; Jung, C.S.; Yoon, H.C.; Wee, S.H.; Pfeiffer, D.U.; et al. Transmission Dynamics of African Swine Fever Virus, South Korea, 2019. Emerg. Infect. Dis. 2021, 27, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vizcaino, J.M.; Mur, L.; Martinez-Lopez, B. African swine fever: An epidemiological update. Transbound. Emerg. Dis. 2012, 59 (Suppl. S1), 27–35. [Google Scholar] [CrossRef]
- Shi, Z.; Liu, C.; Yang, H.; Chen, Y.; Liu, H.; Wei, L.; Liu, Z.; Jiang, Y.; He, X.; Wang, J. Fur Seal Feces-Associated Circular DNA Virus Identified in Pigs in Anhui, China. Virol. Sin. 2021, 36, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Li, L.; Simmonds, P.; Wang, C.; Moeser, A.; Delwart, E. The fecal virome of pigs on a high-density farm. J. Virol. 2011, 85, 11697–11708. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic. Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, L.; Zheng, Y.; Zhang, J.; Guo, B.; Yoon, K.J.; Gauger, P.C.; Harmon, K.M.; Main, R.G.; Li, G. Metagenomic analysis of the RNA fraction of the fecal virome indicates high diversity in pigs infected by porcine endemic diarrhea virus in the United States. Virol. J. 2018, 15, 95. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Zhu, L.; Ma, J.Y.; Zhou, Q.F.; Song, Y.H.; Sun, B.L.; Chen, R.A.; Xie, Q.M.; Bee, Y.Z. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field strains in south China. Virus Genes 2012, 45, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.Q.; Ge, F.F.; Ju, H.B.; Wang, J.; Liu, J.; Ning, K.; Liu, P.H.; Zhou, J.P.; Sun, Q.Y. Whole-genome analysis of porcine epidemic diarrhea virus (PEDV) from eastern China. Arch. Virol. 2014, 159, 2777–2785. [Google Scholar] [CrossRef]
- Song, D.; Zhou, X.; Peng, Q.; Chen, Y.; Zhang, F.; Huang, T.; Zhang, T.; Li, A.; Huang, D.; Wu, Q.; et al. Newly Emerged Porcine Deltacoronavirus Associated With Diarrhoea in Swine in China: Identification, Prevalence and Full-Length Genome Sequence Analysis. Transbound. Emerg. Dis. 2015, 62, 575–580. [Google Scholar] [CrossRef]
- Zuniga, S.; Pascual-Iglesias, A.; Sanchez, C.M.; Sola, I.; Enjuanes, L. Virulence factors in porcine coronaviruses and vaccine design. Virus Res. 2016, 226, 142–151. [Google Scholar] [CrossRef]
- Mora-Diaz, J.C.; Magtoto, R.; Houston, E.; Baum, D.; Carrillo-Avila, J.A.; Temeeyasen, G.; Zimmerman, J.; Pineyro, P.; Gimenez-Lirola, L. Detecting and Monitoring Porcine Hemagglutinating Encephalomyelitis Virus, an Underresearched Betacoronavirus. mSphere 2020, 5, e00199-20. [Google Scholar] [CrossRef]
- Gao, W.; Zhao, K.; Zhao, C.; Du, C.; Ren, W.; Song, D.; Lu, H.; Chen, K.; Li, Z.; Lan, Y.; et al. Vomiting and wasting disease associated with hemagglutinating encephalomyelitis viruses infection in piglets in Jilin, China. Virol. J. 2011, 8, 130. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, K.; Lu, H.; Li, Z.; Lv, X.; Lan, Y.; Guan, J.; He, W.; Gao, F. Genomic characterization and pathogenicity of a porcine hemagglutinating encephalomyelitis virus strain isolated in China. Virus Genes 2018, 54, 672–683. [Google Scholar] [CrossRef]
- Bahoussi, A.N.; Guo, Y.Y.; Shi, R.Z.; Wang, P.H.; Li, Y.Q.; Wu, C.X.; Xing, L. Genetic Characteristics of Porcine Hemagglutinating Encephalomyelitis Coronavirus: Identification of Naturally Occurring Mutations Between 1970 and 2015. Front. Microbiol. 2022, 13, 860851. [Google Scholar] [CrossRef]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef]
- Sabir, J.S.; Lam, T.T.; Ahmed, M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef]
- Tian, P.F.; Jin, Y.L.; Xing, G.; Qv, L.L.; Huang, Y.W.; Zhou, J.Y. Evidence of recombinant strains of porcine epidemic diarrhea virus, United States, 2013. Emerg. Infect. Dis. 2014, 20, 1735–1738. [Google Scholar] [CrossRef]
- Wong, A.C.P.; Lau, S.K.P.; Woo, P.C.Y. Interspecies Jumping of Bat Coronaviruses. Viruses 2021, 13, 2188. [Google Scholar] [CrossRef]
- Hess, R.G.; Bollwahn, W.; Pospischil, A.; Heinritzi, K.; Bachmann, P.A. Current aspects in the etiology of viral diarrheas of swine: Occurrence of infections with the epizootic viral diarrhea (EVD) virus. Berl. Munch. Tierarztl. Wochenschr. 1980, 93, 445–449. [Google Scholar]
- Weiwei, H.; Qinghua, Y.; Liqi, Z.; Haofei, L.; Shanshan, Z.; Qi, G.; Kongwang, H.; Qian, Y. Complete genomic sequence of the coronavirus transmissible gastroenteritis virus SHXB isolated in China. Arch. Virol. 2014, 159, 2295–2302. [Google Scholar] [CrossRef]
- Sanchez, C.M.; Gebauer, F.; Sune, C.; Mendez, A.; Dopazo, J.; Enjuanes, L. Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology 1992, 190, 92–105. [Google Scholar] [CrossRef]
- Li, C.; Lu, H.; Geng, C.; Yang, K.; Liu, W.; Liu, Z.; Yuan, F.; Gao, T.; Wang, S.; Wen, P.; et al. Epidemic and Evolutionary Characteristics of Swine Enteric Viruses in South-Central China from 2018 to 2021. Viruses 2022, 14, 1420. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y. Genomic Characterization of a New PRCV Variant, United States, 2014. Transbound. Emerg. Dis. 2017, 64, 672–674. [Google Scholar] [CrossRef]
- Delmas, B.; Gelfi, J.; L’Haridon, R.; Vogel, L.K.; Sjostrom, H.; Noren, O.; Laude, H. Aminopeptidase N is a major receptor for the entero-pathogenic coronavirus TGEV. Nature 1992, 357, 417–420. [Google Scholar] [CrossRef]
- Yeager, C.L.; Ashmun, R.A.; Williams, R.K.; Cardellichio, C.B.; Shapiro, L.H.; Look, A.T.; Holmes, K.V. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992, 357, 420–422. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Q.; Huang, L.; Yuan, C.; Wang, J.; Yang, Q. An alternative pathway of enteric PEDV dissemination from nasal cavity to intestinal mucosa in swine. Nat. Commun. 2018, 9, 3811. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Liu, P.; Wang, X.; Ma, Y.; Zhong, Q.; Yang, Q. Porcine Epidemic Diarrhea Virus Infection Disrupts the Nasal Endothelial Barrier To Favor Viral Dissemination. J. Virol. 2022, 96, e0038022. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, W.; Shi, Z.; Wang, P.; Zhao, B.; Li, J.; Wei, X.; Wei, L.; Wang, J. Metavirome Analysis Reveals a High Prevalence of Porcine Hemagglutination Encephalomyelitis Virus in Clinically Healthy Pigs in China. Pathogens 2023, 12, 510. https://doi.org/10.3390/pathogens12040510
Sun W, Shi Z, Wang P, Zhao B, Li J, Wei X, Wei L, Wang J. Metavirome Analysis Reveals a High Prevalence of Porcine Hemagglutination Encephalomyelitis Virus in Clinically Healthy Pigs in China. Pathogens. 2023; 12(4):510. https://doi.org/10.3390/pathogens12040510
Chicago/Turabian StyleSun, Weiyao, Zhibin Shi, Pengfei Wang, Bingbing Zhao, Jiaqi Li, Xinyu Wei, Lili Wei, and Jingfei Wang. 2023. "Metavirome Analysis Reveals a High Prevalence of Porcine Hemagglutination Encephalomyelitis Virus in Clinically Healthy Pigs in China" Pathogens 12, no. 4: 510. https://doi.org/10.3390/pathogens12040510
APA StyleSun, W., Shi, Z., Wang, P., Zhao, B., Li, J., Wei, X., Wei, L., & Wang, J. (2023). Metavirome Analysis Reveals a High Prevalence of Porcine Hemagglutination Encephalomyelitis Virus in Clinically Healthy Pigs in China. Pathogens, 12(4), 510. https://doi.org/10.3390/pathogens12040510