
Survey of Genotype Diversity, Virulence, and Antimicrobial Resistance Genes in Mastitis-Causing Streptococcus uberis in Dairy Herds Using Whole-Genome Sequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolates
2.2. Whole-Genome Sequencing
2.2.1. DNA Isolation, Sequencing, and Data Processing
2.2.2. Screening for Virulence Factors
2.2.3. Screening for Antimicrobial Resistance Genes
2.2.4. Multilocus Sequence Typing
2.2.5. Principal Component Analysis and Heatmap
3. Results
3.1. MLST Characterization
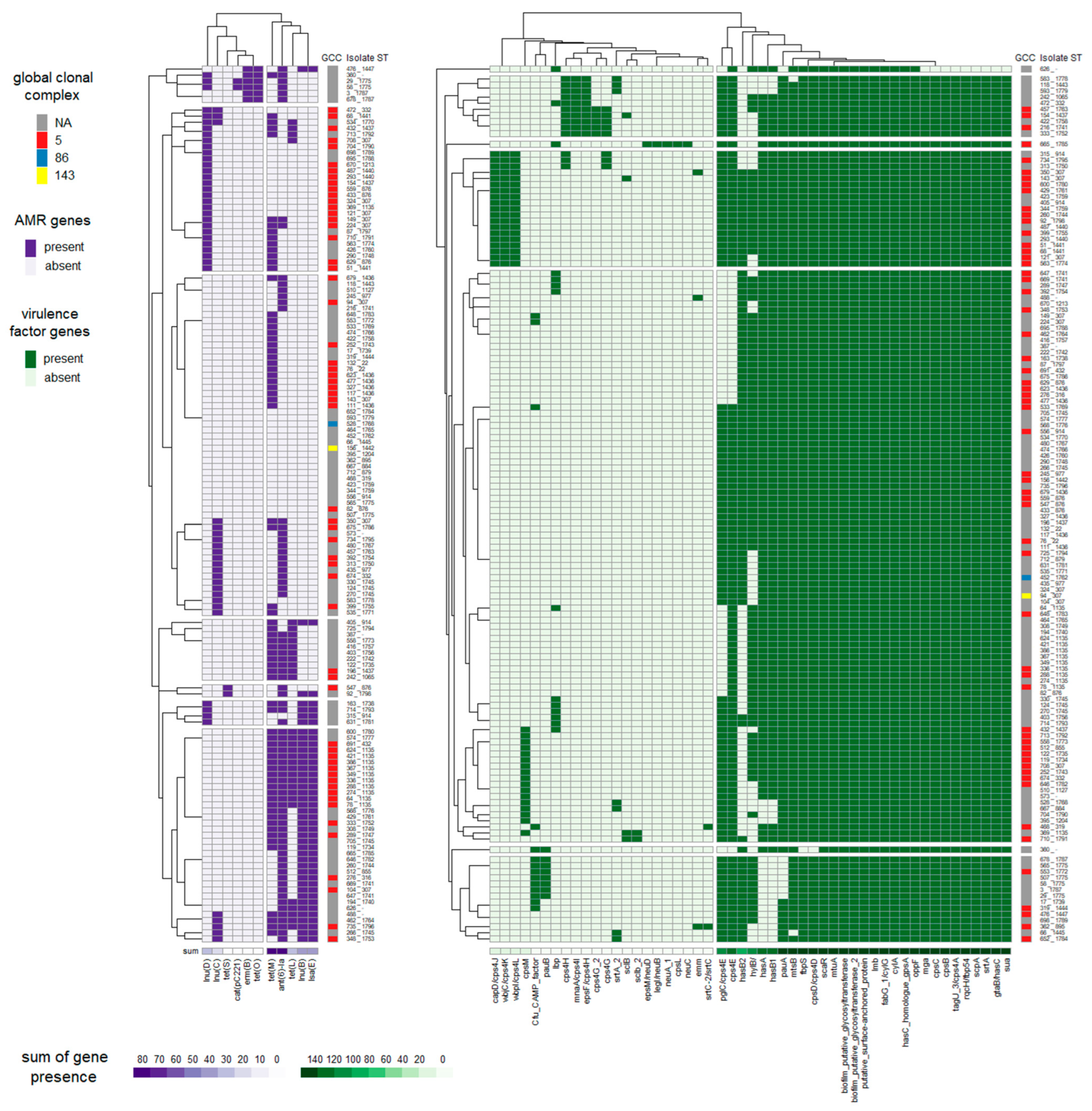
3.2. Identification of Virulence Factors
3.3. Occurrence of Antimicrobial Resistance Genes
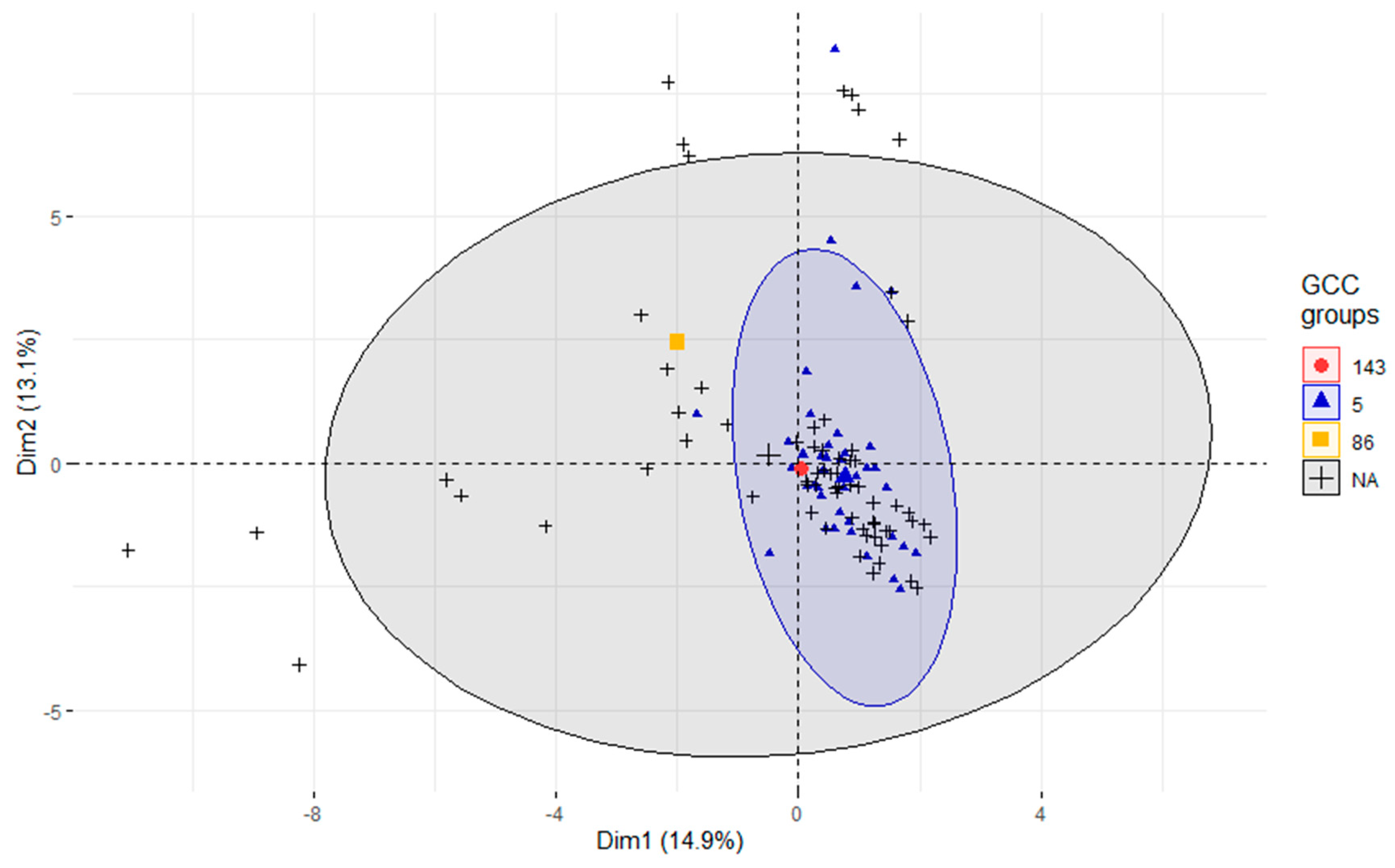
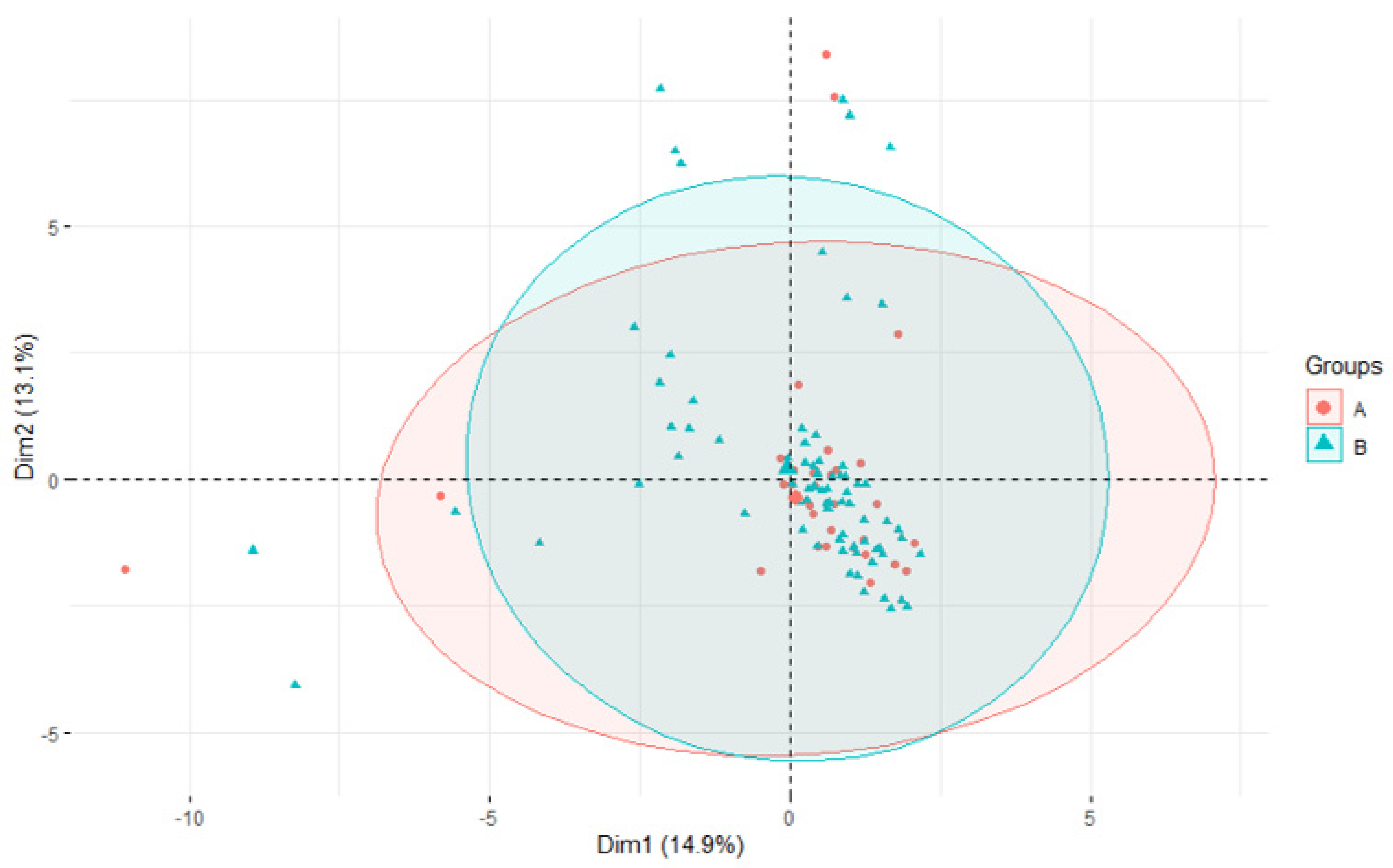
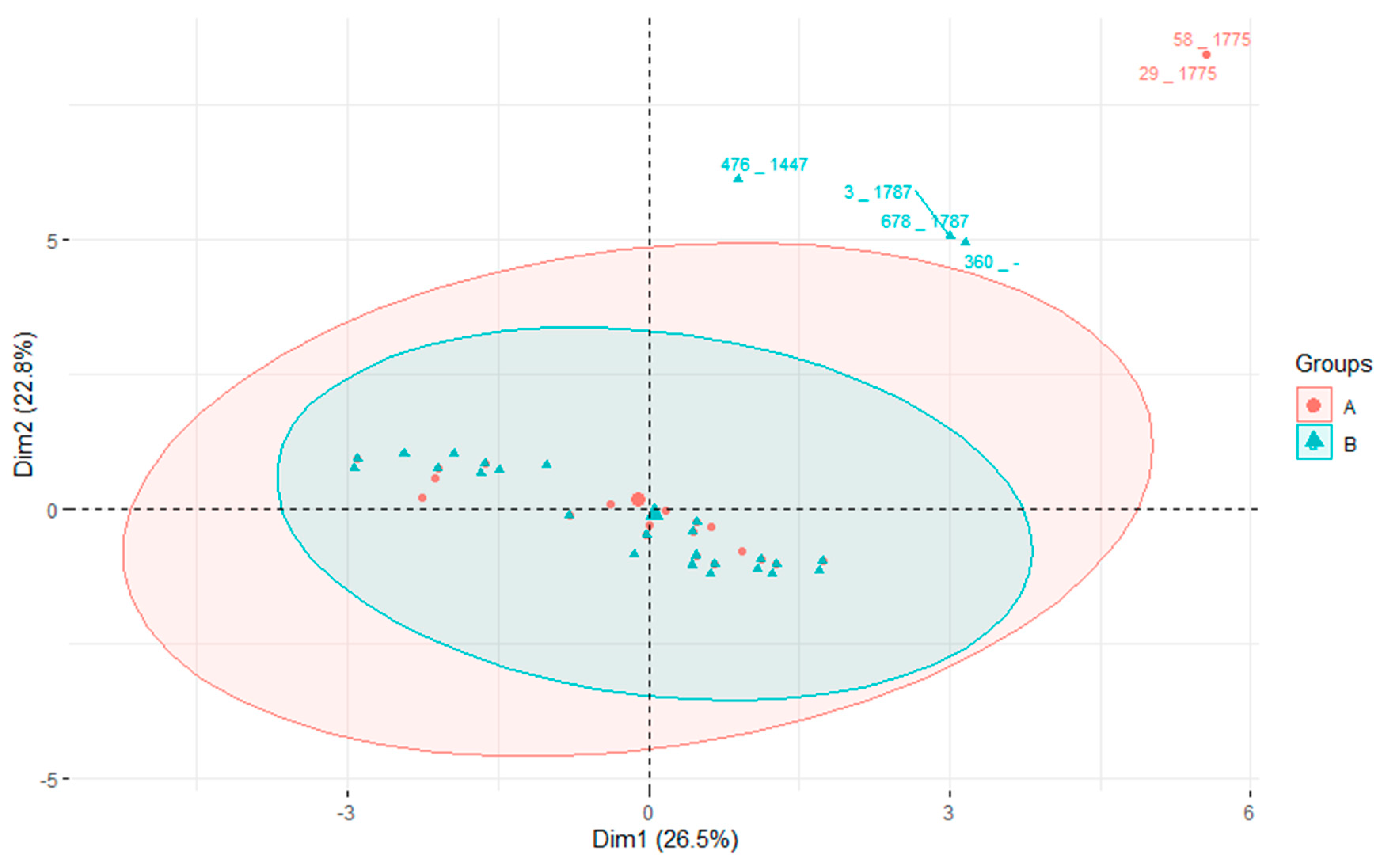
3.4. Association of STs with Virulence and AMR Genes
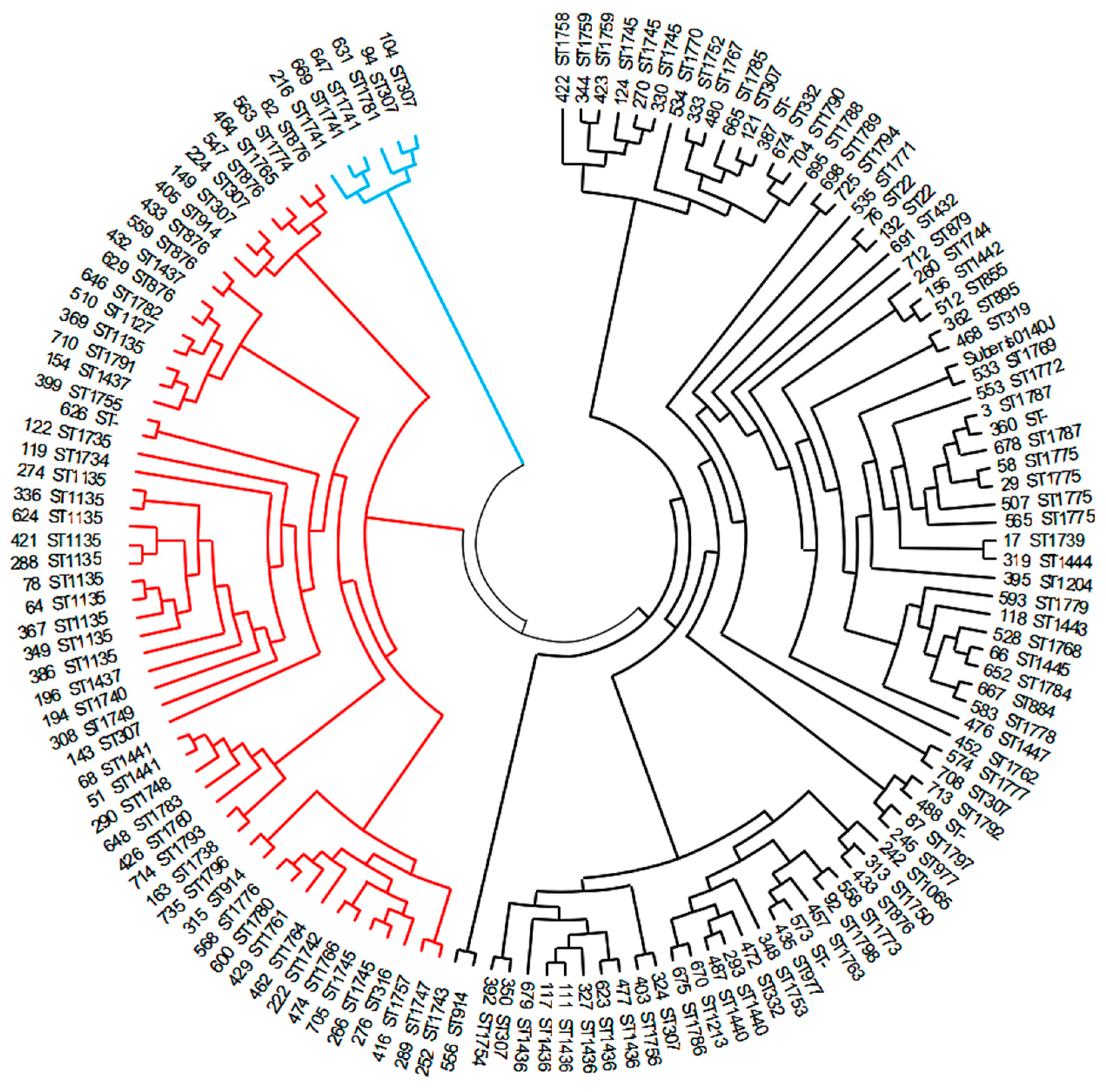
3.5. Phylogeny
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tomita, T.; Meehan, B.; Wongkattiya, N.; Malmo, J.; Pullinger, G.; Leigh, J.; Deighton, M. Identification of Streptococcus uberis multilocus sequence types highly associated with mastitis. Appl. Environ. Microbiol. 2008, 74, 114–124. [Google Scholar] [CrossRef]
- Käppeli, N.; Morach, M.; Zurfluh, K.; Corti, S.; Nüesch-Inderbinen, M.; Stephan, R. Sequence types and antimicrobial resistance profiles of Streptococcus uberis isolated from bovine mastitis. Front. Vet. Sci. 2019, 6, 234. [Google Scholar] [CrossRef]
- Silva, N.C.C.; Yang, Y.; Rodrigues, M.X.; Tomazi, T.; Bicalho, R.C. Whole-genome sequencing reveals high genetic diversity of Streptococcus uberis isolated from cows with mastitis. BMC Vet. Res. 2021, 17, 321. [Google Scholar] [CrossRef]
- Vezina, B.; Al-harbi, H.; Ramay, H.R.; Soust, M.; Moore, R.J.; Olchowy, T.W.J.; Alawneh, J.I. Sequence characterisation and novel insights into bovine mastitis-associated Streptococcus uberis in dairy herds. Sci. Rep. 2021, 11, 3046. [Google Scholar] [CrossRef]
- Zhang, T.; Niu, G.; Boonyayatra, S.; Pichpol, D. Antimicrobial Resistance Profiles and Genes in Streptococcus uberis Associated with Bovine Mastitis in Thailand. Front. Vet. Sci. 2021, 8, 705338. [Google Scholar] [CrossRef]
- Halasa, T.; Huijps, K.; Østerås, O.; Hogeveen, H. Economic effects of bovine mastitis and mastitis management: A review. Vet. Q. 2007, 29, 18–31. [Google Scholar] [CrossRef]
- Saini, V.; McClure, J.T.; Léger, D.; Dufour, S.; Sheldon, A.G.; Scholl, D.T.; Barkema, H.W. Antimicrobial use on Canadian dairy farms. J. Dairy Sci. 2012, 95, 1209–1221. [Google Scholar] [CrossRef]
- Martins, L.; Gonçalves, J.L.; Leite, R.F.; Tomazi, T.; Rall, V.L.M.; Santos, M.V. Association between antimicrobial use and antimicrobial resistance of Streptococcus uberis causing clinical mastitis. J. Dairy Sci. 2021, 104, 12030–12041. [Google Scholar] [CrossRef]
- Wente, N.; Klocke, D.; Paduch, J.H.; Zhang, Y.; Seeth, M.T.; Zoche-Golob, V.; Reinecke, F.; Mohr, E.; Krömker, V. Associations between Streptococcus uberis strains from the animal environment and clinical bovine mastitis cases. J. Dairy Sci. 2019, 102, 9360–9369. [Google Scholar] [CrossRef]
- Pullinger, G.D.; Coffey, T.J.; Maiden, M.C.; Leigh, J.A. Multilocus-sequence typing analysis reveals similar populations of Streptococcus uberis are responsible for bovine intramammary infections of short and long duration. Vet. Microbiol. 2007, 31, 194–204. [Google Scholar] [CrossRef]
- Srithanasuwan, A.; Pangprasit, N.; Suriyasathaporn, W. Comparison of Virulence Patterns Between Streptococcus uberis Causing Transient and Persistent Intramammary Infection. Front. Vet. Sci. 2022, 9, 806674. [Google Scholar] [CrossRef] [PubMed]
- Leelahapongsathon, K.; Schukken, Y.; Srithanasuwan, A.; Suriyasathaporn, W. Molecular epidemiology of Streptococcus uberis intramamary infections: Persistent and transient patterns of infection in a dairy herd. J. Dairy Sci. 2020, 103, 3565–3576. [Google Scholar] [CrossRef] [PubMed]
- Pullinger, G.D.; López-Benavides, M.; Coffey, T.J.; Williamson, J.H.; Cursons, R.T.; Summers, E.; Lacy-Hulbert, J.; Maiden, M.C.; Leigh, J.A. Application of Streptococcus uberis multilocus sequence typing: Analysis of the population structure detected among environmental and bovine isolates from New Zealand and the United Kingdom. Appl. Environ. Microbiol. 2006, 72, 1429–1436. [Google Scholar] [CrossRef]
- Phuektes, P.; Mansell, P.D.; Dyson, R.S.; Hooper, N.D.; Dick, J.S.; Browning, G.F. Molecular epidemiology of Streptococcus uberis isolates from dairy cows with mastitis. J. Clin. Microbiol. 2001, 39, 1460–1466. [Google Scholar] [CrossRef]
- Zouharova, M.; Nedbalcova, K.; Kralova, N.; Slama, P.; Matiaskova, K.; Matiasovic, J. Multilocus Sequence Genotype Heterogeneity in Streptococcus uberis Isolated from Bovine Mastitis in the Czech Republic. Animals 2022, 12, 2327. [Google Scholar] [CrossRef]
- Coffey, T.J.; Pullinger, G.D.; Urwin, R.; Jolley, K.A.; Wilson, S.M.; Maiden, M.C.; Leigh, J.A. First Insights into the Evolution of Streptococcus uberis: A Multilocus Sequence Typing Scheme That Enables Investigation of Its Population Biology. Appl. Environ. Microbiol. 2006, 72, 1420–1428. [Google Scholar] [CrossRef]
- Ward, P.N.; Holden, M.T.; Leigh, J.A.; Lennard, N.; Bignell, A.; Barron, A.; Clark, L.; Quail, M.A.; Woodward, J.; Barrell, B.G.; et al. Evidence for niche adaptation in the genome of the bovine pathogen Streptococcus uberis. BMC Genom. 2009, 10, 54. [Google Scholar] [CrossRef]
- Hassan, A.A.; Khan, I.U.; Abdulmawjood, A.; Lämmler, C. Evaluation of PCR methods for rapid identification and differentiation of Streptococcus uberis and Streptococcus parauberis. J. Clin. Microbiol. 2001, 39, 1618–1621. [Google Scholar] [CrossRef]
- Quijada, N.M.; Rodríguez-L’azaro, D.; Eiros, J.M.; Hern´andez, M. TORMES: An automated pipeline for whole bacterial genome analysis. Bioinformatics 2019, 35, 4207–4212. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. ABRicate Github. Available online: https://github.com/tseemann/abricate (accessed on 8 June 2021).
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, M.; Sousa, A.; Ramirez, M.; Francisco, A.P.; Carriço, J.A.; Vaz, C. PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 2017, 33, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Douglas, V.L.; Fenwick, S.G.; Pfeiffer, D.U.; Williamson, N.B.; Holmes, C.W. Genomic typing of Streptococcus uberis isolates from cases of mastitis, in New Zealand dairy cows, using pulsed-field gel electrophoresis. Vet. Microbiol. 2000, 75, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Wieliczko, R.J.; Williamson, J.H.; Cursons, R.T.; Lacy-Hulbert, S.J.; Woolford, M.W. Molecular typing of Streptococcus uberis strains isolated from cases of bovine mastitis. J. Dairy Sci. 2002, 85, 2149–2154. [Google Scholar] [CrossRef]
- Rahman, A.; Bhattacharjee, A.; Tabassum, T.; Islam, M.A.; Hossain, M. Prevalence and population biology of mastitis-causing Streptococcus uberis using an MLST based approach. J. Adv. Biotechnol. Exp. 2021, 4, 311–321. [Google Scholar] [CrossRef]
- Anonymous. Šlechtění Holštýnského Skotu; Svaz Chovatelů Holštýnského Skotu ČR: Praha, Czech Republic, 2005. (In Czech) [Google Scholar]
- Anonymous. Ročenka 2022–1 Část; Svaz Chovatelů Holštýnského Skotu ČR: Praha, Czech Republic, 2023. (In Czech) [Google Scholar]
- Davies, P.L.; Leigh, J.A.; Bradley, A.J.; Archer, S.C.; Emes, R.D.; Green, M.J. Molecular epidemiology of Streptococcus uberis clinical mastitis in dairy herds: Strain heterogeneity and transmission. J. Clin. Microbiol. 2016, 54, 68–74. [Google Scholar] [CrossRef]
- Wang, L.; Chen, W.; Zhang, L.; Zhu, Y. Genetic diversity of Streptococcus uberis isolates from dairy cows with subclinical mastitis in Southern Xinjiang Province, China. J. Gen. Appl. Microbiol. 2013, 9, 287–293. [Google Scholar] [CrossRef]
- Field, T.R.; Ward, P.N.; Pedersen, L.H.; Leigh, J.A. The hyaluronic acid capsule of Streptococcus uberis is not required for the development of infection and clinical mastitis. Infect. Immun. 2003, 71, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Vélez, J.R.; Cameron, M.; Rodríguez-Lecompte, J.C.; Xia, F.; Heider, L.C.; Saab, M.; McClure, J.T.; Sánchez, J. Whole-Genome Sequence Analysis of Antimicrobial Resistance Genes in Streptococcus uberis and Streptococcus dysgalactiae Isolates from Canadian Dairy Herds. Front. Vet. Sci. 2017, 4, 63. [Google Scholar] [CrossRef] [PubMed]
- Stasiak, M.; Maćkiw, E.; Kowalska, J.; Kucharek, K.; Postupolski, J. Silent Genes: Antimicrobial Resistance and Antibiotic Production. Pol. J. Microbiol. 2021, 70, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Vezina, B.; Rosa, M.N.; Canu, A.; Tola, S. Genomic surveillance reveals antibiotic resistance gene transmission via phage recombinases within sheep mastitis-associated Streptococcus uberis. BMC Vet. Res. 2022, 18, 264. [Google Scholar] [CrossRef]
- Tram, G.; Jennings, M.P.; Blackall, P.J.; Atack, J.M. Streptococcus suis pathogenesis-A diverse array of virulence factors for a zoonotic lifestyle. Adv. Microb. Physiol. 2021, 78, 217–257. [Google Scholar] [CrossRef]
- Lynch, M. Mutation pressure, drift, and the pace of molecular coevolution. Proc. Natl. Acad. Sci. USA 2023, 120, e2306741120. [Google Scholar] [CrossRef]
- Lefébure, T.; Stanhope, M.J. Evolution of the core and pan-genome of Streptococcus: Positive selection, recombination, and genome composition. Genome Biol. 2007, 8, R71. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ST | No of Isolates | No of Farms |
|---|---|---|
| 1135 | 11 | 8 |
| 307 | 9 | 6 |
| 1436 | 6 | 5 |
| 876 | 5 | 4 |
| 1775 | 4 | 4 |
| 1745 | 5 | 3 |
| 914 | 3 | 3 |
| 1437 | 3 | 3 |
| 1741 | 3 | 3 |
| 332 | 2 | 2 |
| 977 | 2 | 2 |
| 1440 | 2 | 2 |
| 1759 | 2 | 2 |
| 1787 | 2 | 2 |
| 22 | 2 | 1 |
| 1441 | 2 | 1 |
| Others * | 77 | 58 |
| AMR Gene | Substance Concerned | No of Isolates | Percentage |
|---|---|---|---|
| ant(6)-Ia | Streptomycin | 75 | 53.6 |
| cat(pC221) | Chloramphenicol | 2 | 1.4 |
| erm(B) | Erythromycin; lincomycin; clindamycin | 6 | 4.3 |
| lnu(B) | Lincomycin; clindamycin | 42 | 30 |
| lnu(C) | Lincomycin | 24 | 17.1 |
| lnu(D) | Lincomycin | 34 | 24.3 |
| lsa(E) | Lincomycin; clindamycin; tiamulin | 42 | 30 |
| tet(L) | Doxycycline; tetracycline | 32 | 22.9 |
| tet(M) | Doxycycline; tetracycline; minocycline | 71 | 50.7 |
| tet(O) | Doxycycline; tetracycline; minocycline | 6 | 4.3 |
| tet(S) | Doxycycline; tetracycline; minocycline | 2 | 1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zouharová, M.; Matiašovic, J.; Gebauer, J.; Matiašková, K.; Nedbalcová, K. Survey of Genotype Diversity, Virulence, and Antimicrobial Resistance Genes in Mastitis-Causing Streptococcus uberis in Dairy Herds Using Whole-Genome Sequencing. Pathogens 2023, 12, 1378. https://doi.org/10.3390/pathogens12121378
Zouharová M, Matiašovic J, Gebauer J, Matiašková K, Nedbalcová K. Survey of Genotype Diversity, Virulence, and Antimicrobial Resistance Genes in Mastitis-Causing Streptococcus uberis in Dairy Herds Using Whole-Genome Sequencing. Pathogens. 2023; 12(12):1378. https://doi.org/10.3390/pathogens12121378
Chicago/Turabian StyleZouharová, Monika, Ján Matiašovic, Jan Gebauer, Katarína Matiašková, and Kateřina Nedbalcová. 2023. "Survey of Genotype Diversity, Virulence, and Antimicrobial Resistance Genes in Mastitis-Causing Streptococcus uberis in Dairy Herds Using Whole-Genome Sequencing" Pathogens 12, no. 12: 1378. https://doi.org/10.3390/pathogens12121378
APA StyleZouharová, M., Matiašovic, J., Gebauer, J., Matiašková, K., & Nedbalcová, K. (2023). Survey of Genotype Diversity, Virulence, and Antimicrobial Resistance Genes in Mastitis-Causing Streptococcus uberis in Dairy Herds Using Whole-Genome Sequencing. Pathogens, 12(12), 1378. https://doi.org/10.3390/pathogens12121378