Abstract
Brucella is an intracellular parasitic bacterium that uses multiple strategies to evade the host’s defense mechanisms. However, how Brucella manipulates the host-induced oxidative stress and relevant biological processes are still poorly understood. In this study, a comparative transcriptome assay of macrophages infected with Brucella abortus S2308 and its rough mutant RB14 was performed to investigate the differentially expressed genes which might be associated with the pathogenic mechanism of Brucella. Our results showed that numerous host pro-oxidative and antioxidative stress genes were differentially expressed in macrophages infected with B. abortus S2308 and mutant RB14 at 4, 8, 24, and 48 h post-infection. Interestingly, we found that several ferroptosis-associated genes were differentially expressed during B. abortus RB14 infection. Moreover, we found that the rough mutant RB14-induced macrophage death was associated with reduced levels of host glutathione and glutathione peroxidase 4, together with increased free iron, lipid peroxidation, and ROS, all of which are important hallmarks of ferroptosis. The ferroptosis occurring during infection with RB14 was reduced by treatment with the inhibitor ferrostatin-1. However, B. abortus S2308 infection did not induce these hallmarks of ferroptosis. Taken together, our results demonstrate that ferroptosis is involved in rough B. abortus infection. Investigating how Brucella manipulates oxidative stress and ferroptosis in its host will be helpful to clarify the pathogenicity of B. abortus.
1. Introduction
Brucellosis is a serious zoonotic infectious disease caused by Brucella, a facultatively intracellular bacterial pathogen. Brucella has no classical virulence factors, such as invasive proteases, exotoxins, capsules, virulence plasmids, etc. The virulence of Brucella relies on its ability to evade the host’s immune system and survive within its macrophages. Brucella also uses its own components to regulate host-cell-related biological processes to promote the infection process. For example, Brucella uses the type IV secretion system (T4SS) to secrete Btp1 protein, which competes with downstream molecules of the toll-like receptors, allowing it to evade the innate immune response []. Another effector protein, BtpB, secreted by Brucella T4SS, also interferes with the immune function of the MyD88 pathway []. Brucella also uses the type IV secretion system (T4SS) to secrete TcpB protein, which affects endoplasmic reticulum stress, allowing it to evade the innate immune response and facilitating its intracellular survival [,]. Research has shown that Brucella induces host cell apoptosis and autophagy, which facilitates its spread and diffusion [,]. However, there are few studies on how B. abortus regulates the oxidative stress genes and relevant biological processes in host cells. Therefore, screening and evaluating the oxidative stress genes and related biological processes during B. abortus infection should be helpful to clarify the novel pathogenic mechanism of B. abortus.
Brucella abortus strain 2308 is a pathogenic strain affecting cattle and humans. RB14, a rough mutant strain lacking the O-antigen of lipopolysaccharide (LPS), is an artificially constructed derivative from the parental B. abortus strain S2308. B. abortus S2308 has evolved many remarkable strategies to evade the host immune response, which allows the establishment of chronic infections, whereas rough mutant RB14 cannot survive in macrophages. Rough Brucella RB14 infection also causes the death of macrophages []. In this study, we compared the transcriptomes of the mouse macrophage cell line RAW 264.7 infected with the B. abortus S2308, or the rough mutant RB14 at 4, 8, 24, and 48 h post-infection (hpi) to clarify the underlying pathogenic mechanism of Brucella.
Our comparative transcriptomic analysis showed that a large number of genes were differentially expressed in RAW 264.7 macrophages infected with B. abortus S2308 and its rough mutant RB14. We focused on those genes related to oxidative stress and programmed cell death, including ferroptosis. We then confirmed the transcriptome data with real-time PCR, which was consistent with the transcriptome data for most of the genes tested. Importantly, our study showed that Brucella rough mutant RB14 but not B. abortus S2308 induced macrophage ferroptosis in the early stage of the infection. These newly identified differentially expressed genes (DEGs) and the novel form of programmed cell death associated with Brucella infection should allow us to clarify the survival and replication strategies of Brucella within macrophages.
2. Materials and Methods
2.1. Reagents
P53 and α-tubulin antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). ACSL4, SLC7A11, and NOX2 antibodies were purchased from Abclonal (Wuhan, China). Horseradish peroxidase (HRP)-conjugated IgG (H + L) secondary antibodies and CellROX Green Reagent were purchased from Thermo Scientific (Waltham, MA, USA). The Gpx4 antibody was obtained from Abcam (Cambridge, MA, USA). The Cytotox 96 nonradioactive cytotoxicity assay kit was purchased from Promega (Fitchburg, USA). Erastin was purchased from MCE (New York, NY, USA). The Lipid Peroxidation (MDA) Assay Kit, and GSSG/GSH quantification kits were purchased from Beyotime (Shanghai, China). Hoechst 33342 and one-step RT-qPCR kits were purchased from Sangon Biotech (Shanghai, China). Ferrostatin-1 (Fer-1) was purchased from Solarbio Science & Technology (Beijing, China). Precast Gel was purchased from Tsingke Biotechnology (Beijing, China). ChamQ Universal SYBR qPCR master mix was purchased from Vazyme Biotech (Nanjing, China). FerroOrange was purchased from Dojindo Molecular Technology (Kyushu, Japan). Gentamicin and other chemicals were from Sangon Biotech (Shanghai, China). All drug concentrations are expressed as the final molar concentration in working buffer.
2.2. Bacterial Strains and Cell Lines
The virulent B. abortus S2308 strain was purchased from American Type Culture Collection (ATCC), and rough mutant RB14 was constructed in our laboratory []. B. abortus were cultured in tryptic soy broth (Difco, BD, Franklin Lakes, NJ, USA) or tryptic soy agar (TSA) at 37 °C with 5% CO2. RAW264.7 murine macrophages were purchased from ATCC and cultured in Dulbecco’s Modified Eagle Medium (DMEM; Hyclone, Logan, UT, USA) with 10% fetal bovine serum (FBS; Gibco, ThermoScientific) at 37 °C with 5% CO2.
2.3. Sample Collection and Preparation
RAW264.7 cells were seeded on 6-well plates at 2 × 106 cells per well, 24 h prior to infection. In order to ensure the similar invasion of Brucella, cells were infected with B. abortus S2308 and rough mutant RB14 in triplicate wells of the 6-well plates at a multiplicity of infection (MOI) of 1000 and 60 by centrifuging bacteria onto cells at 400 g for 5 min at room temperature. Following 1 h of incubation at 37 °C in an atmosphere containing 5% CO2, the cells were washed twice with PBS to remove extracellular bacteria and incubated for an additional 1 h in medium supplemented with 100 μg/mL gentamicin to kill extracellular bacteria. Samples were collected at 4, 8, 24, and 48 hpi for further RNA extraction.
2.4. RNA Isolation, Library Preparation, and Sequencing
RNA degradation and contamination were monitored on 1% agarose gels. RNA purity was checked using the NanoPhotometer spectrophotometer (IMPLEN, Westlake Village, CA, USA). RNA concentration was measured using the Qubit RNA Assay Kit in Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). The clustering of the index-coded samples was performed on a cBot cluster generation system using the TruSeq PE Cluster Kit v3-cBot-HS (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. After cluster generation, the libraries were sequenced on an Illumina Hiseq 2500 platform and 125 bp paired-end reads were generated.
2.5. Data Analysis
Raw data (raw reads) of fastq format were firstly processed through in-house perl scripts. In this step, clean data (clean reads) were obtained by removing reads containing adapter, reads on containing ploy-N, and low-quality reads from raw data. At the same time, the Q20, Q30, and GC content of the clean data were calculated, and normalized to untreated mock controls. All the downstream analyses were based on the clean data with high quality. Cuffdiff provides statistical routines for determining differential expression in digital transcript or gene expression data using a model based on the negative binomial distribution []. Transcripts with an adjusted p-value < 0.05 were assigned as differentially expressed.
2.6. GO Enrichment and Differential Expression Analysis
Gene ontology (GO) analysis for the biological processes was performed to identify the biological function classification of the genes, which describes properties of genes and their products. GO enrichment analysis of differentially expressed genes was implemented using the GOseq R package, in which gene length bias was corrected. GO terms with a corrected p-value less than 0.05 were considered significantly enriched by differential expressed genes.
2.7. Cell Infection and Survival Assays
Cells were seeded on 24-well plates at 3 × 105 cells per well or 6-well plates at 2 × 106 cells, 24 h prior to infection. Cells were infected with B. abortus S2308 and rough mutant RB14. Following 1 h of incubation at 37 °C in an atmosphere containing 5% CO2, the cells were washed twice with PBS to remove extracellular bacteria and incubated for an additional 1 h in medium supplemented with 100 μg/mL gentamicin to kill extracellular bacteria. To monitor B. abortus intracellular survival, the infected cells were lysed with 0.25% Triton X-100 in PBS at specific time points, and serial dilutions of lysates were rapidly plated onto TSA to enumerate colony forming units (CFUs).
2.8. Lactate Dehydrogenase Release Assay
Macrophages were seeded in 96-well plates and infected with S2308 and RB14 as described above. The supernatants were analyzed for lactate dehydrogenase (LDH) level using the CytoTox 96 LDH-release assay as per the manufacturer’s instructions. Percentage LDH release was calculated as 100 × ((Experimental LDH Release—Culture Medium Background)/(Maximum LDH Release—Culture Medium Background)).
2.9. RNA Extraction and Quantitative Real-Time PCR
Total RNA was isolated from RAW 264.7 cells (uninfected or infected with respective B. abortus strain) at different time points using the TRIzol reagent (Ambion, Carlsbad, CA, USA); samples were performed strictly to the manufacturer’s instructions. Genomic contamination was removed with the Turbo DNA-free kit (Ambion, Foster, CA, USA). The resulting RNA was reverse transcribed with the PrimeScript RT reagent kit (TaKaRa, Dalian, China) to produce a cDNA template. The Universal SYBR master mix was used for real-time PCR, according to the manufacturer’s instructions. A total of 1 µL of cDNA, 0.5 µL of forward or reverse primer (10 µM), 8 µL of nuclease-free water and 10 µL of 2× qPCR master mix were added and mixed. The reaction was performed on a Mastercycler ep Realplex system (Eppendorf, Hamburg, Germany). The cycling parameters were as follows: 95 °C for 2 min, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. Each gene was tested in triplicate and the β-actin gene was used as the internal control. The relative transcription level of each gene was calculated using the 2-∆∆Ct method. The primers are listed in Table S2.
2.10. Immunoblot Analysis
At the indicated times, cells were lysed in western blot lysis buffer on ice for 15 min and spun at 16,000× g for 15 min to pellet the insoluble fraction. Soluble fractions were used for immunoblot assays. Cytosolic extracts were separated using polyacrylamide gel, transferred to nitrocellulose membranes (Millipore, Boston, MA, USA). The membranes were then blocked for 1 h in Tris-buffered saline (TBS) solution containing 0.1% Tween-20 (TBST) and 5% non-fat milk, and incubated overnight at 4 °C with primary antibodies (1:1000 dilution). Following overnight incubation, the membranes were washed with TBST and incubated with HRP-conjugated IgG (H + L) secondary antibodies (1:10,000 dilution) at room temperature for 1 h and washed three times with TBST. The protein bands were developed using ECL reagent, visualized using a Tanon 5200 automatic chemiluminescence image analysis system (Tanon, Shanghai, China), and quantified using the ImageJ software 1.0 (National Institutes of Health, Rockville, MD, USA).
2.11. Immunofluorescence Assay
RAW264.7 cells were cultured on 15 mm diameter glass coverslips (Thermo Scientific) in a 24-well plate and infected with B. abortus RB14, as described above. At the designated time points, the cells were washed twice with PBS and incubated with FerroOrange in DMEM at a final concentration of 1 μM. The cells were incubated for 20 min at 37 °C, then the coverslips were incubated with Hoechst 33342 staining solution for 10 min at 37 °C to stain DNA. Finally, the coverslips were mounted on glass slides with Eukitt quick-hardening mounting medium (Sigma-Aldrich, Burlington, MA, USA) and observed under laser scanning confocal microscopy (Nikon D-Eclipse C1, Tokyo, Japan) []. The assay was performed in triplicate.
2.12. Lipid Peroxidation Assay
The lipid peroxidation was determined by detection of the malondialdehyde (MDA) concentration in the cell lysates, which was performed strictly to the manufacturer’s instructions (Beyotime, Shanghai, China). For MDA detection, the thiobarbituric acid included in the kit was added to the supernatants of the cell homogenate to form a TBA-MDA mixture, which was then examined spectrophotometrically at 535 nm. All assays were performed with three independent replicates.
2.13. Measurement of ROS
RAW264.7 cells were plated in a 6-well plate. At designated time points, RAW264.7 cells were resuspended and incubated with CellRox Green in DMEM at a final concentration of 5 μM. The cells were incubated for 30 min at 37 °C, inactivated in 4% (w/v) formaldehyde for 15 min at 37 °C, and resuspended in PBS. ROS were detected using a NovoCyte flow cytometer.
2.14. GSH Assay
The intracellular levels of reductive GSH were determined using a GSSG/GSH quantification kit (Beyotime, Shanghai, China) to detect reductive GSH concentrations in the cell lysates, strictly according to the manufacturer’s instructions. The experiment was performed with three independent replicates.
2.15. Statistical Analysis
All data were imported into GraphPad Prism 8 (Graph Pad Software, Boston, MA, USA) for analysis. The differences in the data were compared using unpaired two-tailed t-tests or two-way ANOVAs with the Sidak’s multiple comparisons test. The data represent the means ± SEM of three independent experiments. ns, no significant difference, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
3. Results and Discussion
3.1. Rough Mutant RB14 Infection Reduced Its Intracellular Survival in RAW264.7 Cells and Increased the Death of Macrophages, in Contrast to B. abortus S2308
RAW264.7 cells were infected with B. abortus S2308 or RB14, and the intracellular growth of B. abortus was assessed at different time points after infection with colony-forming unit (CFU) assays. B. abortus S2308 was killed at 0–12 hpi, and the surviving bacteria began to replicate at 12–48 hpi in the cells. However, the intracellular survival of B. abortus RB14 decreased after it entered the macrophages (Figure 1A). The supernatants of uninfected, B. abortus S2308-infected, and RB14-infected RAW264.7 macrophages were collected at different time points, and the amounts of LDH released from the macrophages were determined. Macrophages infected with rough mutant RB14 showed a much greater percentage release of LDH than macrophages infected with B. abortus S2308 (Figure 1B). These results suggest that rough mutant RB14 infection not only decreased the intracellular survival but also caused higher cell death, compared to the smooth B. abortus S2308 infection in RAW264.7 cells.
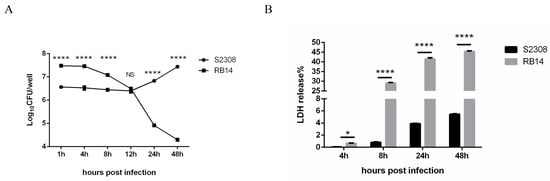
Figure 1.
Intracellular growth of B. abortus in RAW264.7 macrophages and lactate dehydrogenase (LDH) release into the supernatant of macrophage cultures. (A) Intracellular numbers of B. abortus RB14 (MOI = 60) and B. abortus S2308 (MOI = 1000) in RAW264.7 cells at 1, 4, 8, 12, 24, and 48 h post-infection. (B) LDH release measured in supernatants of macrophages after B. abortus S2308 (MOI = 1000) and RB14 infection (MOI = 60). Data are means ± SEM of three independent experiments. Data points and error bars represent the means and SEM of triplicate CFU determinations, respectively. NS, no significant difference; * p < 0.05, **** p < 0.0001.
3.2. Identification of DEGs in Macrophages Infected with B. abortus S2308 and Its Rough Mutant RB14
Based on the intracellular survival results, we designed a comparative transcriptomic analysis of macrophages infected with B. abortus S2308 and its rough mutant RB14 to illustrate the underlying pathogenic genes of B. abortus. Deep sequencing analyses were performed at 4, 8, 24, and 48 hpi. Each library contained approximately 12–15 G raw reads. After the low-quality reads were removed, approximately 80–96 million clean reads from each sample met the criteria for high-quality eukaryotic transcriptome reconstruction, which requires more than 10 million reads to identify new genes. The Q30 score indicated that more than 90% of the reads from all the samples possessed a 99.8% accuracy rate for the sequenced bases (Table 1). Under these criteria, 40, 145, 1703, and 61 DEGs were identified in RAW264.7 cells after infection with rough mutant RB14 or B. abortus S2308 at 4, 8, 24, and 48 hpi, respectively. Compared with their expression in RAW264.7 cells after B. abortus S2308 infection, 21, 111, 931, and 31 genes were upregulated in RAW264.7 cells at 4, 8, 24, and 48 h after rough mutant RB14 infection, respectively; and 19, 34, 772, and 30 genes were downregulated after B. abortus S2308 infection, respectively (Figure 2); the data were normalized to untreated mock controls. To confirm these transcriptome data, we randomly selected 10 genes for real-time PCR analysis (upregulated genes: Edn1, Plk2, Psgt2, Phlda1, Ccl3, and Tnf; downregulated genes: Prdx5, Hgf, Plaur, and Pdp1). The results showed that six of the genes were upregulated and four were downregulated, which is consistent with the transcriptome data (Table 2).

Table 1.
Statistics for RNA-seq datasets.
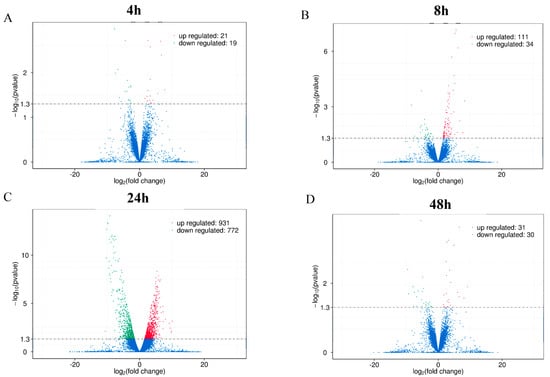
Figure 2.
Volcano plots of differentially expressed genes in RAW264.7 cells infected with B. abortus RB14 or B. abortus S2308. Differentially expressed genes in RAW264.7 cells infected with B. abortus RB14 or B. abortus S2308 at 4 hpi (A), 8 hpi (B), 24 hpi (C), and 48 hpi (D). Red dots indicate upregulated mRNAs, green dots indicate downregulated mRNAs, and blue dots indicate mRNAs with no significant changes.
Table 2.
Verification of transcriptome data with real-time RT-PCR (RB14 versus S2308).
3.3. GO Analysis of Differential Host Gene Expression after Infection with Rough Mutant RB14 and B. abortus S2308
Many host genes were differentially expressed at 4, 8, 24, and 48 hpi in macrophages infected with rough mutant RB14 and B. abortus S2308. The identified genes were related to the immune response, signal transduction, oxidative stress, the inflammatory response, cytokine-mediated signaling pathways, and the regulation of cellular processes (Figure 3). Among the genes differentially expressed post rough mutant RB14 or B. abortus S2308 infection, we focused on those functions that are rarely reported or remain poorly understood in infected cells, and investigated the DEGs related to oxidative stress or novel programmed cell death.
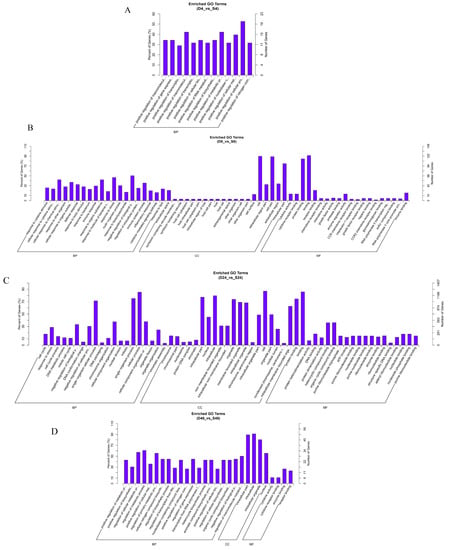
Figure 3.
Comparison of biological gene ontology (GO) enrichment of the differentially expressed genes in RAW264.7 cells infected with B. abortus RB14 or B. abortus S2308. GO analysis of genes differentially expressed in RAW264.7 cells infected with B. abortus RB14 or B. abortus S2308 at 4 hpi (A), 8 hpi (B), 24 hpi (C), and 48 hpi (D). The x-axis indicates the GO terms in one of three subcategories: BP (biological process), CC (cellular component), and MF (molecular function). The left y-axis indicates the percentage of genes (numbers of enriched genes in each term divided by the total number of genes), and the right y-axis indicates the number of genes for each term. D: B. abortus RB14; S: B. abortus S2308.
3.4. Rough Mutant RB14 Caused Stronger Oxidative Stress Than B. abortus S2308
Reactive oxygen species (ROS) are important effector molecules involved in the killing strategies of a number of bacteria [,]. However, Brucella has evolved multiple strategies to regulate host gene expression in order to resist oxidative stress. Therefore, screening and identifying the host DEGS related to oxidative stress could allow new strategies for intracellular survival during Brucella infection to be identified. The expression of numerous genes related to oxidative stress differed in macrophages infected with rough mutant RB14 and those infected with B. abortus S2308 (Table 3). The expression of fewer pro-oxidative stress genes and more antioxidative stress genes were induced in macrophages during B. abortus S2308 infection than during rough mutant RB14 infection, suggesting that antioxidation plays an important role in the pathogenic process of Brucella. Among these pro-oxidative stress genes, Edn1, Plaur, and Hnrnpk were upregulated and Abcg1 was downregulated in macrophages infected with rough mutant RB14 relative to their expression after B. abortus S2308 infection. Several studies have reported that EDN1 stimulates ROS production via the activation of NADPH oxidase (NOX), xanthine oxidase, and mitochondrial cytochrome oxidase [,]. ABCG1 upregulation blunts the activity of pro-oxidant NADPH oxidase and the expression of Nox4, which encodes one of the NADPH oxidase subunits []. Therefore, all these genes are related to NOXs, which produce cellular ROS. Research has shown that NOXs, xanthine oxidase, peroxisomes, cyclooxygenases (COXs), lipoxygenases (LOXs), and cytochrome P450 enzymes are also sources of ROS []. Therefore, B. abortus S2308 may regulate the expression of those genes related to ROS generation more rigorously than rough mutant RB14, better preventing the production of ROS. The upregulation of several genes related to the mitochondria or the production of mitochondrial ROS, such as Plk2, Plk3, Pdp1, Pdk2, eIF5A, Ifit3, and Mtfp1, whose cognate proteins influence the structure or function of the mitochondria, increased the production of ROS in macrophages infected with the rough mutant RB14 relative to that in macrophages infected with B. abortus S2308. Other genes involved in the production of ROS may act through different signaling pathways (Tgm2, Ogt, Ier3, Mmp9, Akap12, Src3, etc.). In summary, the genes involved in ROS production were expressed at higher levels in macrophages infected with RB14 compared to S2308. These results shed light on the different roles played by rough mutant RB14 and B. abortus S2308 in regulating the expression of oxidative-stress-related genes during infection of their hosts.
Table 3.
Oxidative-stress-related genes significantly differentially expressed post-infection (RB14 versus S2308).
Genes encoding several macrophage antioxidant enzymes (Prdx5, Hmox1, Srxn1, and Grx1) showed higher expression during B. abortus S2308 infection than during infection with rough mutant RB14. PRDX5, HO-1, SRXN1, and GRX1 are antioxidant enzymes involved in reducing oxidative stress, and can eliminate the ROS produced in host cells. Several genes whose cognate proteins regulate the expression of antioxidant enzymes were also detected, including Hgf, Met, Csde1, Ccl5, and Hao1. In contrast to infections of rough mutant RB14, the expression of these genes was also upregulated during B. abortus S2308 infection, reducing the production of ROS. For example, CCl5 increased GPX1 expression and reduced intracellular ROS levels, which subsequently increased cell survival in both primary neuron cultures and an overexpression model in SHSY5Y cells []. HGF/MET signaling upregulates the expression of antioxidant enzymes in cardiomyocytes through the p38α pathway []. Therefore, in contrast to rough mutant RB14, B. abortus S2308 may upregulate the expression of host antioxidant enzymes and related genes to abolish the production of ROS, which would allow smooth Brucella to evade the killing effects of ROS. Compared with rough mutant RB14, B. abortus S2308, on the one hand, reduces the expression of the ROS source genes to inhibit the production of ROS, and on the other hand, upregulates antioxidant genes and related genes to eliminate any ROS produced. In summary, the levels of ROS induced in macrophages infected with B. abortus S2308 were much lower than those induced in macrophages infected with rough mutant RB14, and oxidative-stress-related genes were also differentially expressed in cells infected with these two strains. These results shed light on the different roles played by rough mutant RB14 and B. abortus S2308 in regulating the expression of oxidative-stress-related genes during infection of their hosts.
3.5. Rough Mutant RB14 Induced More Expression of Ferroptosis-Associated Genes Than B. abortus S2308
A previous study showed that rough Brucella infections induce macrophage apoptosis and pyroptosis, mediated by caspase 2 [,], whereas smooth Brucella inhibits host cell apoptosis and promotes its own survival and replication in host cells [,]. Our transcriptomic analysis demonstrated that several genes related to apoptosis showed much higher expression during infection with rough mutant RB14 than during B. abortus S2308 infection (Table S1). More importantly, apart from DEGs related to oxidative stress, we also detected several DEGs involved in the novel programmed cell death, ferroptosis (Table 4). Ferroptosis is an iron-dependent form of necrotic cell death, characterized by oxidative damage to phospholipids []. Iron toxicity, lipid peroxidation, and reduced glutathione (GSH) are the pivotal biological features of ferroptosis []. Interestingly, our transcriptomic analysis detected several DEGs involved in iron overload, lipid peroxidation, and reduced GSH. Our research data showed that during rough mutant RB14 infection, the expression of the iron-related genes Fth1 and Lcn2 was downregulated relative to their expression during B. abortus S2308 infection. Fth1 encodes ferritin heavy chain 1, which plays an important role in the maintenance of the cellular iron balance during ferroptosis []. Lipocalin 2 (LCN2) is a critical iron regulatory protein that regulates iron homeostasis under physiological and inflammatory conditions [,]. Therefore, macrophages infected with rough mutant RB14 may induce a greater concentration of free intracellular iron than B. abortus S2308-infected macrophages due to the downregulation of pivotal iron-associated genes. Our results also showed that during rough mutant RB14 infection, the expression of the lipid-peroxidation-related genes Cox4i2 and Alox5 was upregulated relative to their expression in B. abortus S2308-infected macrophages. COX4I2 is considered to increase COX activity, thereby promoting ROS production and levels of MDA, a product of lipid peroxidation []. ALOX5 is a crucial enzyme that mediates lipid peroxidation by producing lipid peroxides []. During rough mutant RB14 infection, the expression of the lipid-peroxidation-related genes Prdx5 and Hmox1 in macrophages was downregulated relative to their expression in macrophages during B. abortus S2308 infection. Research has shown that cytosolic human peroxiredoxin 5 protects yeast cells from the cytotoxicity and lipid peroxidation caused by paraquat []. HMOX1 is an antioxidative gene that also reduces the levels of lipid peroxides and MDA []. The expression of the GSH-related genes Grx1 and Slc7a11 in macrophages was downregulated during infection with rough mutant RB14 relative to their expression during B. abortus S2308 infection. GSH is an important antioxidant with an essential role in maintaining redox homeostasis []. Glutaredoxin 1 (GRX1) acts as the main deglutathionylation enzyme and plays a key role in redox signaling and redox homeostasis []. SLC7A11 is the light chain subunit of the system xc− cystine/glutamate antiporter and plays critical roles in cystine uptake, glutathione synthesis, and ferroptosis resistance [,]. In the present study, the expression of transformation-related protein 53 (TRP53), which acts as an upstream negative regulator of SLC7A11, was also increased during rough mutant RB14 infection relative to its expression in B. abortus S2308-infected macrophages. Therefore, these up- and downregulated DEGs involved in lipid peroxidation may help rough Brucella induce macrophage ferroptosis after infection. In summary, the expression of ferroptosis-associated genes increased in macrophages infected with rough mutant RB14 relative to that in B. abortus S2308-infected macrophages.
Table 4.
Ferroptosis-related genes significantly differentially expressed at 24 h post-infection (RB14 versus S2308).
3.6. Rough Mutant RB14 Induced Macrophage Ferroptosis Soon after Infection
Based on our transcriptome data, we speculate that Brucella rough mutant RB14 infection may induce macrophage ferroptosis soon after infection. Dixon et al., showed that ferroptotic cell death depends on intracellular iron overload, elevated ROS, and lipid peroxidation []. Therefore, we investigated several key hallmarks of ferroptosis during Brucella infection. Interestingly, higher levels of lipid peroxidation and ROS were observed in macrophages after infection with rough mutant RB14 than after infection with B. abortus S2308 (Figure 4A,B). The level of intracellular labile iron also increased after rough mutant RB14 infection (Figure 5A). Research has shown that GPX4 is a central and essential regulator of ferroptosis. It utilizes its GSH substrate to detoxify lipid peroxidation and plays an essential role in inhibiting ferroptosis [,]. The inactivation or depletion of GPX4 induces ferroptosis in a variety of cell types [,]. Therefore, we examined the expression of GPX4 in macrophages after rough mutant RB14 infection. Consistent with this concept, we observed that GPX4 expression was reduced in macrophages infected with rough mutant RB14, as measured with western blotting (Figure 4C). We also examined the levels of GSH, the substrate of GPX4. Consistent with the expression of GPX4, the level of GSH decreased after infection with the rough mutant RB14 (Figure 4D). The expression of several other pivotal ferroptosis-related genes and ferroptosis-regulated genes also showed significant changes after rough mutant RB14 infection, including Acsl4 and Trp53, indicating that macrophages infected with rough mutant RB14 underwent ferroptosis (Figure 4C). To more directly examine the involvement of ferroptosis during Brucella infection, we tested the effect of the ferroptosis inhibitor ferrostatin-1 (Fer-1) on the macrophage death caused by rough mutant RB14 infection. The addition of Fer-1 to RB14-infected macrophage cultures dose-dependently inhibited cell death (Figure 5B). We then confirmed that the addition of Fer-1 at 1–10 µM had no significant toxic effect on the macrophages (Figure 5C). Furthermore, the lipid peroxidation was also reduced following the addition of Fer-1 at 1–10 µM (Figure 5D). These observations indicate that macrophages infected with rough mutant RB14 undergo ferroptosis. During B. abortus S2308 infection, although the production of ROS also increased, lipid peroxidation, GSH, and the expression of the pivotal protein GPX4 showed no significant changes (Figure 4A–D). Further, the level of intracellular labile iron also showed no change after B. abortus S2308 infection (Figure 5A), so B. abortus S2308 did not induce ferroptosis at 24 or 48 hpi. In summary, rough mutant RB14 induced macrophage ferroptosis early after infection, whereas B. abortus S2308 showed no significant increase in the major characteristics associated with ferroptosis in the early period after infection.
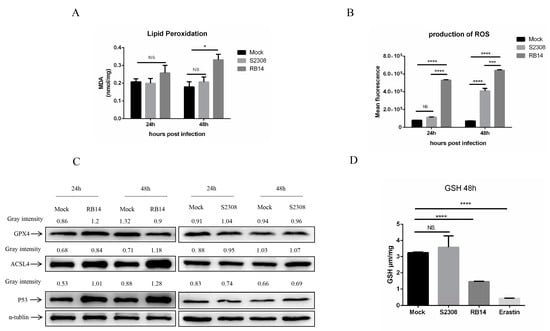
Figure 4.
Rough mutant RB14-induced cell death through ferroptosis. (A) Lipid peroxidation levels in RAW264.7 cells infected with B. abortus RB14 (MOI = 5) and B. abortus S2308 (MOI = 100). (B) Flow-cytometric analysis of ROS production in RAW264.7 cells infected with B. abortus RB14 and B. abortus S2308. (C) Western blotting analysis of the levels of ferroptosis-related proteins in RAW264.7 cells infected with B. abortus RB14 and B. abortus S2308. α-tublin was used as the loading control. (D) GSH levels in RAW264.7 cells infected with B. abortus RB14 and B. abortus S2308 (48 hpi). Erastin (10 μM) is a ferroptosis inducer. Data are means ± SEM of three independent experiments. Data were analyzed with two-way ANOVA. NS, no significant difference; * p < 0.05, *** p < 0.001, **** p < 0.0001.
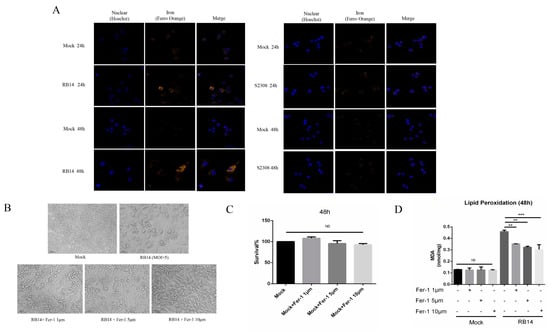
Figure 5.
Inhibiting ferroptosis with a ferroptosis inhibitor reduced the death of macrophages. (A) Analysis of Fe2+ levels in B. abortus RB14- and B. abortus S2308- infected RAW264.7 cells with the fluorescent probe FerroOrange (orange). (B) Cell death induced by RB14 at 48 hpi in macrophages untreated or treated with Fer-1 (bars, 100 µm). (C) Toxicity of ferrostatin-1 in cells after 48 h was observed with a cell viability assay. (D) Lipid peroxidation levels in B. abortus RB14- infected RAW264.7 cells at 48 hpi after treatment with or without ferrostatin-1 (Fer-1); MOI = 5. Ferrostatin-1, an inhibitor of ferroptosis, was added to the macrophage cultures after infection until the indicated times. Data are means ± SEM of three independent experiments. NS, no significant difference; ** p < 0.01, *** p < 0.001.
Brucellosis is a serious zoonotic infectious disease caused by Brucella. It is epidemic throughout the world, and poses a serious threat to public health and livestock development in many countries []. Most of the pathogenic mechanisms of Brucella involve blocking immune receptors, inhibiting phagolysosome fusion and apoptosis, and reducing antigen presentation [,]. However, how Brucella manipulates the oxidative stress and related biological processes of its host remains poorly understood. In this study, we undertook a comparative transcriptomic analysis of the RAW 264.7 cell line infected with B. abortus S2308 or rough mutant RB14 at different time points after infection to investigate the underlying pathogenic mechanisms of B. abortus. We focused on those genes related to oxidative stress and ferroptosis in macrophages.
Endogenous oxidative stress is a consequence of life in an aerobic environment, and bacteria’s interactions with their hosts’ immune systems induce exogenous oxidative stress []. ROS are thought to act via two distinct pathways: causing oxidative damage to bacterial biocompounds and affecting the signaling control of cytokines, autophagy, and apoptosis []. To overcome the deleterious effects of oxidative damage, Brucella has evolved protective, detoxification, and repair mechanisms, including the production of catalase and superoxide dismutase, which directly detoxify ROS, or the production of enzymes that repair oxidative damage to bacterial cellular components []. Brucella has also evolved several mechanisms to influence the expression of host genes, which reduce the production of ROS and oxidative damage. In a previous study, we demonstrated that several host genes are regulated by B. abortus to reduce the production of ROS and facilitate Brucella survival [,,]. However, the host oxidative-stress-related genes regulated by Brucella are not well understood, and further genes and regulatory mechanisms must be identified. The main cellular source of ROS is the mitochondrial respiratory chain and the family of NOXs [,]. Other sources of ROS in macrophages include xanthine oxidase, peroxisomes, COXs, LOXs, and cytochrome P450 enzymes []. The cellular antioxidant defense system balances the production of ROS against the production of antioxidant enzymes, such as the superoxide dismutases (SODs), GSH, and coenzyme Q10 []. The results of the present study show that both antioxidative stress and pro-oxidative stress effects correlated with these gene expression profiles. Several pro-oxidative stress genes (Edn1, Plaur, P2X7, and Hnrnpk) related to NOXs were upregulated in macrophages infected with rough mutant RB14 relative to their expression in B. abortus-S2308-infected macrophages. Moreover, Alox5, Cox4i2, and Ptgs2, which encode proteins that facilitate the production of more ROS, were also upregulated in macrophages infected with rough mutant RB14 relative to their expression in B. abortus-S2308-infected macrophages. These genes are related to other pro-oxidative enzymes, such as COXs, LOXs, and cytochrome P450 enzymes. It is therefore possible that in contrast to rough mutant RB14, smooth B. abortus inhibits the expression of these pro-oxidative genes to reduce the production of ROS. Genes encoding several host antioxidant enzymes or related proteins (Prdx5, Hmox1, Srxn1, Grx1, Hgf, Met, Csde1, Ccl5, and Hao1) showed higher expression during B. abortus S2308 infection than during rough mutant RB14 infection. These antioxidant enzymes reduce the production of ROS, indicating that B. abortus may reduce ROS production by upregulating the expression of antioxidant enzymes or related genes. This hypothesis is consistent with a report that Brucella does not induce an oxidative burst when it invades its host cells []. Therefore, our comparative transcriptomic analysis of the differential expression of oxidative stress genes between B. abortus S2308 and rough mutant RB14 should help us to characterize the underlying pathogenic mechanism during B. abortus infection.
Previous studies have shown that smooth Brucella inhibits macrophage apoptosis, whereas rough Brucella induces macrophage apoptosis, pyroptosis, and oncosis [,,,]. Consistent with those reports, we have demonstrated that rough mutant RB14 caused much greater LDH release from macrophages than B. abortus S2308. We also showed that several DEGs related to apoptosis were more expressed during rough mutant RB14 infection than during B. abortus S2308 infection (Table S2). More importantly, our results indicate for the first time that ferroptosis is also involved in rough mutant RB14 infection. Ferroptosis is an iron-dependent form of regulated cell death caused by unrestricted lipid peroxidation and subsequent membrane damage []. Research has shown that two key initial signals trigger ferroptosis, excessive iron accumulation and the inhibition of GPX4, a protein that prevents lipid peroxidation and detoxifies lipid hydroperoxides [].
The expression of iron-related genes Fth1 and Lcn2 was downregulated in macrophages after rough mutant RB14 infection relative to their expression post B. abortus S2308 infection. FTH1 is the heavy chain of ferritin, a widely expressed and highly conserved protein, consisting of two types of polypeptide chain: the ferritin heavy chain and ferritin light chain. The ferritin heavy chain catalyzes the Fe2+ oxidation reaction, whereas the ferritin light chain plays an important role in the storage of Fe3+. Both chains are essential for the maintenance of iron homeostasis and the prevention of iron overload. FTH1 has also been shown to inhibit ferroptosis through ferritinophagy in Parkinson’s disease []. Therefore, lower expression of Fth1 may accelerate iron accumulation. Lcn2 encodes lipocalin 2, which regulates iron homeostasis and prevents the bacterial reuptake of iron-loaded siderophores []. LCN2 also plays a pivotal role in the inhibition of ferroptosis. LCN2 expression blocks ferroptotic cell death by reducing iron accumulation and subsequent oxidative damage. Therefore, the downregulation of Lcn2 may facilitate ferroptosis by increasing iron accumulation.
During lipid peroxidation, the high levels of polyunsaturated fatty acids (PUFAs) in cellular and organellar membranes are damaged by ROS, and this is an important index of ferroptosis. We identified several DEGs involved in ferroptosis and lipid peroxidation, including Cox4i2, Alox5, Prdx5, Slc7a11, and Grx1. Infection with rough mutant RB14 upregulated lipid-peroxidation-related genes Alox5 and Cox4i2 in macrophages relative to their expression after infection with B. abortus S2308. Research has shown that ROS-mediated lipid peroxidation is mainly attributable to ALOX []. Regulating the activity of ALOX5 has been shown to be a useful way to control ROS- and ferroptosis-induced damage, which promotes degeneration in retinal diseases []. COX4I2 is also reported to increase ROS production and lipid peroxidation, leading to ferroptosis []. Infection with rough mutant RB14 also suppressed the expression of several antioxidant genes (Prdx5, Slc7a11, and Grx1) in macrophages, in contrast to infection with B. abortus S2308. Our data indicate that macrophages infected with rough mutant RB14 showed greater lipid peroxidation at 24 hpi than those infected with B. abortus S2308. Therefore, it is possible that rough mutant RB14 infection induces ferroptosis at 24 hpi, whereas B. abortus S2308 does not. To confirm whether rough mutant RB14 will induce ferroptosis, we investigated the pivotal biological characteristics of ferroptosis in macrophages with infection of the rough RB14 mutant. The production of ROS and lipid peroxidation increased at 24 and 48 h after infection with rough mutant RB14. Iron accumulation in macrophages was also observed with immunofluorescence after rough mutant RB14 infection. The expression of several pivotal genes associated with ferroptosis (Gpx4, Acsl4, Trp53, Slc7a11, and Nox2) also changed after rough mutant RB14 infection. GPX4 is a glutathione- and selenium-dependent glutathione peroxidase that can detoxify lipid hydroperoxides. Research has shown that GPX4 inhibitors (e.g., RSL3, ML162, ML210, FIN56, and FINO2) are also classic ferroptosis activators []. Acyl-CoA synthetase long-chain family member 4 (ACSL4) plays a key role in promoting ferroptosis by incorporating PUFAs into cellular phospholipids [], because PUFAs are the main substrates of lipid peroxidation in the process of ferroptosis, which damages membrane structure and function []. Therefore, GPX4 and ACSL4 regulating lipid peroxidation might influence the ferroptosis in macrophages with rough mutant RB14 infection. A previous study showed that the P53–system xc−–GSH–GPX4 signaling pathway is a main route through which ferroptosis is induced []. System xc− is composed of two protein subunits, SLC7A11 and SLC3A2. SLC7A11 is an amino acid transporter that imports cystine and exports glutamate []. The expression of SLC7A11 and the importation of cystine promote GSH biosynthesis and ferroptosis resistance []. GSH is commonly referred to as the body’s main antioxidant. It is necessary for the activity of GPX4, a selenoenzyme that functions as a central ferroptosis repressor by reducing toxic phospholipid hydroperoxides to nontoxic phospholipid alcohols []. Interestingly, our results not only show that the expression of GPX4 and SLC7A11 decreased, but also that the level of GSH was downregulated in macrophages after infection with rough mutant RB14. The expression of the upstream transcription factor P53 also increased after infection with rough mutant RB14. Research has shown that the transcription of P53 may affect the expression of the SLC7A11 protein and thus influence the production of reduced GSH []. Therefore, it is possible that the P53–system xc−–GSH–GPX4 signaling pathway is involved in the macrophage ferroptosis caused by rough mutant RB14. How these ferroptosis related genes interact with each other is presented in Figure 6. According to our data, rough mutant RB14 infection induced ferroptosis, whereas B. abortus S2308 induced no obvious ferroptotic characteristics in macrophages. A previous study showed that Brucella infection inhibits macrophage apoptosis, promoting the bacterium’s intracellular growth []. Does B. abortus infection inhibit macrophage ferroptosis to promote its intracellular survival and growth in the early period after infection? This question warrants further investigation, together with studies that clarify the complex functions and regulatory mechanisms involved. Research has shown that ferroptotic stress protects macrophages against intracellular E. coli, Salmonella pullorum, and Staphylococcus aureus [], whereas other research has shown that Mycobacterium tuberculosis infection also induces cell ferroptosis, facilitating mycobacterial spread []. Whether ferroptosis in macrophages is an efficient macrophage defense strategy against Brucella, or whether ferroptosis in macrophages is a novel strategy that allows Brucella to evade the host’s immune response requires clarification. The function of ferroptosis during Brucella infection and the underlying regulatory mechanisms also warrant further study.
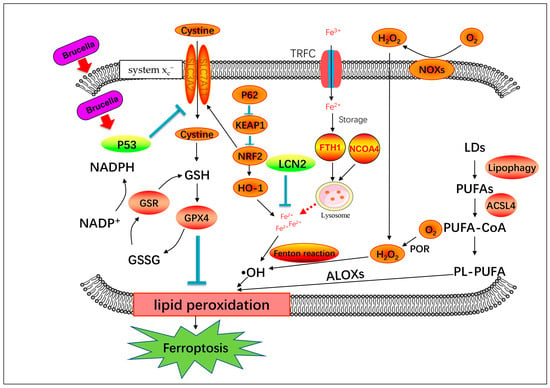
Figure 6.
Schematic diagram of genes involved in ferroptosis process.
Overall, our results demonstrate that a large number of novel genes related to oxidative stress and ferroptosis are regulated by B. abortus, which should allow us to identify further immunity-evading strategies used by Brucella. More importantly, this study showed that ferroptosis, a novel type of programmed cell death, is involved in infection by rough Brucella. This knowledge should extend our understanding of the pathogenic mechanisms of Brucella.
System Xc−mediated cystine uptake and subsequent GSH production and GPX4 activation may be involved in Brucella-induced ferroptosis. The generation of polyunsaturated phospholipids (by ACSL4 and LPCAT3) and subsequent activation of ALOX may have a main role in promoting lipid peroxidation. This process requires hydrogen peroxide (H2O2) production from an iron-mediated Fenton reaction or the activation of POR, NOX, P62/KEAP1/NRF2/HO-1, or other pathways.
4. Conclusions
Our study revealed that the expression of numerous host pro-oxidative stress genes was downregulated in B. abortus S2308-infected macrophages at 4 and 8 hpi relative to their expression in rough mutant RB14, most of which encode oxidases or oxygenases. At 24 and 48 hpi, B. abortus S2308 upregulated the expression of the host’s antioxidative stress genes, especially several genes encoding antioxidant enzymes. Our results also demonstrate for the first time that ferroptosis is involved in rough B. abortus infection. While B. abortus S2308 infection did not induce obvious ferroptosis at an early time post-infection. The key findings, that Brucella manipulates oxidative stress and ferroptosis during infection for its intracellular survival, will help us to elucidate the pathogenicity of B. abortus. However, the mechanism by which different phenotype strains of Brucella regulate ferroptosis and the function of ferroptosis on infection need to be further clarified, which will help us to illustrate the pathogenicity of B. abortus.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pathogens12101189/s1, Table S1: Significantly regulated apoptosis-related genes postinfection (RB14 versus S2308); Table S2: Specific primers of candidate genes for RT-qPCR used in this study.
Author Contributions
S.Y. and C.D. conceived of and designed the experiments. H.H. performed the experiments and analyzed the data. H.H. and M.T. prepared the figures and wrote the manuscript. G.Z., Y.Y. and X.G. contributed reagents, materials, analysis tools, and discussions. S.Y. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The National Natural Science Foundation of China (32002318, 31972723), National Key Research and Development Program of China (2021YFD1800402), and China Postdoctoral Science Foundation (2021M700167, 2022T150709).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data that support the findings of this study are available from National Center for Biotechnology Information at https://www.ncbi.nlm.nih.gov/sra/PRJNA905888 (accessed on 17 January 2023), reference number: PRJNA905888.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Salcedo, S.P.; Marchesini, M.I.; Lelouard, H.; Fugier, E.; Jolly, G.; Balor, S.; Muller, A.; Lapaque, N.; Demaria, O.; Alexopoulou, L.; et al. Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 2008, 4, e21. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, S.P.; Marchesini, M.I.; Degos, C.; Terwagne, M.; Von Bargen, K.; Lepidi, H.; Herrmann, C.K.; Santos Lacerda, T.L.; Imbert, P.R.C.; Pierre, P.; et al. BtpB, a novel Brucella TIR-containing effector protein with immune modulatory functions. Front. Cell. Infect. Microbiol. 2013, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Khan, M.; Magnani, D.D.; Harms, J.S.; Durward, M.; Radhakrishnan, G.K.; Liu, Y.-P.; Splitter, G.A. Brucella induces an unfolded protein response via TcpB that supports intracellular replication in macrophages. PLoS Pathog. 2013, 9, e1003785. [Google Scholar] [CrossRef] [PubMed]
- Byndloss, M.X.; Tsai, A.Y.; Walker, G.T.; Miller, C.N.; Young, B.M.; English, B.C.; Seyffert, N.; Kerrinnes, T.; de Jong, M.F.; Atluri, V.L.; et al. Brucella abortus Infection of Placental Trophoblasts Triggers Endoplasmic Reticulum Stress-Mediated Cell Death and Fetal Loss via Type IV Secretion System-Dependent Activation of CHOP. mBio 2019, 10, e01538-19. [Google Scholar] [CrossRef]
- Starr, T.; Child, R.; Wehrly, T.D.; Hansen, B.; Hwang, S.; López-Otin, C.; Virgin, H.W.; Celli, J. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 2012, 11, 33–45. [Google Scholar] [CrossRef]
- Pei, J.; Kahl-McDonagh, M.; Ficht, T.A. Brucella dissociation is essential for macrophage egress and bacterial dissemination. Front. Cell. Infect. Microbiol. 2014, 4, 23. [Google Scholar] [CrossRef]
- Tian, M.; Yin, Y.; Lian, Z.; Li, Z.; Song, M.; Hu, H.; Guan, X.; Ding, C.; Wang, S.; Li, T.; et al. A rough Brucella mutant induced macrophage death depends on secretion activity of T4SS, but not on cellular Txnip- and Caspase-2-mediated signaling pathway. Vet. Microbiol. 2020, 244, 108648. [Google Scholar] [CrossRef]
- Zhang, M.; Han, X.; Liu, H.; Tian, M.; Ding, C.; Song, J.; Sun, X.; Liu, Z.; Yu, S. Inactivation of the ABC transporter ATPase gene in Brucella abortus strain 2308 attenuated the virulence of the bacteria. Vet. Microbiol. 2013, 164, 322–329. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Kan, X.; Yin, Y.; Song, C.; Tan, L.; Qiu, X.; Liao, Y.; Liu, W.; Meng, S.; Sun, Y.; Ding, C. Newcastle-disease-virus-induced ferroptosis through nutrient deprivation and ferritinophagy in tumor cells. iScience 2021, 24, 102837. [Google Scholar] [CrossRef]
- López-Pérez, W.; Sai, K.; Sakamachi, Y.; Parsons, C.; Kathariou, S.; Ninomiya-Tsuji, J. TAK1 inhibition elicits mitochondrial ROS to block intracellular bacterial colonization. Proc. Natl. Acad. Sci. USA 2021, 118, e2023647118. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.F.; Pidwill, G.R.; Carnell, O.T.; Surewaard, B.G.J.; Shamarina, D.; Sutton, J.A.F.; Jeffery, C.; Derré-Bobillot, A.; Archambaud, C.; Siggins, M.K.; et al. Commensal bacteria augment Staphylococcus aureus infection by inactivation of phagocyte-derived reactive oxygen species. PLoS Pathog. 2021, 17, e1009880. [Google Scholar] [CrossRef]
- Callera, G.E.; Tostes, R.C.; Yogi, A.; Montezano, A.C.I.; Touyz, R.M. Endothelin-1-induced oxidative stress in DOCA-salt hypertension involves NADPH-oxidase-independent mechanisms. Clin. Sci. 2006, 110, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S.; Wang, C.; Wang, D. Endothelin-1-Induced Microvascular ROS and Contractility in Angiotensin-II-Infused Mice Depend on COX and TP Receptors. Antioxidants 2019, 8, 193. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Fan, J.; Li, Y.; Wu, W.; Yan, Q.; Zheng, Q. ABCG1 Attenuates Oxidative Stress Induced by H2O2 through the Inhibition of NADPH Oxidase and the Upregulation of Nrf2-Mediated Antioxidant Defense in Endothelial Cells. Anal. Cell. Pathol. 2020, 2020, 2095645. [Google Scholar] [CrossRef]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.-H.; Yen, C.-H.; Hsieh, T.-H.; Kao, T.-J.; Chiu, J.-Y.; Chiang, Y.-H.; Hoffer, B.J.; Chang, W.-C.; Chou, S.-Y. CCL5 via GPX1 activation protects hippocampal memory function after mild traumatic brain injury. Redox Biol. 2021, 46, 102067. [Google Scholar] [CrossRef]
- Arechederra, M.; Carmona, R.; González-Nuñez, M.; Gutiérrez-Uzquiza, Á.; Bragado, P.; Cruz-González, I.; Cano, E.; Guerrero, C.; Sánchez, A.; López-Novoa, J.M.; et al. Met signaling in cardiomyocytes is required for normal cardiac function in adult mice. Biochim. Biophys. Acta 2013, 1832, 2204–2215. [Google Scholar] [CrossRef]
- Chen, F.; He, Y. Caspase-2 mediated apoptotic and necrotic murine macrophage cell death induced by rough Brucella abortus. PLoS ONE 2009, 4, e6830. [Google Scholar] [CrossRef][Green Version]
- Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nuñez, G.; He, Y.; Yin, X.-M.; O’Riordan, M.X.D. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity 2015, 43, 451–462. [Google Scholar]
- Ferrero, M.C.; Fossati, C.A.; Baldi, P.C. Smooth Brucella strains invade and replicate in human lung epithelial cells without inducing cell death. Microbes Infect. 2009, 11, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Wei, P.; Zhao, Y.; Guan, Z.; Yang, L.; Sun, W.; Wang, S.; Peng, Q. Brucella infection inhibits macrophages apoptosis via Nedd4-dependent degradation of calpain2. Vet. Microbiol. 2014, 174, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Amaral, E.P.; Costa, D.L.; Namasivayam, S.; Riteau, N.; Kamenyeva, O.; Mittereder, L.; Mayer-Barber, K.D.; Andrade, B.B.; Sher, A. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J. Exp. Med. 2019, 216, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Chen, X.; Tan, Q.; Zhou, H.; Xu, J.; Gu, Q. Inhibiting Ferroptosis through Disrupting the NCOA4-FTH1 Interaction: A New Mechanism of Action. ACS Cent. Sci. 2021, 7, 980–989. [Google Scholar] [CrossRef]
- Mertens, C.; Kuchler, L.; Sola, A.; Guiteras, R.; Grein, S.; Brüne, B.; von Knethen, A.; Jung, M. Macrophage-Derived Iron-Bound Lipocalin-2 Correlates with Renal Recovery Markers Following Sepsis-Induced Kidney Damage. Int. J. Mol. Sci. 2020, 21, 7527. [Google Scholar] [CrossRef]
- Mertens, C.; Schnetz, M.; Rehwald, C.; Grein, S.; Elwakeel, E.; Weigert, A.; Brüne, B.; Jung, M. Iron-Bound Lipocalin-2 from Tumor-Associated Macrophages Drives Breast Cancer Progression Independent of Ferroportin. Metabolites 2021, 11, 180. [Google Scholar] [CrossRef]
- Chang, B.; Guan, H.; Wang, X.; Chen, Z.; Zhu, W.; Wei, X.; Li, S. Cox4i2 Triggers an Increase in Reactive Oxygen Species, Leading to Ferroptosis and Apoptosis in HHV7 Infected Schwann Cells. Front. Mol. Biosci. 2021, 8, 660072. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Nguyên-Nhu, N.T.; Knoops, B. Mitochondrial and cytosolic expression of human peroxiredoxin 5 in Saccharomyces cerevisiae protect yeast cells from oxidative stress induced by paraquat. FEBS Lett. 2003, 544, 148–152. [Google Scholar] [CrossRef]
- Ge, M.-H.; Tian, H.; Mao, L.; Li, D.-Y.; Lin, J.-Q.; Hu, H.-S.; Huang, S.-C.; Zhang, C.-J.; Mei, X.-F. Zinc attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury by activating Nrf2/GPX4 defense pathway. CNS Neurosci. Ther. 2021, 27, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Gorelenkova Miller, O.; Mieyal, J.J. Critical Roles of Glutaredoxin in Brain Cells-Implications for Parkinson’s Disease. Antioxid. Redox Signal. 2019, 30, 1352–1368. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, Y.; Wang, X.; Tian, H.; Wang, Y.; Jin, J.; Shan, Z.; Liu, Y.E.; Cai, Z.; Tong, X.; et al. Stem Cell Factor SOX2 Confers Ferroptosis Resistance in Lung Cancer via Upregulation of SLC7A11. Cancer Res. 2021, 81, 5217–5229. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 12. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Zhang, Y.; Swanda, R.V.; Nie, L.; Liu, X.; Wang, C.; Lee, H.; Lei, G.; Mao, C.; Koppula, P.; Cheng, W.; et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 2021, 12, 1589. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- D’Anastasio, R.; Staniscia, T.; Milia, M.L.; Manzoli, L.; Capasso, L. Origin, evolution and paleoepidemiology of brucellosis. Epidemiol. Infect. 2011, 139, 149–156. [Google Scholar] [CrossRef]
- Campos, P.C.; Gomes, M.T.R.; Guimarães, G.; Franco, M.M.S.C.; Marim, F.M.; Oliveira, S.C. Brucella abortus DNA is a major bacterial agonist to activate the host innate immune system. Microbes Infect. 2014, 16, 979–984. [Google Scholar] [CrossRef]
- Skendros, P.; Pappas, G.; Boura, P. Cell-mediated immunity in human brucellosis. Microbes Infect. 2011, 13, 134–142. [Google Scholar] [CrossRef]
- Gaupp, R.; Ledala, N.; Somerville, G.A. Staphylococcal response to oxidative stress. Front. Cell. Infect. Microbiol. 2012, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell. Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Hornback, M.L.; Roop, R.M., 2nd. The Brucella abortus xthA-1 gene product participates in base excision repair and resistance to oxidative killing but is not required for wild-type virulence in the mouse model. J. Bacteriol. 2006, 188, 1295–1300. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Li, P.; Guan, X.; Lian, Z.; Yin, Y.; Shi, W.; Ding, C.; Yu, S. Brucella Infection Regulates Thioredoxin-Interacting Protein Expression to Facilitate Intracellular Survival by Reducing the Production of Nitric Oxide and Reactive Oxygen Species. J. Immunol. 2020, 204, 632–643. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Yin, Y.; Zuo, D.; Guan, X.; Ding, C.; Yu, S. Brucella induces heme oxygenase-1 expression to promote its infection. Transbound. Emerg. Dis. 2021, 69, 2697–2711. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, M.; Li, P.; Bao, Y.; Guan, X.; Lian, Z.; Yin, Y.; Ding, C.; Yu, S. Brucella infection regulates peroxiredoxin-5 protein expression to facilitate intracellular survival by reducing the production of nitric oxide and reactive oxygen species. Biochem. Biophys. Res. Commun. 2019, 516, 82–88. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Sarniak, A.; Lipińska, J.; Tytman, K.; Lipińska, S. Endogenous mechanisms of reactive oxygen species (ROS) generation. Adv. Hyg. Exp. Med. 2016, 70, 1150–1165. [Google Scholar] [CrossRef]
- Köhler, S.; Porte, F.; Jubier-Maurin, V.; Ouahrani-Bettache, S.; Teyssier, J.; Liautard, J.-P. The intramacrophagic environment of Brucella suis and bacterial response. Vet. Microbiol. 2002, 90, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Ficht, T.A. Brucella abortus rough mutants are cytopathic for macrophages in culture. Infect. Immun. 2004, 72, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Bronner, D.N.; O’Riordan, M.X.; He, Y. Caspase-2 mediates a Brucella abortus RB51-induced hybrid cell death having features of apoptosis and pyroptosis. Front. Cell. Infect. Microbiol. 2013, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 2021, 218, e20210518. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Lu, J.; Hao, X.; Li, H.; Zhang, G.; Liu, X.; Li, X.; Zhao, C.; Kuang, W.; Chen, D.; et al. FTH1 Inhibits Ferroptosis through Ferritinophagy in the 6-OHDA Model of Parkinson’s Disease. Neurotherapeutics 2020, 17, 1796–1812. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.R.; Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Siderophores in Iron Metabolism: From Mechanism to Therapy Potential. Trends Mol. Med. 2016, 22, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef]
- Lee, J.-J.; Chang-Chien, G.-P.; Lin, S.; Hsiao, Y.-T.; Ke, M.-C.; Chen, A.; Lin, T.-K. 5-Lipoxygenase Inhibition Protects Retinal Pigment Epithelium from Sodium Iodate-Induced Ferroptosis and Prevents Retinal Degeneration. Oxid. Med. Cell. Longev. 2022, 2022, 1792894. [Google Scholar] [CrossRef]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Cui, G.; Lu, Q.; Yang, L.; Guan, Z.; Sun, W.; Zhao, Y.; Wang, S.; Peng, Q. A20 promotes Brucella intracellular growth via inhibition of macrophage cell death and activation. Vet. Microbiol. 2015, 175, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Fang, L.; Chen, L.; Wang, X.; Jiang, J.; Gao, L. Ferroptotic stress promotes macrophages against intracellular bacteria. Theranostics 2022, 12, 2266–2289. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).