
Selective Depletion of ZAP-Binding CpG Motifs in HCV Evolution
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
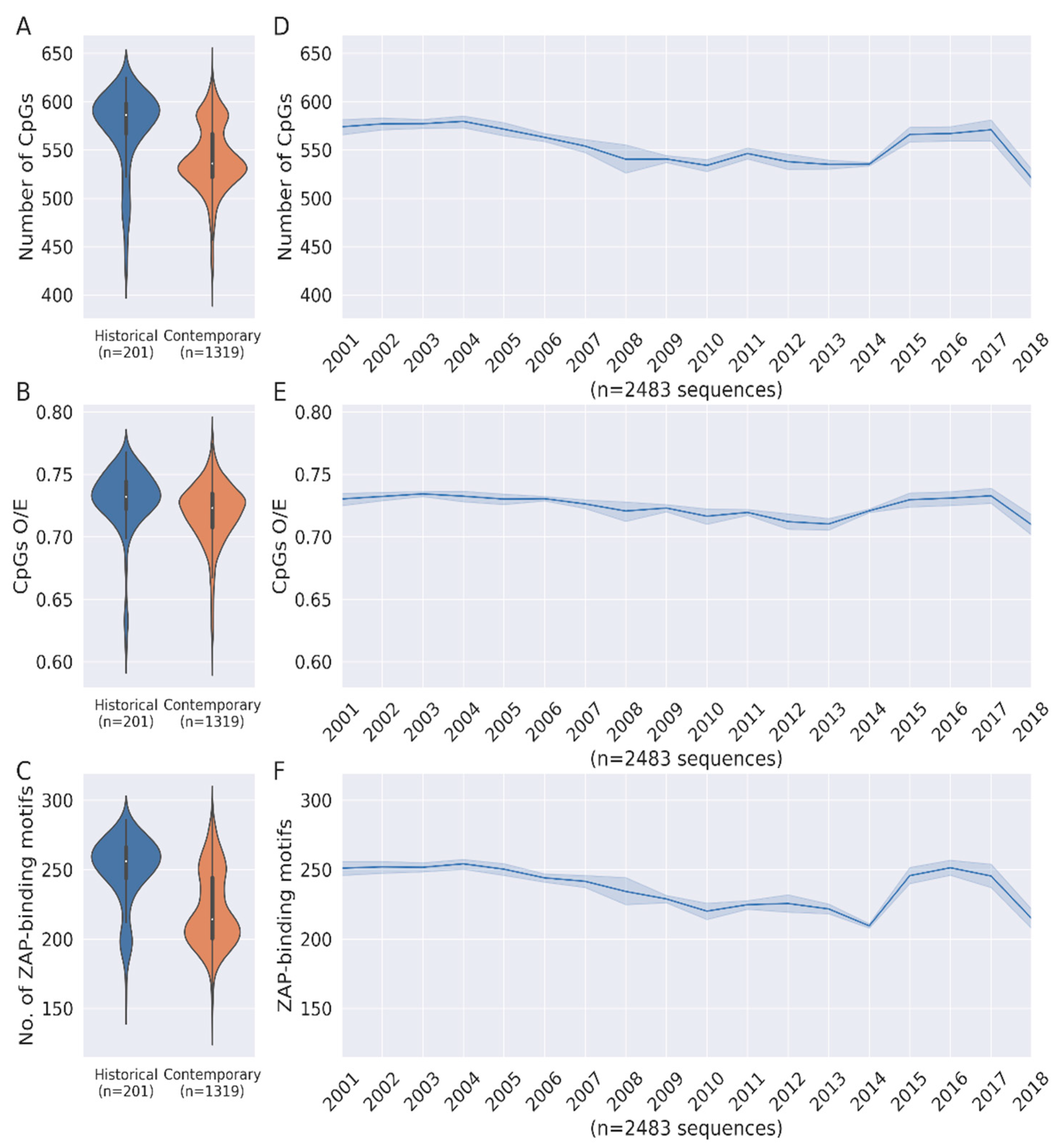
3.1. Loss of CpG Content and ZAP-Binding Motifs in HCV Genome
3.2. Correlation between Loss of ZAP Binding Motif and CpG Loss
3.3. CpG Depeletion in HCV Genomes Are Primarily Driven by the Loss of ZAP-Binding Motifs
3.4. Selective Conservation of Specific ZAP-Binding Motifs during HCV Evolution
3.5. Enhanced Loss of CpGs from the HCV Core Gene from Outside ZAP-Binding Motifs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Institutes of Health. National Institutes of Health Consensus Development Conference Statement: Management of Hepatitis C 2002 (June 10–12, 2002). Gastroenterology 2002, 123, 2082–2099. [Google Scholar] [CrossRef]
- Sievert, W.; Altraif, I.; Razavi, H.A.; Abdo, A.; Ahmed, E.A.; Alomair, A.; Amarapurkar, D.; Chen, C.H.; Dou, X.; el Khayat, H.; et al. A Systematic Review of Hepatitis C Virus Epidemiology in Asia, Australia and Egypt. Liver Int. 2011, 31, 61–80. [Google Scholar] [CrossRef]
- Blach, S.; Terrault, N.A.; Tacke, F.; Gamkrelidze, I.; Craxi, A.; Tanaka, J.; Waked, I.; Dore, G.J.; Abbas, Z.; Abdallah, A.R.; et al. Global Change in Hepatitis C Virus Prevalence and Cascade of Care between 2015 and 2020: A Modelling Study. Lancet Gastroenterol. Hepatol. 2022, 7, 396–415. [Google Scholar] [CrossRef]
- Hepatitis, C. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-C (accessed on 28 September 2022).
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded Classification of Hepatitis C Virus into 7 Genotypes and 67 Subtypes: Updated Criteria and Genotype Assignment Web Resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef]
- Simmonds, P.; McOmish, F.; Yap, P.L.; Dow, B.C.; Follett, E.A.C.; Seed, C.; Keller, A.J.; Cobain, T.J.; Krusius, T.; Kolho, E.; et al. Geographical Distribution of Hepatitis C Virus Genotypes in Blood Donors: An International Collaborative Survey. J. Clin. Microbiol. 1994, 32, 884–892. [Google Scholar] [CrossRef]
- Kumar, A.; Goyal, N.; Saranathan, N.; Dhamija, S.; Saraswat, S.; Menon, M.B.; Vivekanandan, P. The Slowing Rate of CpG Depletion in SARS-CoV-2 Genomes Is Consistent with Adaptations to the Human Host. Mol. Biol. Evol. 2022, 39, msac029. [Google Scholar] [CrossRef]
- Wasson, M.K.; Borkakoti, J.; Kumar, A.; Biswas, B.; Vivekanandan, P. The CpG Dinucleotide Content of the HIV-1 Envelope Gene May Predict Disease Progression. Sci. Rep. 2017, 7, 8162. [Google Scholar] [CrossRef]
- Upadhyay, M.; Vivekanandan, P. Depletion of CpG Dinucleotides in Papillomaviruses and Polyomaviruses: A Role for Divergent Evolutionary Pressures. PLoS ONE 2015, 10, e0142368. [Google Scholar] [CrossRef]
- Upadhyay, M.; Samal, J.; Kandpal, M.; Vasaikar, S.; Biswas, B.; Gomes, J.; Vivekanandan, P. CpG Dinucleotide Frequencies Reveal the Role of Host Methylation Capabilities in Parvovirus Evolution. J. Virol. 2013, 87, 13816–13824. [Google Scholar] [CrossRef]
- Hoelzer, K.; Shackelton, L.A.; Parrish, C.R. Presence and Role of Cytosine Methylation in DNA Viruses of Animals. Nucleic Acids Res. 2008, 36, 2825–2837. [Google Scholar] [CrossRef]
- Cheng, X.; Virk, N.; Chen, W.; Ji, S.; Ji, S.; Sun, Y.; Wu, X. CpG Usage in RNA Viruses: Data and Hypotheses. PLoS ONE 2013, 8, e74109. [Google Scholar] [CrossRef]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG Dinucleotide Suppression Enables Antiviral Defence Targeting Non-Self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef]
- Luo, X.; Wang, X.; Gao, Y.; Zhu, J.; Liu, S.; Gao, G.; Gao, P. Molecular Mechanism of RNA Recognition by Zinc-Finger Antiviral Protein. Cell Rep. 2020, 30, 46–52.e4. [Google Scholar] [CrossRef]
- Shen, J.; Wang, S.; Zhang, Y.J.; Wu, H.C.; Kibriya, M.G.; Jasmine, F.; Ahsan, H.; Wu, D.P.H.; Siegel, A.B.; Remotti, H.; et al. Exploring Genome-Wide DNA Methylation Profiles Altered in Hepatocellular Carcinoma Using Infinium HumanMethylation 450 BeadChips. Epigenetics 2013, 8, 34–43. [Google Scholar] [CrossRef]
- Bryan-Marrugo, O.L.; Ramos-Jiménez, J.; Barrera-Saldaña, H.; Rojas-Martínez, A.; Vidaltamayo, R.; Rivas-Estilla, A.M. History and Progress of Antiviral Drugs: From Acyclovir to Direct-Acting Antiviral Agents (DAAs) for Hepatitis C. Med. Univ. 2015, 17, 165–174. [Google Scholar] [CrossRef]
- Ferrarese, A.; Germani, G.; Gambato, M.; Russo, F.P.; Senzolo, M.; Zanetto, A.; Shalaby, S.; Cillo, U.; Zanus, G.; Angeli, P.; et al. Hepatitis C Virus Related Cirrhosis Decreased as Indication to Liver Transplantation since the Introduction of Direct-Acting Antivirals: A Single-Center Study. World J. Gastroenterol. 2018, 24, 4403–4411. [Google Scholar] [CrossRef]
- Zeuzem, S.; Mizokami, M.; Pianko, S.; Mangia, A.; Han, K.H.; Martin, R.; Svarovskaia, E.; Dvory-Sobol, H.; Doehle, B.; Hedskog, C.; et al. NS5A Resistance-Associated Substitutions in Patients with Genotype 1 Hepatitis C Virus: Prevalence and Effect on Treatment Outcome. J. Hepatol. 2017, 66, 910–918. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Ray Ayb, R.B.; Ray, R. Hepatitis C Virus Core Protein: Intriguing Properties and Functional Relevance. FEMS Microbiol. Lett. 2001, 202, 149–156. [Google Scholar] [CrossRef]
- Kunkel, M.; Lorinczi, M.; Rijnbrand, R.; Lemon, S.M.; Watowich, S.J. Self-Assembly of Nucleocapsid-Like Particles from Recombinant Hepatitis C Virus Core Protein. J. Virol. 2001, 75, 2119–2129. [Google Scholar] [CrossRef]
- Preciado, M.V.; Valva, P.; Escobar-Gutierrez, A.; Rahal, P.; Ruiz-Tovar, K.; Yamasaki, L.; Vazquez-Chacon, C.; Martinez-Guarneros, A.; Carpio-Pedroza, J.C.; Fonseca-Coronado, S.; et al. Hepatitis C Virus Molecular Evolution: Transmission, Disease Progression and Antiviral Therapy. World J. Gastroenterol. 2014, 20, 15992–16013. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Holmes, E.C.; Cha, T.A.; Chan, S.W.; McOmish, F.; Irvine, B.; Beall, E.; Yap, P.L.; Kolberg, J.; Urdea, M.S. Classification of Hepatitis C Virus into Six Major Genotypes and a Series of Subtypes by Phylogenetic Analysis of the NS-5 Region. J. Gen. Virol. 1993, 74 Pt 11, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Echeverría, N.; Moratorio, G.; Cristina, J.; Moreno, P. Hepatitis C Virus Genetic Variability and Evolution. World J. Hepatol. 2015, 7, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Alinejad-Rokny, H.; Anwar, F.; Waters, S.A.; Davenport, M.P.; Ebrahimi, D. Source of CpG Depletion in the HIV-1 Genome. Mol. Biol. Evol. 2016, 33, 3205–3212. [Google Scholar] [CrossRef] [PubMed]
- Sankar, S.; Borkakoti, J.; Ramamurthy, M.; Nandagopal, B.; Vivekanandan, P.; Gopalan, S. Identification of Tell-Tale Patterns in the 3′ Non-Coding Region of Hantaviruses That Distinguish HCPS-Causing Hantaviruses from HFRS-Causing Hantaviruses. Emerg. Microbes Infect. 2018, 7, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, B.D.; Levine, A.J.; Bhanot, G.; Rabadan, R. Patterns of Evolution and Host Gene Mimicry in Influenza and Other RNA Viruses. PLoS Pathog. 2008, 4, e1000079. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Extreme Genomic CpG Deficiency in SARS-CoV-2 and Evasion of Host Antiviral Defense. Mol. Biol. Evol. 2020, 37, 2699–2705. [Google Scholar] [CrossRef]
- Gray, R.R.; Tanaka, Y.; Takebe, Y.; Magiorkinis, G.; Buskell, Z.; Seeff, L.; Alter, H.J.; Pybus, O.G. Evolutionary Analysis of Hepatitis C Virus Gene Sequences from 1953. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20130168. [Google Scholar] [CrossRef]
- Geddawy, A.; Ibrahim, Y.F.; Elbahie, N.M.; Ibrahim, M.A. Direct Acting Anti-Hepatitis C Virus Drugs: Clinical Pharmacology and Future Direction. J. Transl. Int. Med. 2017, 5, 8–17. [Google Scholar] [CrossRef]
- Kitrinos, K.M.; Nelson, J.A.E.; Resch, W.; Swanstrom, R. Effect of a Protease Inhibitor-Induced Genetic Bottleneck on Human Immunodeficiency Virus Type 1 Env Gene Populations. J. Virol. 2005, 79, 10627–10637. [Google Scholar] [CrossRef][Green Version]
- Contreras, A.M.; Hiasa, Y.; He, W.; Terella, A.; Schmidt, E.V.; Chung, R.T. Viral RNA Mutations Are Region Specific and Increased by Ribavirin in a Full-Length Hepatitis C Virus Replication System. J. Virol. 2002, 76, 8505–8517. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Santolini, E.; Migliaccio, G.; la Monica, N. Biosynthesis and Biochemical Properties of the Hepatitis C Virus Core Protein. J. Virol. 1994, 68, 3631–3641. [Google Scholar] [CrossRef] [PubMed]
- Cristofari, G.; Ivanyi-Nagy, R.; Gabus, C.; Boulant, S.; Lavergne, J.P.; Penin, F.; Darlix, J.L. The Hepatitis C Virus Core Protein Is a Potent Nucleic Acid Chaperone That Directs Dimerization of the Viral (+) Strand RNA in Vitro. Nucleic Acids Res. 2004, 32, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.C.; Dellos, S.R.; Lingappa, J.R. Identification of Residues in the Hepatitis C Virus Core Protein That Are Critical for Capsid Assembly in a Cell-Free System. J. Virol. 2005, 79, 6814–6826. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, S.; Kumar, A.; Samal, J.; Gupta, E.; Vivekanandan, P.; Menon, M.B. Selective Depletion of ZAP-Binding CpG Motifs in HCV Evolution. Pathogens 2023, 12, 43. https://doi.org/10.3390/pathogens12010043
Mukherjee S, Kumar A, Samal J, Gupta E, Vivekanandan P, Menon MB. Selective Depletion of ZAP-Binding CpG Motifs in HCV Evolution. Pathogens. 2023; 12(1):43. https://doi.org/10.3390/pathogens12010043
Chicago/Turabian StyleMukherjee, Sanket, Akhil Kumar, Jasmine Samal, Ekta Gupta, Perumal Vivekanandan, and Manoj B. Menon. 2023. "Selective Depletion of ZAP-Binding CpG Motifs in HCV Evolution" Pathogens 12, no. 1: 43. https://doi.org/10.3390/pathogens12010043
APA StyleMukherjee, S., Kumar, A., Samal, J., Gupta, E., Vivekanandan, P., & Menon, M. B. (2023). Selective Depletion of ZAP-Binding CpG Motifs in HCV Evolution. Pathogens, 12(1), 43. https://doi.org/10.3390/pathogens12010043