
Development of Novel Anti-Leishmanials: The Case for Structure-Based Approaches

Abstract
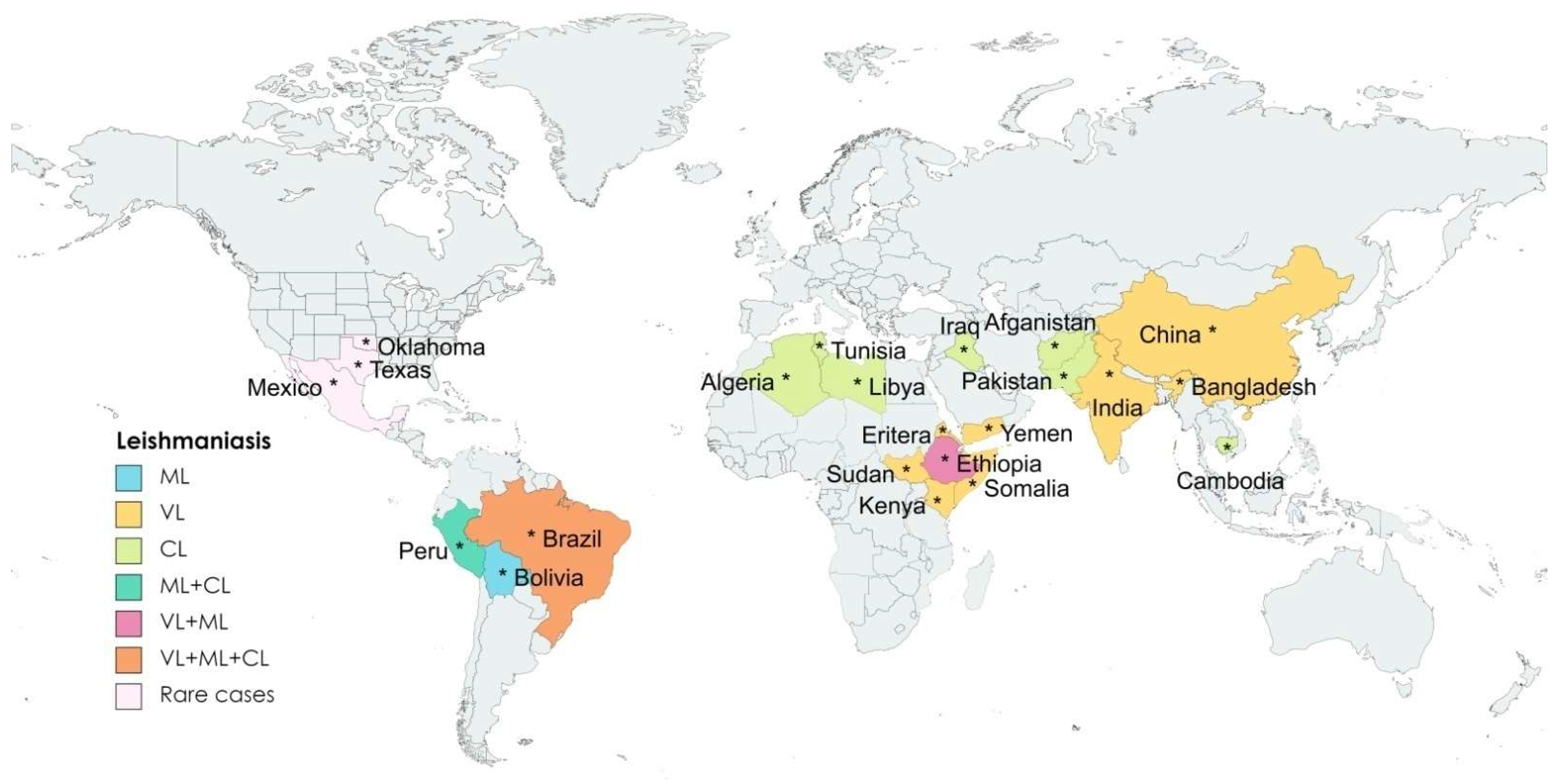
:1. Background
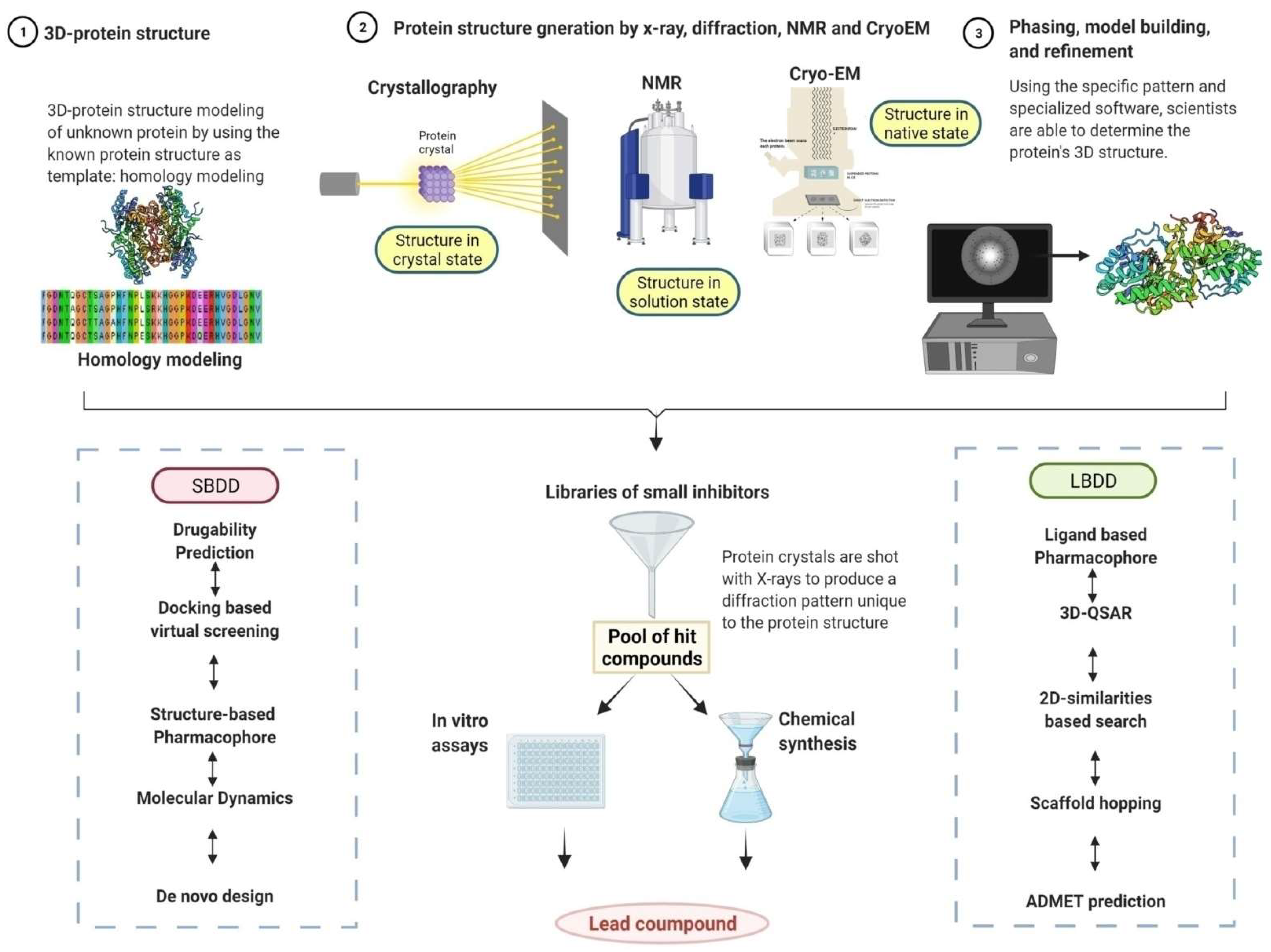
2. Emergence of Artificial Intelligence (AI) in Anti-leishmanial Drug Discovery
3. Parasite-Specific Essential Metabolic Pathways and Possible Drug Targets
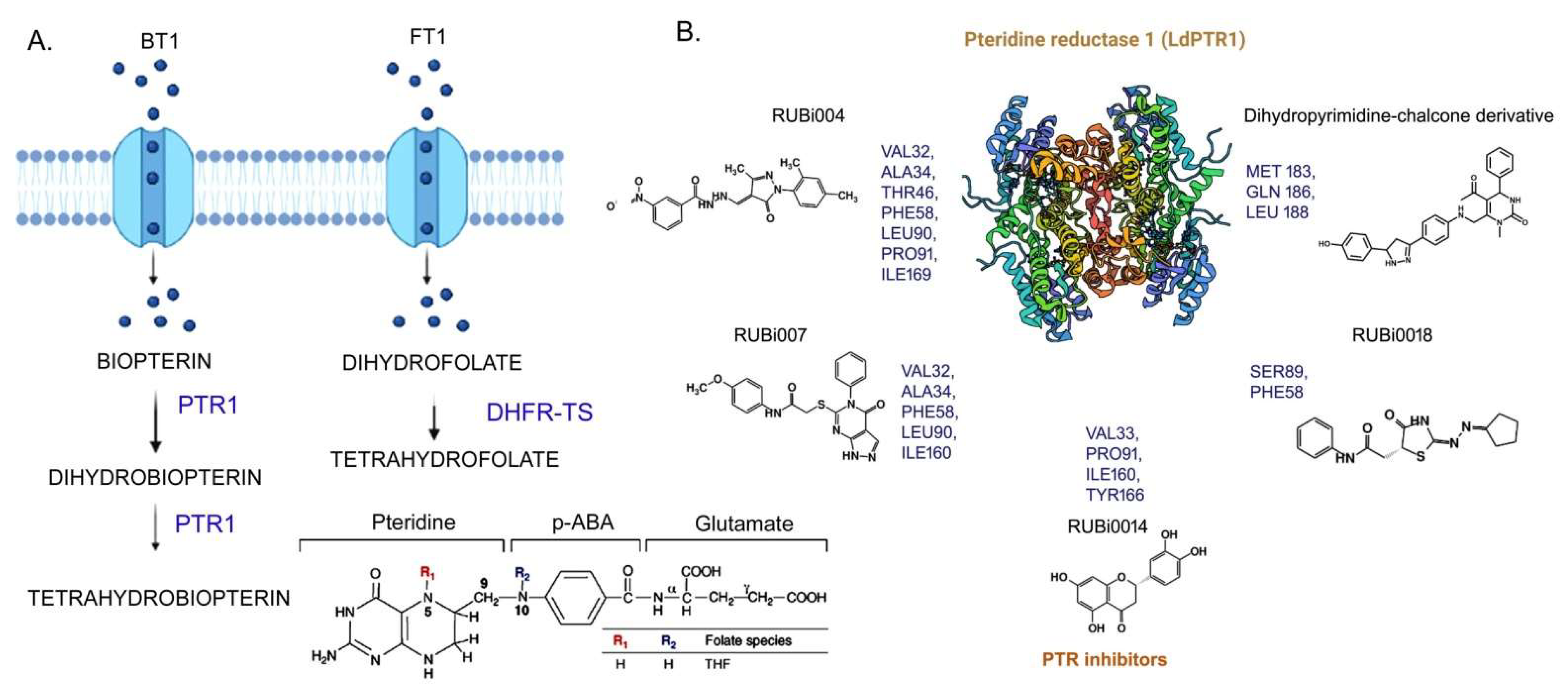
3.1. Folate Biosynthesis Pathway
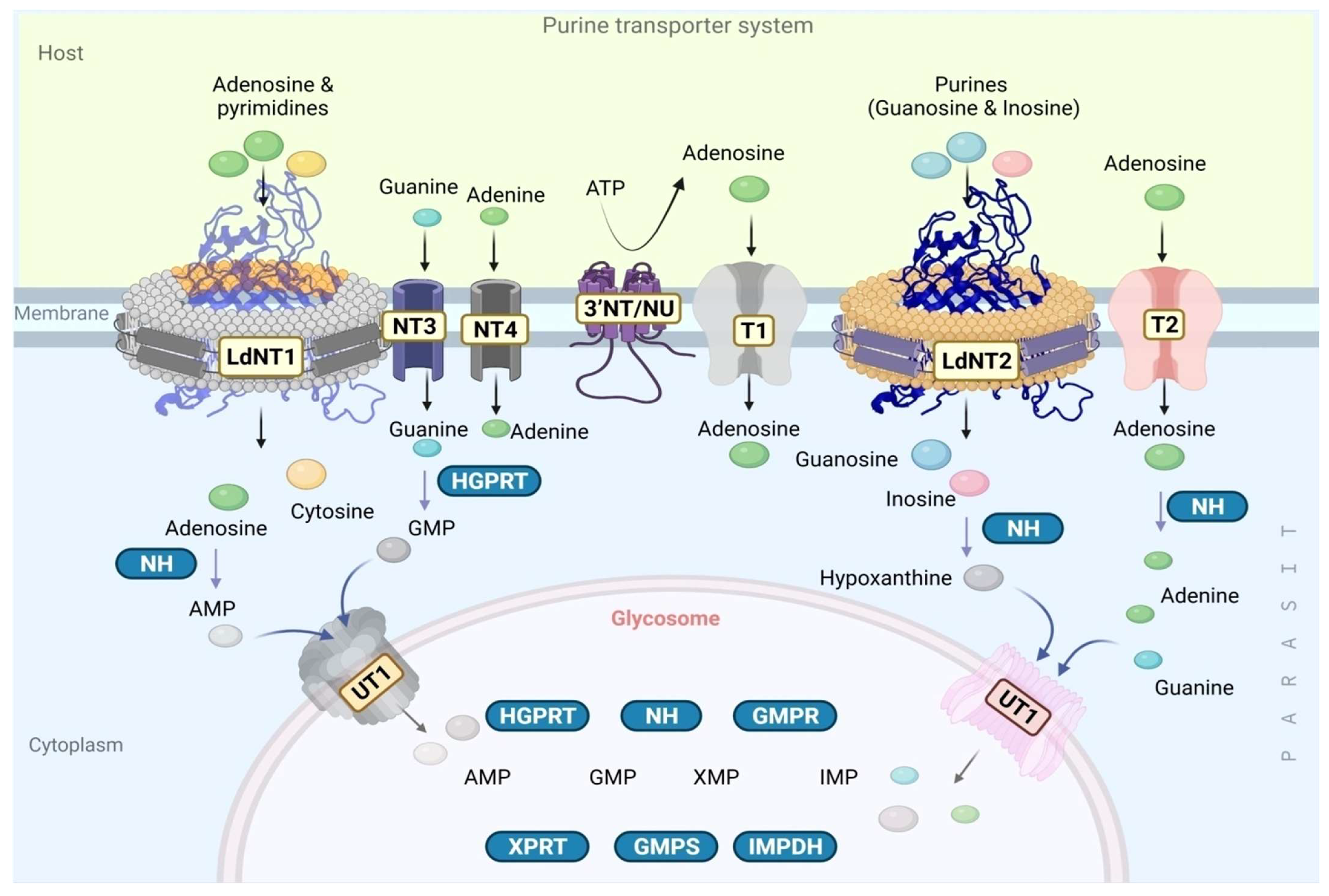
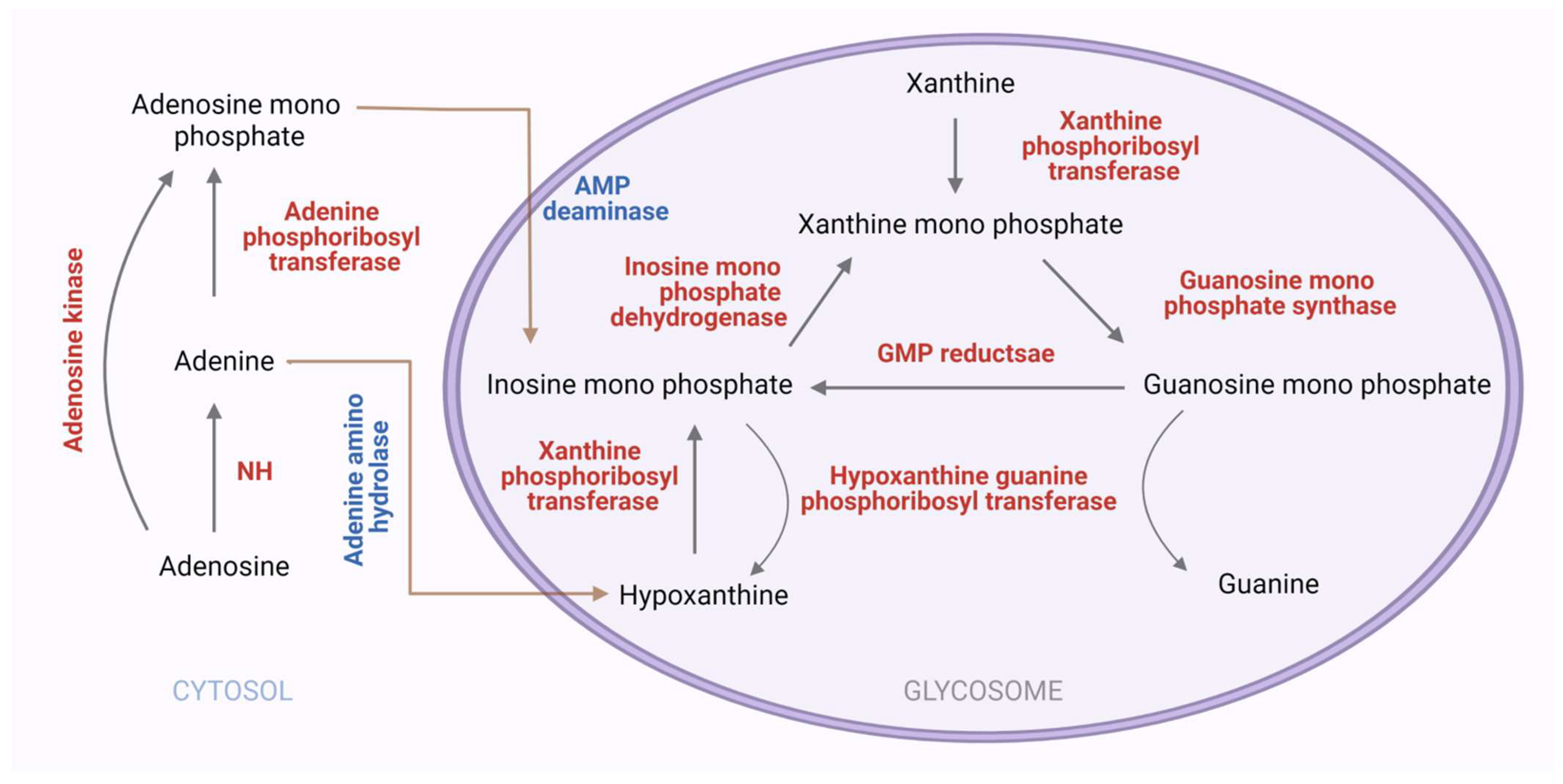
3.2. The Purine Salvage Pathway
3.2.1. Extracellular Nucleotide Metabolism in Leishmania by Ecto-Nucleotidase
3.2.2. The Purine Transporter Systems
3.2.3. Purine Salvage Enzymes
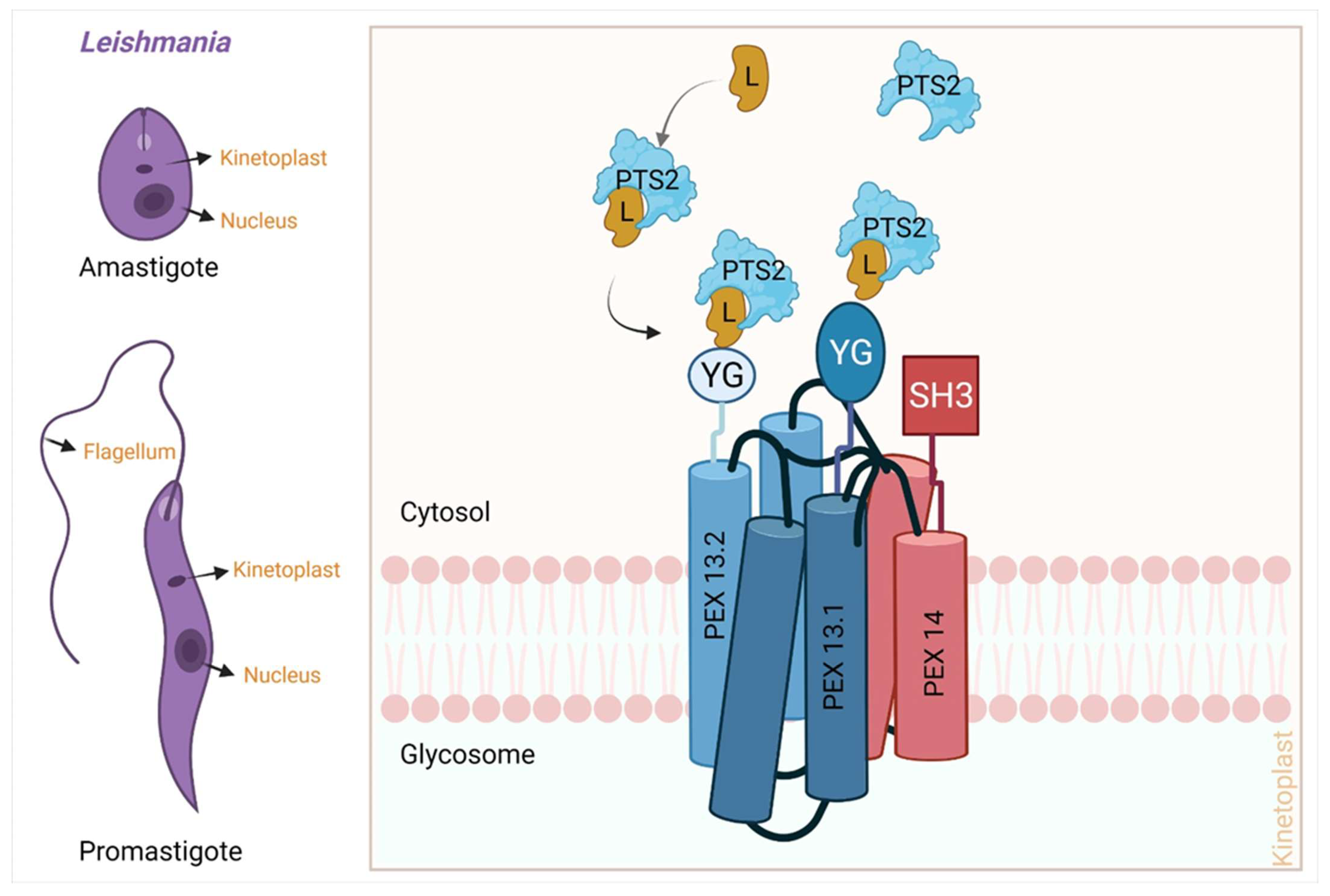
3.3. Peroxisomal Import Pathway
- (a)
- Recognition of cargo in the cytosol
- (b)
- Loading of cargo or receptor complex onto the peroxisomal membrane
- (c)
- Cargo translocation over the membrane
- (d)
- Release of cargo complex into the peroxisomal membrane
- (e)
- Receptor recycling
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Van Griensven, J.; Ritmeijer, K.; Lynen, L.; Diro, E. Visceral leishmaniasis as an AIDS defining condition: Towards consistency across WHO guidelines. PLoS Negl. Trop. Dis. 2014, 8, e2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zijlstra, E.E. Biomarkers in Post-kala-azar Dermal Leishmaniasis. Front. Cell. Infect. Microbiol. 2019, 9, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization = Organisation mondiale de la, S., Weekly Epidemiological Record 2021, 96, 35 [full issue]. Weekly Epidemiological Record = Relevé Épidémiologique Hebdomadaire 2021, 96, 401–420. Available online: https://apps.who.int/iris/handle/10665/352020 (accessed on 5 May 2022).
- Kedzierski, L.; Sakthianandeswaren, A.; Curtis, J.M.; Andrews, P.C.; Junk, P.C.; Kedzierska, K. Leishmaniasis: Current treatment and prospects for new drugs and vaccines. Curr. Med. Chem. 2009, 16, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Neto, V.V.; Cunha Junior, E.F.; Faioes, V.D.S.; Martins, T.P.; Silva, R.L.; Leon, L.L.; Santos, E.C.T. Leishmaniasis treatment: Update of possibilities for drug repurposing. Front. Biosci. 2018, 23, 967–996. [Google Scholar]
- Roatt, B.M.; de Oliveira Cardoso, J.M.; De Brito, R.C.F.; Coura-Vital, W.; de Oliveira Aguiar-Soares, R.D.; Reis, A.B. Recent advances and new strategies on leishmaniasis treatment. Appl. Microbiol. Biotechnol. 2020, 104, 8965–8977. [Google Scholar] [CrossRef]
- Chakraborty, A.K.; Majumder, H.K. Mode of action of pentavalent antimonials: Specific inhibition of type I DNA topoisomerase of Leishmania donovani. Biochem. Biophys. Res. Commun. 1988, 152, 605–611. [Google Scholar] [CrossRef]
- Marsden, P.D. The discovery of urea stibamine. Rev. Soc. Bras. Med. Trop. 1986, 19, 115. [Google Scholar] [CrossRef] [Green Version]
- Mehrizi, T.Z.; Rezayat, S.M.; Ardestani, M.S.; Shahmabadi, H.E.; Ramezani, A. A Review Study about the Effect of Chitosan Nanocarrier on Improving the Efficacy of Amphotericin B in the Treatment of Leishmania from 2010 to 2020. Curr. Drug Deliv. 2021, 18, 1234–1243. [Google Scholar] [CrossRef]
- Dutcher, J.D. The Discovery and Development of Amphotericin B. Dis. Chest 1968, 54 (Suppl. 1), 296–298. [Google Scholar] [CrossRef]
- Noyman, Y.; Levi, A.; Ben Amitai, D.; Reiss-Huss, S.; Sabbah, F.; Hodak, E.; Mimouni, T.; Friedland, R. Treating pediatric cutaneous Leishmania tropica with systemic liposomal amphotericin B: A retrospective, single-center study. Dermatol. Ther. 2022, 35, e15185. [Google Scholar] [CrossRef]
- Apa, H.; Devrim, I.; Bayram, N.; Deveci, R.; Demir-Özek, G.; Cartı, U. Liposomal amphotericin B versus pentavalent antimony salts for visceral Leishmania in children. Turk. J. Pediatr. 2013, 55, 378. [Google Scholar]
- Moen, M.D.; Lyseng-Williamson, K.A.; Scott, L.J. Liposomal amphotericin B: A review of its use as empirical therapy in febrile neutropenia and in the treatment of invasive fungal infections. Drugs 2009, 69, 361–392. [Google Scholar] [CrossRef]
- Bulté, D.; Van Bockstal, L.; Dirkx, L.; Kerkhof, M.V.D.; De Trez, C.; Timmermans, J.-P.; Hendrickx, S.; Maes, L.; Caljon, G. Miltefosine enhances infectivity of a miltefosine-resistant Leishmania infantum strain by attenuating its innate immune recognition. PLoS Neglected Trop. Dis. 2021, 15, e0009622. [Google Scholar] [CrossRef]
- Muschiola, C.; Berger, M.R.; Schuler, B.; Scherf, H.R.; Garzon, F.T.; Zeller, W.J.; Unger, C.; Eibl, H.J.; Schmähl, D. Alkyl phosphocholines: Toxicity and anticancer properties. Lipids 1987, 22, 930–934. [Google Scholar] [CrossRef]
- Coser, E.M.; Ferreira, B.A.; Branco, N.; Yamashiro-Kanashiro, E.H.; Lindoso, J.A.L.; Coelho, A.C. Activity of paromomycin against Leishmania amazonensis: Direct correlation between susceptibility in vitro and the treatment outcome in vivo. Int. J. Parasitol. Drugs Drug Resist. 2020, 14, 91–98. [Google Scholar] [CrossRef]
- Carter, C.H. Paromomycin (Humatin) in the treatment of intestinal amebiasis: A preliminary dose-range study. Antibiot. Med. Clin. Ther. 1959, 6, 586–588. [Google Scholar]
- Maia, A.C.R.G.; Porcino, G.N.; Detoni, M.L.; Quellis, L.R.; Emídio, N.B.; Marconato, D.G.; Messias, W.F.; Soldati, L.L.; Faria-Pinto, P.; Capriles, P.V.D.S.Z.; et al. Leishmania infantum amastigote nucleoside triphosphate diphosphohydrolase 1 (NTPDase 1): Its inhibition as a new insight into mode of action of pentamidine. Exp. Parasitol. 2019, 200, 1–6. [Google Scholar] [CrossRef]
- Sands, M.; Kron, M.A.; Brown, R.B. Pentamidine: A Review. Clin. Infect. Dis. 1985, 7, 625–6344. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Broni, E.; Dankwa, B.; Enninful, K.S.; Kwarko, G.B.; Darko, L.; Durvasula, R.; Kempaiah, P.; Rathi, B.; Iii, W.A.M.; et al. Outwitting an Old Neglected Nemesis: A Review on Leveraging Integrated Data-Driven Approaches to Aid in Unraveling of Leishmanicides of Therapeutic Potential. Curr. Top. Med. Chem. 2020, 20, 349–366. [Google Scholar] [CrossRef]
- Halder, A.K.; Dias Soeiro Cordeiro, M.N. Advanced in Silico Methods for the Development of Anti- Leishmaniasis and Anti-Trypanosomiasis Agents. Curr. Med. Chem. 2020, 27, 697–718. [Google Scholar] [CrossRef]
- Oliveira, L.F.; Schubach, A.O.; Martins, M.M.; Passos, S.L.; Oliveira, R.V.; Marzochi, M.C.; Andrade, C.A. Systematic review of the adverse effects of cutaneous leishmaniasis treatment in the New World. Acta Trop. 2011, 118, 87–96. [Google Scholar] [CrossRef]
- Singh, K.; Garg, G.; Ali, V. Current Therapeutics, Their Problems and Thiol Metabolism as Potential Drug Targets in Leishmaniasis. Curr. Drug Metab. 2016, 17, 897–919. [Google Scholar] [CrossRef]
- Ghorbani, M.; Farhoudi, R. Leishmaniasis in humans: Drug or vaccine therapy? Drug Des. Dev. Ther. 2018, 12, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef]
- Sheppard, D.; Lipkin, M.; Harris, C.; Catana, C.; Stouten, P. Strategies for Small Molecule Library Design. Curr. Pharm. Des. 2014, 20, 3314–3322. [Google Scholar] [CrossRef]
- Pennington, L.D.; Muegge, I. Holistic drug design for multiparameter optimization in modern small molecule drug discovery. Bioorganic Med. Chem. Lett. 2021, 41, 128003. [Google Scholar] [CrossRef]
- Sarnpitak, P.; Mujumdar, P.; Taylor, P.; Cross, M.; Coster, M.; Gorse, A.-D.; Krasavin, M.; Hofmann, A. Panel docking of small-molecule libraries—Prospects to improve efficiency of lead compound discovery. Biotechnol. Adv. 2015, 33, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Eder, J.; Sedrani, R.; Wiesmann, C. The discovery of first-in-class drugs: Origins and evolution. Nat. Rev. Drug Discov. 2014, 13, 577–587. [Google Scholar] [CrossRef]
- Srivastava, S.; Shankar, P.; Mishra, J.; Singh, S. Possibilities and challenges for developing a successful vaccine for leishmaniasis. Parasites Vectors 2016, 9, 277. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, L.L.G.; Andricopulo, A.D. Chemoinformatics Strategies for Leishmaniasis Drug Discovery. Front. Pharmacol. 2018, 9, 1278. [Google Scholar] [CrossRef] [Green Version]
- Njogu, P.; Guantai, E.M.; Pavadai, E.; Chibale, K. Computer-Aided Drug Discovery Approaches against the Tropical Infectious Diseases Malaria, Tuberculosis, Trypanosomiasis, and Leishmaniasis. ACS Infect. Dis. 2015, 2, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Rashid, U.; Sultana, R.; Shaheen, N.; Hassan, S.F.; Yaqoob, F.; Ahmad, M.J.; Iftikhar, F.; Sultana, N.; Asghar, S.; Yasinzai, M.; et al. Structure based medicinal chemistry-driven strategy to design substituted dihydropyrimidines as potential antileishmanial agents. Eur. J. Med. Chem. 2016, 115, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Casgrain, P.-A.; Martel, C.; McMaster, W.R.; Mottram, J.C.; Olivier, M.; Descoteaux, A. Cysteine Peptidase B Regulates Leishmania mexicana Virulence through the Modulation of GP63 Expression. PLoS Pathog. 2016, 12, e1005658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marreiros, B.C.; Sena, F.V.; Sousa, F.M.; Oliveira, A.S.F.; Soares, C.M.; Batista, A.P.; Pereira, M.M. Structural and Functional insights into the catalytic mechanism of the Type II NADH:quinone oxidoreductase family. Sci. Rep. 2017, 7, 42303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamidala, R.; Majumdar, P.; Jha, K.K.; Bathula, C.; Agarwal, R.; Chary, M.T.; Majumder, H.K.; Munshi, P.; Sen, S. Identification of Leishmania donovani Topoisomerase 1 inhibitors via intuitive scaffold hopping and bioisosteric modification of known Top 1 inhibitors. Sci. Rep. 2016, 6, 1–13. [Google Scholar]
- Stevanović, S.; Perdih, A.; Senćanski, M.; Glišić, S.; Duarte, M.; Tomás, A.M.; Sena, F.V.; Sousa, F.M.; Pereira, M.M.; Solmajer, T. In Silico Discovery of a Substituted 6-Methoxy-quinalidine with Leishmanicidal Activity in Leishmania infantum. Molecules 2018, 23, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Singh, N.; Agnihotri, P.; Mishra, S.; Singh, S.P.; Kolli, B.K.; Chang, K.P.; Sahasrabuddhe, A.A.; Siddiqi, M.I.; Pratap, J.V. Discovery of novel inhibitors for Leishmania nucleoside diphosphatase kinase (NDK) based on its structural and functional characterization. J. Comput. Mol. Des. 2017, 31, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.R.; Karade, S.S.; Srivastava, V.K.; Rana, A.K.; Gupta, C.; Sahasrabuddhe, A.A.; Pratap, J. Structure of Leishmania donovani coronin coiled coil domain reveals an antiparallel 4 helix bundle with inherent asymmetry. J. Struct. Biol. 2016, 195, 129–138. [Google Scholar] [CrossRef]
- Parihar, P.S.; Singh, A.; Karade, S.S.; Sahasrabuddhe, A.A.; Pratap, J.V. Structural insights into kinetoplastid coronin oligomerization domain and F-actin interaction. Curr. Res. Struct. Biol. 2021, 3, 268–276. [Google Scholar] [CrossRef]
- Nayarisseri, A.; Khandelwal, R.; Tanwar, P.; Madhavi, M.; Sharma, D.; Thakur, G.; Speck-Planche, A.; Singh, S.K. Artificial Intelligence, Big Data and Machine Learning Approaches in Precision Medicine & Drug Discovery. Curr. Drug Targets 2021, 22, 631–655. [Google Scholar] [CrossRef]
- Thakur, A.; Mishra, A.P.; Panda, B.; Rodríguez, C.S.; Gaurav, I.; Majhi, B. Application of Artificial Intelligence in Pharmaceutical and Biomedical Studies. Curr. Pharm. Des. 2020, 26, 3569–3578. [Google Scholar] [CrossRef]
- Yu, K.H.; Beam, A.L.; Kohane, I.S. Artificial intelligence in healthcare. Nat. Biomed. Eng. 2018, 2, 719–731. [Google Scholar] [CrossRef]
- Miller, D.D.; Brown, E.W. Artificial Intelligence in Medical Practice: The Question to the Answer? Am. J. Med. 2018, 131, 129–133. [Google Scholar] [CrossRef]
- Dias, R.; Torkamani, A. Artificial intelligence in clinical and genomic diagnostics. Genome Med. 2019, 11, 70. [Google Scholar] [CrossRef] [Green Version]
- Mann, M.; Kumar, C.; Zeng, W.-F.; Strauss, M.T. Artificial intelligence for proteomics and biomarker discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef]
- Biswas, N.; Chakrabarti, S. Artificial Intelligence (AI)-Based Systems Biology Approaches in Multi-Omics Data Analysis of Cancer. Front. Oncol. 2020, 10, 588221. [Google Scholar] [CrossRef]
- Xu, Y.; Su, G.-H.; Ma, D.; Xiao, Y.; Shao, Z.-M.; Jiang, Y.-Z. Technological advances in cancer immunity: From immunogenomics to single-cell analysis and artificial intelligence. Signal Transduct. Target. Ther. 2021, 6, 312. [Google Scholar] [CrossRef]
- Montesinos-López, O.A.; Montesinos-López, A.; Pérez-Rodríguez, P.; Barrón-López, J.A.; Martini, J.W.R.; Fajardo-Flores, S.B.; Gaytan-Lugo, L.S.; Santana-Mancilla, P.C.; Crossa, J. A review of deep learning applications for genomic selection. BMC Genom. 2021, 22, 19. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Weskamp, N.; Schneider, G. Artificial intelligence in drug discovery: Recent advances and future perspectives. Expert Opin. Drug Discov. 2021, 16, 949–959. [Google Scholar] [CrossRef]
- Hessler, G.; Baringhaus, K.-H. Artificial Intelligence in Drug Design. Molecules 2018, 23, 2520. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, K.; Siddiqi, M.I. Recent trends in artificial intelligence-driven identification and development of anti-neurodegenerative therapeutic agents. Mol. Divers. 2021, 25, 1517–1539. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2020, 26, 80–93. [Google Scholar] [CrossRef]
- Wei, Y.; Zhou, J.; Wang, Y.; Liu, Y.; Liu, Q.; Luo, J.; Wang, C.; Ren, F.; Huang, L. A Review of Algorithm & Hardware Design for AI-Based Biomedical Applications. IEEE Trans. Biomed. Circuits Syst. 2020, 14, 145–163. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Zare, M.; Akbarialiabad, H.; Parsaei, H.; Asgari, Q.; Alinejad, A.; Bahreini, M.S.; Hosseini, S.H.; Ghofrani-Jahromi, M.; Shahriarirad, R.; Amirmoezzi, Y.; et al. A machine learning-based system for detecting leishmaniasis in microscopic images. BMC Infect. Dis. 2022, 22, 48. [Google Scholar] [CrossRef]
- Concu, R.; Dea-Ayuela, M.; Perez-Montoto, L.; Prado-Prado, F.; Uriarte, E.; Bolás-Fernández, F.; Podda, G.; Pazos, A.; Munteanu, C.; Ubeira, F.; et al. 3D entropy and moments prediction of enzyme classes and experimental-theoretic study of peptide fingerprints in Leishmania parasites. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2009, 1794, 1784–1794. [Google Scholar] [CrossRef]
- Jamal, S.; Scaria, V. Cheminformatic models based on machine learning for pyruvate kinase inhibitors of Leishmania mexicana. BMC Bioinform. 2013, 14, 329. [Google Scholar] [CrossRef] [Green Version]
- Bamorovat, M.; Sharifi, I.; Rashedi, E.; Shafiian, A.; Sharifi, F.; Khosravi, A.; Tahmouresi, A. A novel diagnostic and prognostic approach for unresponsive patients with anthroponotic cutaneous leishmaniasis using artificial neural networks. PLoS ONE 2021, 16, e0250904. [Google Scholar] [CrossRef]
- Herrera-Acevedo, C.; Perdomo-Madrigal, C.; Muratov, E.N.; Scotti, L.; Scotti, M.T. Discovery of Alternative Chemotherapy Options for Leishmaniasis through Computational Studies of Asteraceae. ChemMedChem 2020, 16, 1234–1245. [Google Scholar] [CrossRef]
- Raj, S.; Sasidharan, S.; Balaji, S.N.; Saudagar, P. An overview of biochemically characterized drug targets in metabolic pathways of Leishmania parasite. Parasitol. Res. 2020, 119, 2025–2037. [Google Scholar] [CrossRef]
- Dias, M.V.B.; Santos, J.C.; Libreros-Zúñiga, G.A.; Ribeiro, J.A.; Chavez-Pacheco, S.M. Folate biosynthesis pathway: Mechanisms and insights into drug design for infectious diseases. Future Med. Chem. 2018, 10, 935–959. [Google Scholar] [CrossRef] [PubMed]
- Elizabeth, K.E. Folate: Its Biological Interactions and Strategies to Achieve Sufficiency without Causing Excess. Am. J. Public Health 2021, 111, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Hu, X.; Wu, S.; Xiao, H.; Cai, H.; Xie, Z.; Huang, Y.; Jing, X. Biological Characterization of Folate-Decorated Biodegradable Polymer–Platinum (II) Complex Micelles. Mol. Pharm. 2012, 9, 3200–3208. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Sung, L.; Hegemann, J.D.; Chu, J. Disrupting Transcription and Folate Biosynthesis Leads to Synergistic Suppression of Escherichia coli Growth. ChemMedChem 2022, 17, e202200075. [Google Scholar] [CrossRef]
- Gorelova, V.; Bastien, O.; De Clerck, O.; Lespinats, S.; Rébeillé, F.; Van Der Straeten, D. Evolution of folate biosynthesis and metabolism across algae and land plant lineages. Sci. Rep. 2019, 9, 5731. [Google Scholar] [CrossRef] [Green Version]
- Kok, D.E.; Steegenga, W.T.; McKay, J.A. Folate and epigenetics: Why we should not forget bacterial biosynthesis. Epigenomics 2018, 10, 1147–1150. [Google Scholar] [CrossRef] [Green Version]
- El Fadili, A.; Kündig, C.; Roy, G.; Ouellette, M. Inactivation of the Leishmania tarentolae Pterin Transporter (BT1) and Reductase (PTR1) Genes Leads to Viable Parasites with Changes in Folate Metabolism and Hypersensitivity to the Antifolate Methotrexate. J. Biol. Chem. 2004, 279, 18575–18582. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Leblanc, É.; Chang, C.F.; Papadopoulou, B.; Bray, T.; Whiteley, J.M.; Lin, S.X.; Ouellette, M. Pterin and Folate Reduction by theLeishmania tarentolaeH Locus Short-Chain Dehydrogenase/Reductase PTR1. Arch. Biochem. Biophys. 1997, 342, 197–202. [Google Scholar] [CrossRef]
- Papadopoulou, B.; Roy, G.; Mourad, W.; Leblanc, E.; Ouellette, M. Changes in folate and pterin metabolism after disruption of the Leishmania H locus short chain dehydrogenase gene. J. Biol. Chem. 1994, 269, 7310–7315. [Google Scholar] [CrossRef]
- Ellenberger, T.E.; Beverley, S.M. Biochemistry and regulation of folate and methotrexate transport in Leishmania major. J. Biol. Chem. 1987, 262, 10053–10058. [Google Scholar] [CrossRef]
- Scott, D.A.; Coombs, G.H.; Sanderson, B.E. Folate utilisation by Leishmania species and the identification of intracellular derivatives and folate-metabolising enzymes. Mol. Biochem. Parasitol. 1987, 23, 139–149. [Google Scholar] [CrossRef]
- Vickers, T.J.; Beverley, S.M. Folate metabolic pathways in Leishmania. Essays Biochem. 2011, 51, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Hardy, L.W.; Matthews, W.; Nare, B.; Beverley, S.M. Biochemical and Genetic Tests for Inhibitors of Leishmania Pteridine Pathways. Exp. Parasitol. 1997, 87, 158–170. [Google Scholar] [CrossRef]
- Vadloori, B.; Sharath, A.K.; Prabhu, N.P.; Maurya, R. Homology modelling, molecular docking, and molecular dynamics simulations reveal the inhibition of Leishmania donovani dihydrofolate reductase-thymidylate synthase enzyme by Withaferin-A. BMC Res. Notes 2018, 11, 246. [Google Scholar] [CrossRef] [Green Version]
- Kimuda, M.P.; Laming, D.; Hoppe, H.C.; Bishop, T. Identification of Novel Potential Inhibitors of Pteridine Reductase 1 in Trypanosoma brucei via Computational Structure-Based Approaches and in Vitro Inhibition Assays. Molecules 2019, 24, 142. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Bray, T.; Ouellette, M.; Zhao, M.; Ferre, R.A.; Matthews, D.; Whiteley, J.M.; Varughese, K.I. Structure of pteridine reductase (PTR1) from Leishmania tarentolae. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 1539–1544. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, R.; Chen, Y.-P.P. Probing the structure of Leishmania major DHFR TS and structure based virtual screening of peptide library for the identification of anti-leishmanial leads. J. Mol. Model. 2012, 18, 4089–4100. [Google Scholar] [CrossRef]
- Maganti, L.; Manoharan, P.; Ghoshal, N. Probing the structure of Leishmania donovani chagasi DHFR-TS: Comparative protein modeling and protein–ligand interaction studies. J. Mol. Model. 2010, 16, 1539–1547. [Google Scholar] [CrossRef]
- Gueiros-Filho, F.J.; Beverley, S.M. Selection against the dihydrofolate reductase-thymidylate synthase (DHFR-TS) locus as a probe of genetic alterations in Leishmania major. Mol. Cell. Biol. 1996, 16, 5655–5663. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.; Aloso, G.; Langer, P.J.; Beverley, S.M. Sequence of the dihydrofolate reductase-thymidylate synthase (DHFR-TS) gene of Leishmania amazonensis. Nucleic Acids Res. 1990, 18, 2819. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, I.H. Inhibitors of dihydrofolate reductase in leishmania and trypanosomes. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2002, 1587, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, B.V.F.; Teles, A.L.B.; da Silva, S.G.; Brito, C.C.B.; de Freitas, H.F.; Pires, A.B.L.; Froes, T.Q.; Castilho, M.S. Dual and selective inhibitors of pteridine reductase 1 (PTR1) and dihydrofolate reductase-thymidylate synthase (DHFR-TS) from Leishmania chagasi. J. Enzym. Inhib. Med. Chem. 2019, 34, 1439–1450. [Google Scholar] [CrossRef] [Green Version]
- Veras, P.; Brodskyn, C.; Balestieri, F.; De Freitas, L.; Ramos, A.; Queiroz, A.; Barral, A.; Beverley, S.; Barral-Netto, M. A dhfr-ts- Leishmania major Knockout Mutant Cross-protects against Leishmania amazonensis. Mem. Inst. Oswaldo Cruz 1999, 94, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, M.V.D.V.; Van der Veken, P.; Goeminne, A.; Haemers, A.; Augustyns, K. Inhibitors of the Purine Salvage Pathway: A Valuable Approach for Antiprotozoal Chemotherapy? Curr. Med. Chem. 2010, 17, 2456–2481. [Google Scholar] [CrossRef]
- Freitas-Mesquita, A.L.; Meyer-Fernandes, J.R. Ecto-nucleotidases and Ecto-phosphatases from Leishmania and Trypanosoma Parasites. Subcell Biochem. 2013, 74, 217–252. [Google Scholar] [CrossRef]
- Da Silva, W.; da Rocha Torres, N.; de Melo Agripino, J.; da Silva, V.H.; de Souza, A.C.A.; Ribeiro, I.C.; de Oliveira, T.A.; de Souza, L.A.; Andrade, L.K.R.; de Moraes, J.V.B.; et al. ENTPDases from Pathogenic Trypanosomatids and Purinergic Signaling: Shedding Light towards Biotechnological Applications. Curr. Top. Med. Chem. 2021, 21, 213–226. [Google Scholar]
- Gbenle, G.O.; Dwyer, D.M. Purification and properties of 3′-nucleotidase of Leishmania donovani. Biochem. J. 1992, 285, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Debrabant, A.; Ghedin, E.; Dwyer, D.M. Dissection of the functional domains of the Leishmania surface membrane 3’-nucleotidase/nuclease, a unique member of the class I nuclease family. J. Biol. Chem. 2000, 275, 16366–16372. [Google Scholar] [CrossRef] [Green Version]
- Debrabant, A.; Gottlieb, M.; Dwyer, D.M. Isolation and characterization of the gene encoding the surface membrane 3′-nucleotidase/nuclease of Leishmania donovani. Mol. Biochem. Parasitol. 1995, 71, 51–63. [Google Scholar] [CrossRef]
- Boitz, J.M.; Ullman, B.; Jardim, A.; Carter, N.S. Purine salvage in Leishmania: Complex or simple by design? Trends Parasitol. 2012, 28, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Fulwiler, A.L.; Boitz, J.M.; Yates, P.A.; Carter, N.S.; Ullman, B. Characterization of amplicons that suppress the conditional lethal growth phenotype of a Leishmania donovani mutant lacking normal purine salvage mechanisms. Mol. Biochem. Parasitol. 2011, 175, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Boitz, J.M.; Ullman, B. A Conditional Mutant Deficient in Hypoxanthine-guanine Phosphoribosyltransferase and Xanthine Phosphoribosyltransferase Validates the Purine Salvage Pathway of Leishmania donovani. J. Biol. Chem. 2006, 281, 16084–16089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Rajasekariah, G.; Martin, S.K. A nonspecific nucleoside hydrolase from Leishmania donovani: Implications for purine salvage by the parasite. Gene 2001, 280, 153–162. [Google Scholar] [CrossRef]
- Krenitsky, T.A.; Miller, R.L. Purine Salvage Enzymes in Leishmania Donovani and Trichomonas Vaginalis. In Purine Metabolism in Man-IV; Springer: Boston, MA, USA, 1984; pp. 215–218. [Google Scholar] [CrossRef]
- Davies, M.J.; Ross, A.M.; Gutteridge, W.E. The enzymes of purine salvage in Trypanosoma cruzi, Trypanosoma brucei and Leishmania mexicana. Parasitology 1983, 87, 211–217. [Google Scholar] [CrossRef]
- LaFon, S.W.; Nelson, D.J.; Berens, R.L.; Marr, J. Purine and pyrimidine salvage pathways in Leishmania donovani. Biochem. Pharmacol. 1982, 31, 231–238. [Google Scholar] [CrossRef]
- Krenitsky, T.A.; Koszalka, G.W.; Tuttle, J.V.; Adamczyk, D.L.; Elion, G.B.; Marr, J.J. Purine Salvage Enzymes in Man and Leishmania donovani. In Purine Metabolism in Man-III; Springer: Boston, MA, USA, 1980; pp. 51–56. [Google Scholar] [CrossRef]
- Landfear, S.M.; Ullman, B.; Carter, N.S.; Sanchez, M.A. Nucleoside and Nucleobase Transporters in Parasitic Protozoa. Eukaryot. Cell 2004, 3, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Carter, N.S.; Landfear, S.; Ullman, B. Nucleoside transporters of parasitic protozoa. Trends Parasitol. 2001, 17, 142–145. [Google Scholar] [CrossRef]
- Phillips, C.L.; Ullman, B.; Brennan, R.G.; Hill, C.P. Crystal structures of adenine phosphoribosyltransferase from Leishmania donovani. EMBO J. 1999, 18, 3533–3545. [Google Scholar] [CrossRef] [Green Version]
- Parihar, P.S.; Pratap, J.V. The L. donovani Hypoxanthine-guanine phosphoribosyl transferase (HGPRT) oligomer is distinct from the human homolog. Biochem. Biophys. Res. Commun. 2020, 532, 499–504. [Google Scholar] [CrossRef]
- Patel, B.; Patel, D.; Parmar, K.; Chauhan, R.; Singh, D.D.; Pappachan, A.L. donovani XPRT: Molecular characterization and evaluation of inhibitors. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2017, 1866, 426–441. [Google Scholar] [CrossRef]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef] [Green Version]
- Shimozawa, N.; Suzuki, Y.; Tomatsu, S.; Nakamura, H.; Kono, T.; Takada, H.; Tsukamoto, T.; Fujiki, Y.; Orii, T.; Kondo, N. A novel mutation, R125X in peroxisome assembly factor-1 responsible for zellweger syndrome. Hum. Mutat. 1998, 11, S134–S136. [Google Scholar] [CrossRef]
- Masuno, M.; Shimozawa, N.; Suzuki, Y.; Kondo, N.; Orii, T.; Tsukamoto, T.; Osumi, T.; Fujiki, Y.; Imaizumi, K.; Kuroki, Y. Assignment of the Human Peroxisome Assembly Factor-1 Gene (PXMP3) Responsible for Zellweger Syndrome to Chromosome 8q21.1 by Fluorescence in Situ Hybridization. Genomics 1994, 20, 141–142. [Google Scholar] [CrossRef]
- Santos, M.J.; Hoefler, S.; Moser, A.B.; Moser, H.W.; Lazarow, P.B. Peroxisome assembly mutations in humans: Structural heterogeneity in Zellweger syndrome. J. Cell. Physiol. 1992, 151, 103–112. [Google Scholar] [CrossRef]
- Shimozawa, N.; Tsukamoto, T.; Suzuki, Y.; Orii, T.; Shirayoshi, Y.; Mori, T.; Fujiki, Y. A Human Gene Responsible for Zellweger Syndrome That Affects Peroxisome Assembly. Science 1992, 255, 1132–1134. [Google Scholar] [CrossRef]
- Schmitt, K.; Molzer, B.; Stöckler, S.; Tulzer, G.; Tulzer, W. Zellweger syndrome, neonatal adrenoleukodystrophy or infantile Refsum’s disease in a case with generalized peroxisome defect? Wien Klin Wochenschr. 1993, 105, 320–322. [Google Scholar]
- Van Maldergem, L.; Moser, A.B.; Vincent, M.-F.; Roland, D.; Reding, R.; Otte, J.-B.; Wanders, R.J.; Sokal, E. Orthotopic liver transplantation from a living-related donor in an infant with a peroxisome biogenesis defect of the infantile Refsum disease type. J. Inherit. Metab. Dis. 2005, 28, 593–600. [Google Scholar] [CrossRef]
- Mandel, H.; Berant, M.; Meiron, D.; Aizin, A.; Oiknine, J.; Brook, J.G.; Aviram, M. Plasma lipoproteins and monocyte-macrophages in a peroxisome-deficient system: Study of a patient with infantile refsum disease. J. Inherit. Metab. Dis. 1992, 15, 774–784. [Google Scholar] [CrossRef]
- Opperdoes, F. The glycosome of trypanosomes and Leishmania. Biochem. Soc. Trans. 1990, 18, 729–731. [Google Scholar] [CrossRef]
- Shih, S.; Hwang, H.-Y.; Carter, D.; Stenberg, P.; Ullman, B. Localization and Targeting of the Leishmania donovaniHypoxanthine-Guanine Phosphoribosyltransferase to the Glycosome. J. Biol. Chem. 1998, 273, 1534–1541. [Google Scholar] [CrossRef] [Green Version]
- Demaret, T.; Courtoy, G.E.; Ravau, J.; Van Der Smissen, P.; Najimi, M.; Sokal, E.M. Accurate and live peroxisome biogenesis evaluation achieved by lentiviral expression of a green fluorescent protein fused to a peroxisome targeting signal 1. Histochem. Cell Biol. 2020, 153, 295–306. [Google Scholar] [CrossRef]
- Jansen, R.L.M.; van der Klei, I.J. The peroxisome biogenesis factors Pex3 and Pex19: Multitasking proteins with disputed functions. FEBS Lett. 2019, 593, 457–474. [Google Scholar] [CrossRef] [Green Version]
- Farre, J.C.; Mahalingam, S.S.; Proietto, M.; Subramani, S. Peroxisome biogenesis, membrane contact sites, and quality control. EMBO Rep. 2019, 20, e46864. [Google Scholar] [CrossRef]
- Lutje, V.; Probyn, K.; Seixas, J.; Bergman, H.; Villanueva, G. Chemotherapy for second-stage human African trypanosomiasis: Drugs in use. Cochrane Database Syst. Rev. 2021, 12, CD015374. [Google Scholar] [CrossRef]
- Banerjee, H.; LaPointe, P.; Eitzen, G.; Rachubinski, R.A. A Small Molecule Inhibitor of Pex3–Pex19 Interaction Disrupts Glycosome Biogenesis and Causes Lethality in Trypanosoma brucei. Front. Cell Dev. Biol. 2021, 9, 1403–1411. [Google Scholar] [CrossRef]
- Jardim, A.; Rager, N.; Liu, W.; Ullman, B. Peroxisomal targeting protein 14 (PEX14) from Leishmania donovani Molecular, biochemical, and immunocytochemical characterization. Mol. Biochem. Parasitol. 2002, 124, 51–62. [Google Scholar] [CrossRef]
- Cyr, N.; Smith, T.K.; Boisselier, É.; Leroux, L.P.; Kottarampatel, A.H.; Davidsen, A.; Salesse, C.; Jardim, A. The hydrophobic region of the Leishmania peroxin 14: Requirements for association with a glycosome mimetic membrane. Biochem. J. 2018, 475, 511–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakya, A.K.; Pratap, J.V. The coiled-coil domain of glycosomal membrane-associated Leishmania donovani PEX14: Cloning, overexpression, purification and preliminary crystallographic analysis. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2020, 76, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Aggarwal, G.; Berriman, M.; Sisk, E.; Rajandream, M.-A.; Adlem, E.; Aert, R.; et al. The Genome of the Kinetoplastid Parasite, Leishmania Major. Science 2005, 309, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Discovery | Mode of Action | Side Effect | Remark |
|---|---|---|---|---|
| Pentavalent antimonial [7] | Brahmachari, 1940 [8] | Proposed: Trypanothione Reductase and macromolecule biosynthesis inhibitor | Cardiotoxicity, injection pains | Development of drug resistance |
| Amphotericin B (1992) [9] | Repurposed antifungals for neutropenic patients [10]. | Cell membrane permeabilization via a complex with sterols and ergosterols | Renal toxicity, hypokalemia | Dose-limiting |
| Liposomal amphotericinB (2009) [11,12] | Used in antifungal infections, e.g., Cryptococcal meningitis [13] | Targeted drug delivery to infected macrophages to kill amastigotes therein | Minor fever, chills, arthralgia, and rarely renal toxicity | Expensive cold-chain storage required |
| Miltefosine (2002) [14] | Repurposed anticancer drug 1980s [15]. | Modulate cell surface receptors and inositol metabolism, apoptotic cell death, cytochrome C oxidase inhibitor | Teratogenicity, GI, and hepato–renal toxicity | Expensive |
| Paromomycin (1960) [16] | Used to treat certain intestinal parasites [17] | An aminoglycoside binds to 30S ribosomal subunit, inhibiting protein biosynthesis, decreasing membrane potential | Nephro- and ototoxicity, reversible high tone audiometric shift | Expensive |
| Pentamidine (1940) [18] | Repurposed from anti trypanosomiasis [19] | Mitochondrial topoisomerase II and transcription inhibitor | Gastrointestinal disorder, hypotension, diabetes mellitus | Expensive |
| Protein or Enzyme | Comparative Analysis of Leishmania Andhuman Homologs | Specific Inhibitors: Discovery and Approach | |||
|---|---|---|---|---|---|
| Sequence Identity (%) Leishmania vs. Human | Structural Classification @ | Leishmania | Human | ||
| Leishmania | Human | ||||
| DHFR-TS (Dihydro folate reductase-thymidylate synthase) L. donovani, L. infantum, L. amazonensis, L. major | Dihydro folate reductase (60–61.2%) Thymidylate synthase (60–61%) | N/A | Reductase a Transferase b | Structure-based prediction only: Withaferin-A (withanolide) which binds to the active sites of human DHFR and human TS, but binds to a site other than an active site in L. donovani DHFR-TS [75] | Activity based-Withaferin-A discovery against DHFR and TS enzymes |
| PTR1 (Pteridine reductase) L. major | L-xylulosereductase (36.8%) | Oxidoreductase c | N/A | 1-Structure-based: benzimidazole/benzoxazole derivatives [61] 2-Structure-based: RUBi004, RUBi007, RUBi014, RUBi018 [76] | N/A |
| PTR1 (Pteridine reductase) L. donovani | Dehydrogenase and reductase SDR family member 4 -like 2 (48.8%) | Oxidoreductase d | N/A | N/A | N/A |
| PTR1 (Pteridine reductase) L. tarentole | 3-oxoacyl-[acyl-carrier-protein] reductase (37.7%) | Oxidoreductase e | N/A | N/A | N/A |
| Protein or Enzyme | Comparative Analysis from Leishmania and Human Homologs | Specific Inhibitors: Discovery and Approach | |||
|---|---|---|---|---|---|
| Sequence Identity (%) Leishmania vs. Human | Structural Classification @ | Leishmania | Humans | ||
| Leishmania | Human | ||||
| NT4 (Nucleobasetransporter 4) | Nucleobase transporter 4 (30.2%) | N/A | N/A | N/A | N/A |
| APRT (Adenine phosphor ribosyltransferase) | Adenine phosphor-ribosyltransferase (35.12%) | Transferase a | Transferase b | N/A | Structure-based: iminoaltritolbis-phosphates (L-DIAB and D-DIAB) in Plasmodium falciparum [26] |
| HGPRT (Hypoxanthine phosphoribosyltransferase) | Hypoxanthine-guanine phosphoribosyltransferase (34%) | Transferase c | Transferase d | N/A | Structure-based: Immucillin GP in humans [27] |
| XPRT (Xanthine phosphoribosyltransferase) | Xanthine phosphoribosyltransferase (33.8%) | N/A (only modeled with hHGPRT) | N/A | Structure-based: Ld-XPRT inhibitors (dGDP and cGMP) [28] | N/A |
| AAH (Adenine aminohydrolase) | Adenosine deaminase (23.3%) | N/A | Aminohydrolase e | N/A | N/A |
| GDA (Guanine deaminase) | Guanine deaminase (34.9%) | N/A | Aminohydrolase f | Enzyme kinetics-based: N6-methyladenine (6-methylaminopurine [6-MA] [29] | N/A |
| GMPS (Guanosine mono phosphate synthase) | GMP synthase (48.1%) | N/A | Ligase g | N/A | N/A |
| (AK) Adenosine Kinase | Adenosine Kinase (36.7–41.4%) | N/A | Kinase h | N/A | N/A |
| ADSS (Adenylosuccinate Synthetase) | Adenylosuccinate Synthetase (28.5–32.4%) | Ligase i | Ligase j | N/A | N/A |
| ASL (Adenylosuccinate Lyase) | Adenylosuccinate Lyase (25.3%) | Lyase k | N/A | N/A | N/A |
| AMPDA (Adenosine mono Phosphate deaminase) | AMP deaminase (47.5%) | N/A | Hydrolase l | N/A | N/A |
| Protein and Enzyme | Comparative Analysis of Leishmania and Human Homologs | Specific Inhibitors: Discovery and Approach | |||
|---|---|---|---|---|---|
| Sequence Identity (%) Leishmania vs. Human | Structural Classification @ | Leishmania | Human | ||
| Leishmania | Human | ||||
| PEX3 (Peroxisomal targeting signal-1 receptor 3) [114,115,116] | A-kinase anchor protein 9 (24.1%) | N/A | N/A | N/A | N/A |
| PEX5 (Peroxisomal biogenesis factor 5) [114,115,116] | Peroxisomal targeting signal 1 receptor (26–31%) | N/A | Peroxins a | N/A | N/A |
| PEX11 (Peroxisomal biogenesis factor 11) [114,115,116] | Probable UDP-sugar transporter protein SLC35A5 (34.1%) | N/A | N/A | N/A | N/A |
| PEX13 (Peroxisomal biogenesis factor 13) [114,115,116] | Peroxisomal membrane protein PEX13 (43.7%) | N/A | Peroxins b | N/A | N/A |
| PEX14 (Peroxisomal membrane protein) PEX14 [114,115,116] | Peroxisomal membrane protein PEX14 (43.1%) | N/A | Peroxins c | N/A | N/A |
| PEX-7 (Peroxisomal biogenesis factor 7) [114,115,116] | Peroxisomal biogenesis factor 7 PEX7 (30.21%) | N/A | N/A | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soni, M.; Pratap, J.V. Development of Novel Anti-Leishmanials: The Case for Structure-Based Approaches. Pathogens 2022, 11, 950. https://doi.org/10.3390/pathogens11080950
Soni M, Pratap JV. Development of Novel Anti-Leishmanials: The Case for Structure-Based Approaches. Pathogens. 2022; 11(8):950. https://doi.org/10.3390/pathogens11080950
Chicago/Turabian StyleSoni, Mohini, and J. Venkatesh Pratap. 2022. "Development of Novel Anti-Leishmanials: The Case for Structure-Based Approaches" Pathogens 11, no. 8: 950. https://doi.org/10.3390/pathogens11080950
APA StyleSoni, M., & Pratap, J. V. (2022). Development of Novel Anti-Leishmanials: The Case for Structure-Based Approaches. Pathogens, 11(8), 950. https://doi.org/10.3390/pathogens11080950